

161
( https://spnavigator.ru/document/a0de0447-ebec-49e0-8079-8318f0749098)
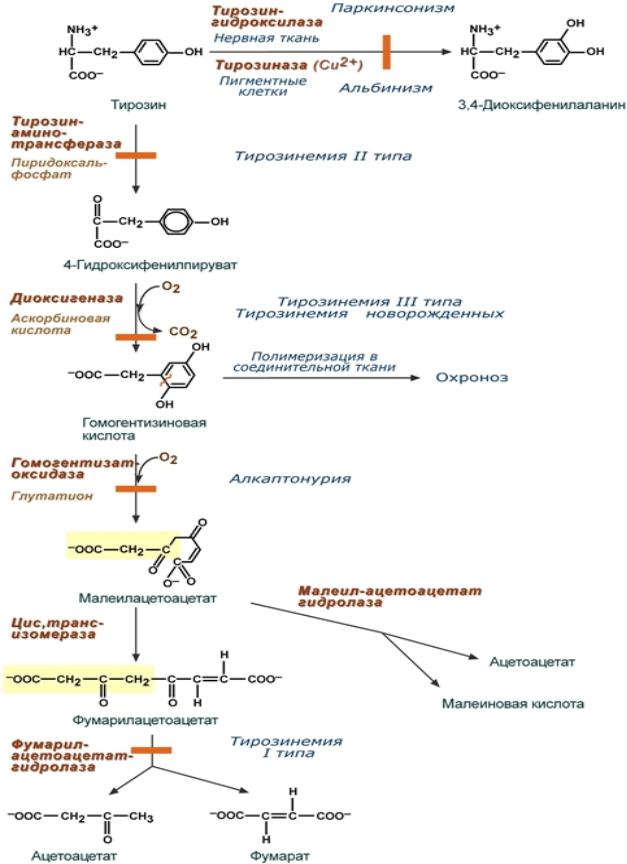
Первый вектор: мутагенность. Фумарилацетоацетат ингибирует ДНК – гликозилазы эукариотических клеток, т.е. ингибирует ферменты эксцизионной репарации ДНК. Другим механизмом является истощение запасов глутатиона в клетках, что также вносит вклад в развитие повреждений ДНК (глутатион является компонентом антиоксидантной системы и защищает клетки от окислительного стресса и свободно - радикальных реакций). Третьим фактором является усиление активности ERK – киназы (extracellular signal kinase), которая опосредует пролиферацию и движение клеток путём эпигенетических изменений (регуляции транскрипции соответствующих генов).
162
( https://spnavigator.ru/document/a0de0447-ebec-49e0-8079-8318f0749098 )
Всё это является факторами развития гепатоцеллюлярной карциномы.
Существуют описания развития данного вида рака у детей 5 – летнего возраста. В
других случаях, накопление фумарилацетоацетата приводит к грубым структурным изменениям в печени с исходом в цирроз печени.
Другой вектор связан с накоплением сукцинилацетона. Сукцинилацетон является антагонистам гамма – левулиновой кислоты, которая является предшественником порфиринов и ключевым метаболитом синтеза гема.
В результате снижается уровень гема, необходимого для синтеза цитохрома/гемопротеинов, и увеличивает уровень железа в митохондриях с развитием митохондриальной токсичности. Само отсутствие гема является достаточно серьёзным испытанием для клеток, а накопление высокого количества левулиновой кислоты является ещё одним ударом для нервной системы, т.к. в
больших количествах проявляет нейротоксичность.
Примечательно и то, что сукцинилоацетон подвергается выведению через почки.
Ешё более примечательно, что подвергаясь реабсорбции в проксимальных канальцах, сукцинилоацетат вызывает их дисфункцию с последующим формированием гипофосфатемического рахита и иных вариаций тубулярной патологии.
163
Накопление токсических метаболитов не может не сказаться на процессах развития нервной системы. Как и в случае нарушений обмена мочевины, здесь мы увидим явления угнетения нервной системы. Во многом это связано с тем, что токсичные метаболиты конкурентно ингибируют транспортные системы клеток,
которые предназначены для переноса аминокислот. Заболевание дебютирует рано,
в виде печёночной недостаточности (нарушения гемостаза, гипогликемия.
