

ПРАКТИЧЕСКАЯ РАБОТА №12 (1 семестр) Тема Понятие о наследственных болезнях. Основы медико-генетического консультирования
.pdfТЕМА ЗАНЯТИЯ: ПОНЯТИЕ О НАСЛЕДСТВЕННЫХ БОЛЕЗНЯХ. ОСНОВЫ МЕДИКО – ГЕНЕТИЧЕСКОГО КОНСУЛЬТИРОВАНИЯ.
Цель занятия:
Ознакомиться с классификацией и генетической основой наследственных болезней.
Освоить принципы медико-генетического консультирования.
Вопросы для самоподготовки:
1. Классификации наследственных болезней.
2.Характеристика (генотипическая и фенотипическая) основных хромосомных синдромов (аутосомных: Дауна, Патау, Эдвардса, трисомии хромосомы 8, «кошачьего крика», Вольфа-Хиршхорна).
3.Классификация моногенных болезней.
4.Характеристика наиболее распространённых наследственных болезней обмена веществ (фенилкетонурия, алкаптонурия, галактоземия, муковисцидоз).
5.Мультифакториальные болезни (с наследственной предрасположенностью). Характеристика генетических и внешнесредовых факторов, являющихся причинами появления болезней. Понятие «порога подверженности».
6. Понятие и классификация врожденных пороков развития.
7.Наследственные болезни с нетрадиционным наследованием: митохондриальные болезни, болезни импринтинга (синдром Прадера-Вилли, Энгельмана); экспансии тринуклеотидных повторов (синдром ломкой Х-хромосомы Мартина-Белла). Генотипическая и фенотипическая характеристика.
8.Принципы профилактики наследственных болезней.
9.Медико-генетическое консультирование как учреждение и врачебное заключение. Виды, этапы проведения, задачи.
10.Пренатальная |
диагностикиа. |
Методы (неинвазивные и инвазивные): УЗИ, |
амниоцентез, биопсия хориона, |
кордоцентез, фетоскопия, определение маркёров: |
|
альфа-фетопротеина, хорионического гонадотропина и др.). Показания к проведению, значение.
Наследственные болезни, будучи частью общей наследственной изменчивости человека как биологического вида, обеспечивают приспособление его к изменяющимся условиям внешней среды. Использование молекулярных подходов позволило не только картировать гены человека, но и идентифицировать в них основные типы мутаций, обусловливающих развитие наследственных заболеваний. В настоящее время описано более 100 хромосомных синдромов, более 4500 моногенных болезней, мультифакториальные болезни составляют 93% всех случаев хронической неинфекционной патологии человека. Наследственные болезни являются причиной нетрудоспособности населения и инвалидности.
Медико-генетическое консультирование - это один из видов специализированной медицинской помощи населению, направленный на предупреждение рождения детей с наследственными болезнями. В России первая медико-генетическая консультация была организована в 1929 году С.Н. Давиденковым в Институте нервно-психической профилактики. В настоящее время эта служба охватывает 4 уровня: районный (городской), региональный, межрегиональный и федеральный.
Методы пренатальной (дородовой) диагностики:
1.Инвазивные (получение плодного материала):
–амниоцентез. Сроки проведения – 13-22 недели беременности. Пункция через брюшную стенку с извлечением 10-20 мл; забор амниотической жидкости для биохимического, цитогенетического анализа;
-биопсия хориона. Сроки проведения – 8-10 недель беременности. Исследования те же, что и при амниоцентезе;
-фетоскопия. Сроки проведения 16-22 недели беременности. Осмотр плода эндоскопом, введённым в амниотическую полость через переднюю стенку матки. Осуществляют биопсию кожи плода, плаценты. Применяется редко;
-кордоцентез. Взятие крови плода из сосудов пуповины под контролем УЗИ.
Проводится на 20-22 неделях беременности с целью биохимического анализа,
атак же для внутриутробного лечения плода;
-определение маркёров (альфа-фетопротеин, хорионический гонадотропин и др.) в амниотической жидкости плода.
2.Неинвазивные:
-УЗИ (ультразвуковое исследование). Сроки проведения, начиная с 18-22 недели беременности;
выявляются пороки развития плода;
- определение сывороточных маркёров (альфа-фетопротеин, хорионический гонадотропин и др. в сыворотке крови матери.