гепатоспленомегалией, повышением ферментов и билирубина) и рахитоподобных изменений скелета в первые месяцы жизни (синдром Фанкони).
11.2.2. Тирозинемия 2 типа характеризуется менее злокачественным течением.
Она связана с нарушением реакции, катализируемой тирозин – трансферазой и накоплением непосредственно тирозина. Тирозинемия 2 типа получила название синдрома Ригнера – Ханхарта, и характеризуется сочетанием энцефалопатии,
умственной отсталости, фотофобии, язвенного кератита, кератоза ладоней и стоп.
Поражение нервной системы при данном типе в плане патогенеза не до конца изученоникем,патогенезязвенныхпораженийроговицыпокатоженесовсемясен.
Однако, интересным представляется кератоз. Тирозин составляет примерно 25%
состава кератиновых белков. В условиях тирозинемии продукция этих белков увеличивается, что лежит в основе кератоза.
11.2.3. Тирозинемия 3 – ого типа. Выделяют тирозинемию 3 - ого типа,
связанного с более «проксимальным» и менее фатальным нарушением обмена тирозина. Речь идёт о структурных изменениях фермента гидроксифенил-
пируват диоксигеназы. Данное заболевание также, как и другие типы тирозинемии, характеризуется поражением нервной системы, если не соблюдать диету. Как показывает жизнь, это не самый плохой вариант.
164
( https://biokhimija.ru/narushenie-aminokislot/tirozin.html )
165
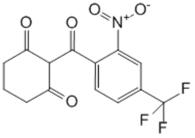
11.2.4. Что по лечению?
В отношении консервативной терапии детей с тирозинозом предложено использование нитизинона. Этот препарат является спасением для детей с тирозинемией 1 – ого типа, так как при применении нитизинона происходит блок на уровне гидроксифенил-пируват диоксигеназы. По большому счёту, мы переводим тирощинемию 1 –ого типа в тирозинемию 3 – ого типа. Профит заключается в прекращении синтеза токсичных метаболитов (малеилацетоацетата,
фумарилацетоацетата, сукцинилацетоацетата и сукцинилацетона). Это,
безусловно, снижает выраженность заболевания и повышает эффективность обычных диетических мероприятий.
(нитизинон)
11. 3. Нарушение метаболизма аминокислот с разветвлённой цепью.
Существуют аминокислоты, способные доставить проблем уже одной схемой своего метаболизма, непостижимого для заучивания. Речь идёт об аминокислотах с разветвлённой цепью, имеющих в своей структуре гидрофобные компоненты – о
лейцине, валине и изолейцине, а также их «ниже лежащих» метаболитов. Сами по себе аминокислоты являются алифатическими и незаменимыми. В силу наличия метильных групп, данные аминокислоты, входя в состав протеиновых цепей,
обеспечивают поддержание в белковых молекулах т.к.н. гидрофобных связей,
обеспечивающих дополнительную стабилизацию нативной (третичной +)
структуры белка.
166
https://biokhimija.ru/narushenie-aminokislot/lejcin-izolejcin.html
167
11.3.1. Лейциноз. (болезнь кленового сиропа)
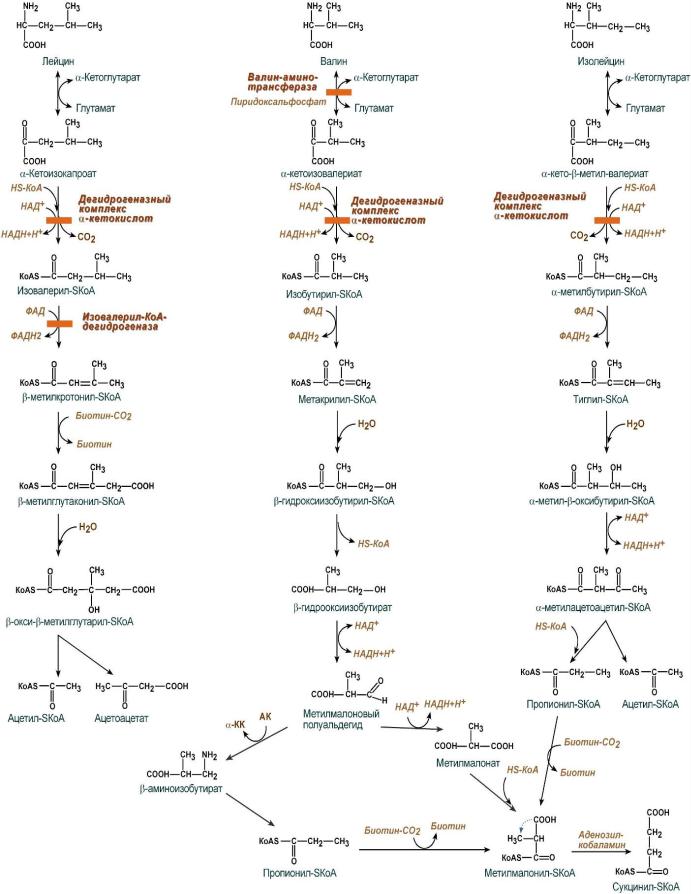
Дефект в структуре дегидрогеназы а – кетокислот с разветвлённой цепью (обзовём его ДКРЦ) порождает грубые метаболические нарушения в виде ацидемии. Вмире выделяют 5 типов лейциноза, однако для нас важно знать следующее:
Манифестация заболевания происходит в первые дни жизни и проявляется нарушениями со стороны нервной системы в виде возбуждения ребёнка,дистонии,
судорожного синдрома, необъяснимой рвотой. У такого ребёнка берут анализ крови и видят кетонемический ацидоз. Первой мыслью в этих условиях будет спросить, не связано ли это с нарушением углеводного обмена. Однако,
патологических изменений содержания глюкозы, как правило, нет. Врачи ищут причинутакихклинических«приколов», но времяидёт. К3-4суткам возбуждение сменяется угнетением ЦНС. Бывает, что лишь накопление лейцина и его метаболитов в моче с формированием запахом кленового сиропа указывает на истинную причину проблемы.
Иные варианты нарушений могут быть на более низких уровнях метаболизма.
11.3.2. Пропионовая ацидемия заключается в блоке реакции, катализируемой пропионил – КоА – карбоксилазой. В результате накапливаются вышеуказанные аминокислоты, а также метионин, треонин (их метаболизм также сопряжён с
образованием пропионил – КоА).
168
Манифестирует заболевание тяжёлым кетоацидозом. Он связан с накоплением кетокислот – метаболитов пропионовой кислоты, собственно пропионовой кислоты, и истощением буферных систем. Примечательно, что при данном заболевании обнаруживается гипогликемия и вторичная гипераммониемия, что затрудняет дифференциальную диагностику.
Для пропионовой ацидемии характерно развитие тубулопатия, т.к. пропионовая кислота экскретируется почечными канальцами и конкурентно ингибирует транспортные системы почечных канальцев. Поэтому весьма заканомерно развитие дегидратации (угнетение реабсорбции натрия и воды). Примечательно,
что при данной форме описывают угнетение костного мозга, а при длительном течении формирование кардиомиопатии. Ахда, поражение ЦНС никто не отменял,
здесь также наблюдается синдром угнетения.
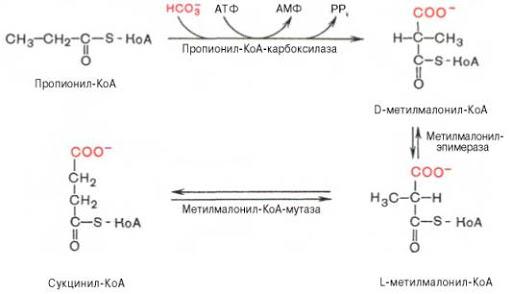
11.3.3. Метилмалоновая ацидемия. Особого внимания заслуживает метилмалоновая ацидемия – результат дефекта метилмалонилКоА- мутазы.