Задачи медико-генетического консультирования:
Постановка точного диагноза наследственной болезни
Определение типа наследования болезни и величины повторного риска в конкретной семье
Объяснение смысла генетического прогноза консультирующимся
Пропаганда медико-генетических знаний
Задание №1. Основные понятия.
Пользуясь учебной литературой, запишите и объясните значение терминов по теме
1.Амниоцентез
2.Анэнцефалия
3.Арахнодактилия
4.Брахидактилия
5.Гипертелоризм
6.Гипотелоризм
7.Долихоцефалия
8.Кордоцентез
9.Медико-генетическое консультирование
10.Макроглоссия
11.Макросомия
12.Микроцефалия
13.Микрогения
14.Микрогнатия
15.Монголоидный разрез глаз
16.Полидактилия
17.Пренатальная диагностика
18.Прогения
19.Прогнатия
20.Синдактилия
21.Стопа-качалка
22.Фетоскопия
23.Хорионбиопсия
24.Экзофтальм
25.Эпикант
Задание №2. Изучите и заучите таблицу №1 «Классификация
наследственных болезней».
|
Таблица 1 |
|
|
|
|
Название болезни |
% встречаемости |
|
|
|
|
Хромосомные болезни |
1% |
|
|
|
|
Моногенные болезни |
3% |
|
|
|
|
Врождённые пороки развития |
2,5 – 3,5% |
|
|
|
|
Мультифакториальные болезни |
92 – 93% |
|
|
|
|
С нетрадиционным типом наследования |
|
|
|
|
|
Задание №3. Изучите таблицу №2 с описанием синдромов. Используйте
эти данные, чтобы заполнить таблицу №3.
|
|
|
|
Таблица №2 |
|
|
«Хромосомные синдромы» |
|
|
||
|
|
|
|
|
|
Название |
Формы синдромов |
Частота |
|
Кариотип синдрома |
|
синдрома |
|
встречаемости |
|
|
|
|
|
|
|
|
|
Болезнь Дауна |
1:550 – 700 новорождённых среди умственно отсталых детей |
|
|||
|
выявляется 10-12 % больных синдромом Дауна |
|
|||
|
Простая трисомия по |
95% всех случаев |
|
47, +21 (ХХ, ХУ) |
|
|
хромосоме 21. |
|
|
|
|
|
|
|
|
|
|
|
Транслокация 21 |
4% всех случаев |
|
46, t 21/15 (ХХ, ХУ); |
|
|
хромосомы на другие |
|
|
46, t21/14 (ХХ, ХУ); |
|
|
(чаще на 15, реже на 14, |
|
|
46, t21/21 (ХХ, ХУ); |
|
|
ещё реже на 21, 22, У- |
|
|
46, t21/22 (ХХ, ХУ); |
|
|
хромосому). |
|
|
|
|
|
Мозаичный вариант |
1% всех случаев |
|
|
|
|
синдрома. |
|
|
|
|
|
|
|
|
|
|
Синдром |
1: 6000 новорождённых |
|
|
||
Патау |
Простая трисомия по |
75 % случаев |
|
47, +13 (ХХ, ХУ) |
|
|
хромосоме 13 |
|
|
|
|
|
Транслокация (чаще |
20 % случаев |
|
|
|
|
робертсоновская); |
|
|
|
|
|
|
|
|
|
|
|
Мозаичный вариант |
5% |
|
|
|
|
синдрома |
|
|
|
|
Синдром |
Частота встречаемости 1: 7000. |
||
Эдвардса |
- Простая трисомия по |
90 % случаев |
|
|
хромосоме 18 |
|
47, +18 (ХХ, ХУ) |
|
- мозаичная |
10 % случаев |
|
|
|
|
|
Синдром |
Частота встречаемости 1: 50000. |
||
трисомии |
Трисомия по |
16 % случаев |
47, +8 (ХХ, ХУ) |
по 8 |
хромосоме 8 |
|
|
хромосоме |
- мозаичный вариант |
84 % случаев |
|
|
- |
|
|
|
|
|
|
Синдром |
Синдром делеции |
Частота |
46, 5р- (ХХ, ХУ) |
кошачьего |
короткого плеча |
встречаемости |
|
крика |
хромосомы 5 (утрата |
1 : 50000. |
|
|
сегмента р15). |
|
|
Синдром |
Синдром делеции |
Частота |
46, 4р- (ХХ, ХУ) |
Вольфа- |
короткого плеча |
встречаемости |
|
Хиршхорна |
хромосомы 4 (сегмент |
1: 100000 |
|
|
р16). |
|
|
Синдром |
Встречается с частотой 1:500 – 700; |
||
Клайнфельтера |
Классическая форма |
80% всех случаев |
47, ХХУ, |
|
синдрома |
синдрома |
|
|
|
|
48,ХХХУ, |
|
мозаичные варианты: |
20% |
|
|
|
редко встречается |
46,ХУ/ 47, ХХУ и др. |
Синдром |
|
|
|
Синдром трипло-Х |
встречается с |
47,ХХХ |
|
трисомии по Х |
новорождённых |
частотой 1:770 |
|
хромосоме |
девочек и протекает |
|
|
|
бессимптомно в раннем |
|
|
|
детском возрасте |
|
|
Синдром |
Частота встречаемости 1:1430 новорождённых девочек. |
||
Шерешевского |
Классический вариант– |
55% всех случаев |
|
-Тернера |
Возможны частичные |
синдрома |
45,Х0 |
|
моносомии и |
|
|
|
мозаичные варианты. |
|
|
|
|
|
|
Задание №4. Используя предложенный материал, заполните таблицу №3
«Фенотипические признаки синдромов».