Интересно это тем, что метилмалоновая кислота является промежуточным метаболитом, т.к. является результатом окисления не только аминокислот, но и жирных кислот с нечётным числом атомов углерода (они проходят стадию метаболизма через метилмалониловую кислоту),
Примечательно, что существуют и другие причины метилмалоновой ацидемии,
например, дефицит витамина В 12, т.к. Аденозилкобаламин, метаболит витамина
B12, является кофактором данного фермента. Такая картина часто сопровождается наличием мегалобластной анемии и фуникулярного миелоза (поражение задних корешков спинного мозга), см. дефицит В12.
В отношении врождённых форм, картина такая же, как и для пропионовой ацидемии и лейциноза.
Как же ориентироваться в таких сложных процессах?
Дело в том, что данная группа заболеваний характеризуется сходной и стереотипной клинической картиной в виде энцефалопатии (синдрома угнетения,
судорог, задержки психомоторного развития),
169
рвоты, потерей веса или слабыми прибавками. Всё это обусловлено кетоацидозом, который связан с накоплением кетокислот, в случае каждого заболевания – своим спектром. Прояснить ситуацию может оценка экскреции органических кислот с мочой, говорят также и о повышенном уровне аминокислот в крови.
Важно помнить, что «катаболический стресс» в результате воспалительных заболеваний, травм, голодание, и любые факторы, провоцирующие скачок концентрации глюкокортикостероидов и катехоламинов, будут провоцировать метаболический криз органических ацидемий в результате усиления интенсивности процессов их метаболизма и накоплением метаболитов выше уровня ферментативного блока. Такие кризы проявляются с различными клиническими фишками,ноглавное,что ихобъединяет –развитиеэнцефалопатии в виде судорог и угнетения сознания. Многие забывают, что врождённые нарушения обмена аминокислот (как и нарушения орнитинового цикла) могут бытьпричинами метаболическойкомыу детей.Икаждыйновыйметаболический криз может стать для ребёнка последним…
11. 4. Нарушение обмена метионина.
Метионин играет очень важную роль в ряде биохимическихпроцессов в силу того,
что является донором метильной группы. Это используются в синтезе таких соединений, как:
-адреналин и норадреналин
-холин (компонент фосфолипидов клеточных мембран)
-карнитин (штука, переносящая жирные кислоты)
-тимин, цитизин
-вспомни сам
Осуществляется это при условии активации метионина, т.е. образования S –
аденозин-метионина (активной формы)
170
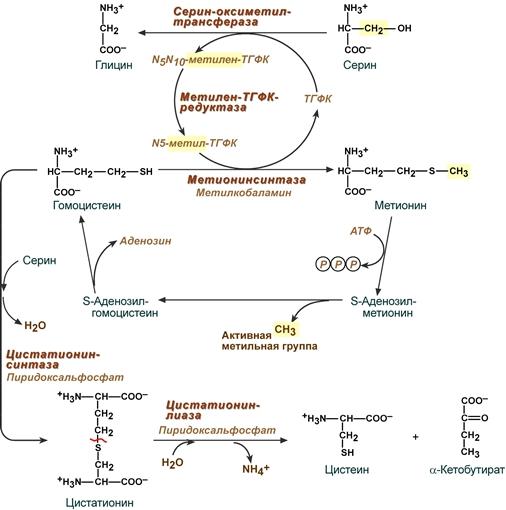
Метаболизм метионина заключается в образовании S – аденозин – гомоцистеина.
Далее, под действием S-AГ – гидролазы, мы получаем гомоцистеин. У него два пути: либо стать метионином (метионинсинтаза В 12 – зависимая), либо превратиться в цистатион и подвергнутся дальнейшему метаболизму.
Гомоцистеинемия является результатом ферментативного блока цистатион – В – синтазы, в некоторых случаях – дефекта метионинсинтазы и метионин – редуктазы. Первый вариант более характерен для врождённой ситуацииии, два последующих – чаще
приобретённые.
Причинами
приобретённой гомоцистеинемии являются:
-авитаминоз В6, В9, В12
-гипотиреоз
-применение цитостатической терапии (метотрексат), применение изониазида,
карбамазепина, метилксантинов (в том числе кофеина), фенитоина
- курение