|
|
|
|
|
|
Таблица 3 |
|
|
|
|
|
|
|
|
|
Синдром |
Размер и |
Пропорции |
Особенности |
Форма |
Форма и |
|
Другие |
|
форма |
тела |
строения лица |
глаз |
расположение |
|
признаки |
|
черепа |
|
|
|
ушей |
|
|
|
|
|
|
|
|
|
|
Болезнь Дауна |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Синдром Патау |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Синдром |
|
|
|
|
|
|
|
Эдвардса |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Синдром |
|
|
|
|
|
|
|
трисомии по 8 |
|
|
|
|
|
|
|
хромосоме |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Синдром |
|
|
|
|
|
|
|
кошачьего |
|
|
|
|
|
|
|
крика |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Синдром |
|
|
|
|
|
|
|
Вольфа- |
|
|
|
|
|
|
|
Хиршхорна |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Синдром |
|
|
|
|
|
|
|
Клайнфельтера |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Синдром |
|
|
|
|
|
|
|
трисомии по Х |
|
|
|
|
|
|
|
хромосоме |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Синдром |
|
|
|
|
|
|
|
Шерешевского- |
|
|
|
|
|
|
|
Тернера |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
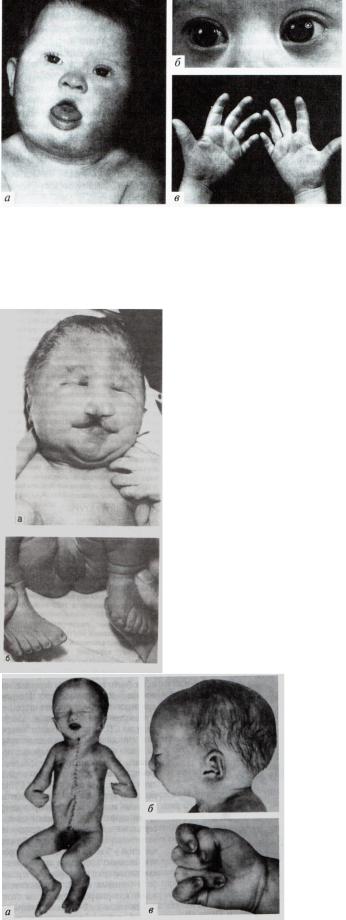
Хромосомные синдромы. Аутосомные синдромы.
1.Синдром Дауна
Фенотип: брахицефалическая форма черепа с увеличением переднее–заднего размера и утолщением затылка, избыток кожи на затылке, плоский профиль лица, эпикант (вертикальная кожная складка у внутреннего угла глазной щели), монголоидный разрез глазных щелей , помутнение хрусталика, косоглазие, короткий нос с широким плоским переносьем, полуоткрытый рот с толстыми губами и высунутым языком
(макроглоссия), узкое и короткое нёбо. Руки короткие и широкие, поперечная складка ладоней. Часто наблюдаются врождённые пороки сердца (дефект межжелудочковой перегородки, открытый артериальный проток), деформация скелета. Умственная отсталость (имбицильность, дебильность.)
2.Синдром Патау
Фенотип: микроцефалия, тригоноцефалия (расширение черепа в затылочной и сужение в лобной части), узкие глазные щели, широкое основание носа, низко посаженные деформированные уши, микрофтальмия (малые размеры глазного яблока), микрогнатия (малые размеры верхней челюсти), расщелина губы и нёба, полидактилия, пороки внутренних органов (головного мозга, сердца и сосудов, почек, пищеварения, половых органов). Дети погибают обычно в течение первых трёх месяцев жизни.
3.Синдром Эдвардса
Фенотип: задержка роста, множественные аномалии развития: долихоцефалический череп (преобладание продольных размеров головы над поперечным) с выступающим затылком, «птичий» профиль лица, короткие и горизонтально расположенные глазные щели; маленькие, деформированные низко расположенные уши, избыточная кожа на затылке; микростомия (узкая ротовая щель); флексорное сгибание кисти с наложением указательного пальца на III, а V на IV; «стопа – качалка» (с провисающим сводом и выступающей кзади пяткой); синдактилия;
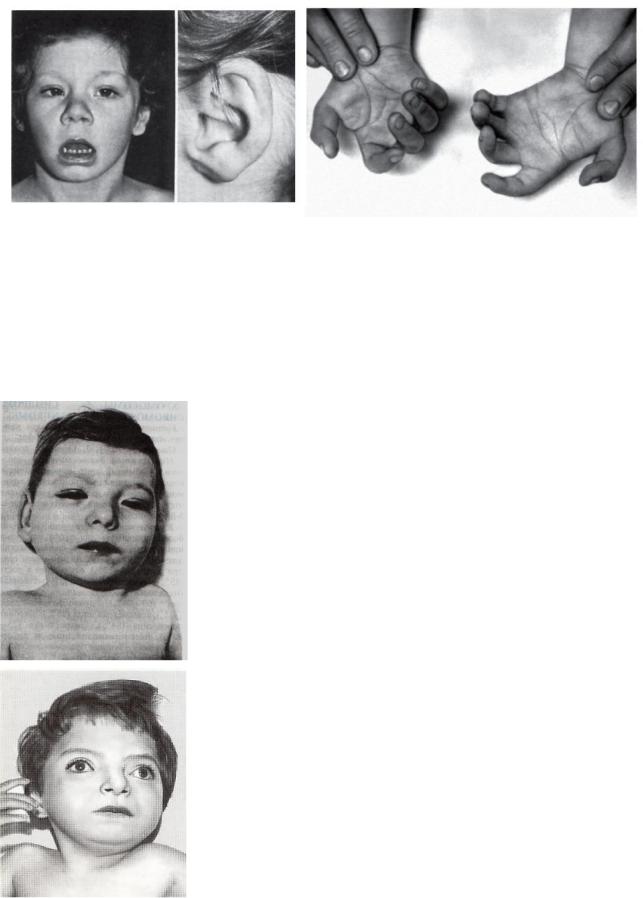
врождённые пороки сердца и крупных сосудов. Продолжительность жизни резко снижена.
4.Трисомия по хромосоме 8
Фенотип: множественные изменения опорно–двигательного аппарата: туловище длинное, слияние рёбер и позвонков, сколиоз, кифоз, spina bifida в грудном или в поясничном отделах (расщепление позвоночного столба); ограничены движения в суставах. Отмечается выраженный черепно-лицевой дисморфизм: большой квадратный череп, выступающий лоб, гипертелоризм, косоглазие, микрогнатия, вывернутая нижняя губа, короткая шея, низко расположенные деформированные уши. Пороки внутренних органов: подковообразная почка, гидронефроз, гипоплазия гениталий. Задержка физического и интеллектуального развития.
5.Синдром «кошачьего крика»
Фенотип: умственное и физическое недоразвитие, микроцефалия, лунообразное лицо, эпикант, антимонголоидный разрез глазных щелей, косоглазие, неправильное расположение зубов (передние резцы выступают вперёд). Продолжительность жизни снижена, только 14% больных доживают до 10 лет. Наиболее характерным является специфический плач, напоминающий кошачье мяуканье, что обусловлено изменениями гортани.
6.Синдром ВольфаХиршхорна
Фенотип: при рождении снижен вес. Характерна задержка физического и психомоторного развития. Умеренная микроцефалия, клювовидный нос, деформированные низко расположенные уши, высокий лоб с глубокой кожной складкой, косоглазие, эпикант, гипертелоризм ( увеличения расстояния между внутренними краями глазниц), пороки внутренних органов. Обычно больные умирают в возрасте до одного года.
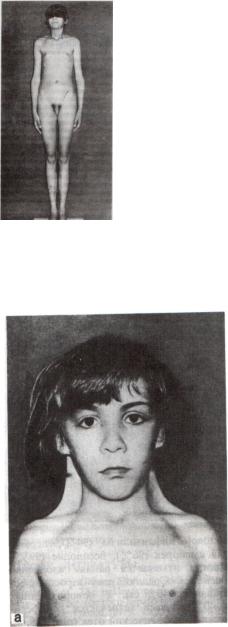
Гоносомные синдромы.
1.Синдром Клаинфельтера
Фенотип: высокий рост, узкие плечи, широкий таз, скудное оволосение, гинекомастия, гипоплазия тестикул, бесплодие, в некоторых случаях умственная отсталость.
2.Синдром Шерешевского-Тернера
Фенотип: низкий рост, «щитообразная» грудная клетка, крыловидные складки на шее, широкая переносица, гипертелоризм, недоразвитие первичных и вторичных половых признаков, первичная аменорея, бесплодие, склонность к психогенным реакциям.
3.Синдром трипло-Х или полисомии по Х-хромосоме
Около 30% больных сохраняют детородную функцию. Клинически больные имеют недоразвитые яичники, гипоплазию матки, преждевременный климакс, склонность к психогенным реакциям, неспецифические соматические дисморфии разной степени выраженности. Наблюдается прямая зависимость тяжести клинических проявлений от числа Х-хромосом.
Задание 4. Изучите таблицу №4: «Наследственные болезни обмена веществ».
|
|
Таблица 4 |
|
|
|
Название |
Биохимический дефект |
Фенотипические |
болезни |
|
признаки |
Фенилкетонурия |
Вызвана нарушением функционирования |
Первые проявления болезни |
|
фермента фенилаланингидроксилазы, |
в возрасте 2-6 месяцев: |
аутосомно- |
превращающей фенилаланин (ФА) в |
вялость, судороги, |
рецессивный тип |
тирозин. Вследствие этого ФА сначала |
характерный «мышиный» |
наследования |
накапливается в крови, а затем |
запах, гипопигментация |
Частота |
происходит накопление промежуточных |
кожи, волос, радужки, |
встречаемости |
продуктов (фенилпировиноградной, |
задержка психомоторного |
в России 1:12000 |
фенилуксусной и др. кислот), |
развития. При отсутствие |
(Москва) |
оказывающих токсическое действие на |
лечения развивается |
|
ЦНС, нарушающих функции печени, |
тяжёлая умственная |
|
обмена белков, метаболизм гормонов. |
отсталость. |
|
|
|
Галактоземия |
Нарушение углеводного обмена. |
Новорождённый ребёнок не |
аутосомно- |
Болезнь возникает при недостатке |
переносит грудное |
рецессивный тип |
фермента, расщепляющего лактозу |
вскармливание. У него |
наследования |
(молочный сахар) до галактозы. |
появляется желтуха, рвота, |
1:100000 |
|
понос, |
новорождённых. |
|
гепатоспленомегалия с |
|
|
исходом в цирроз печени, |
|
|
задержка психомоторного |
|
|
развития, возможна смерть |
|
|
|
Алкоптонурия |
Связана с обменом тирозина. В отсутствие |
Суставные хрящи темнеют |
аутосомно- |
фермента оксидазы гомогентизиновой |
и постепенно развиваются |
рецессивный тип |
кислоты она накапливается и у пожилых |
артриты. |
наследования |
людей откладывается в суставные хрящи. |
|
Встречается |
|
|
редко 3-5:1000000 |
|
|
|
|
|
Муковисцедоз |
Сопровождается множественными |
Происходит закупорка |
Генная мутация |
поражениями желез внешней секреции, |
дыхательных путей, |
в 7-ой хромосоме. |
что проявляется выделением секретов |
протоков поджелудочной |
Аутосомно- |
повышенной вязкости |
железы. Возникают болезни |
рецессивный тип. |
|
дыхательной системы и |
1:2500 |
|
желудочно-кишечного |
новорождённых |
|
тракта. У больных |
|
|
выраженная гипотрофия, |
|
|
авитаминозы |