

Генетика и МГК. Ситуационные задачи с решением
.pdfЗиготы: ХХX, XXY, XX, XY
Лечение.
Трисомии по Х хромосоме часто связаны с различными речевыми задержками и задержками в развитии. Интеллект, как правило, в пределах нормы. IQ может быть на 10- 15 пунктов ниже нормы. Девочки начинают разговаривать в возрасте около 12-18
месяцев. При чтении, родители могут обратить свое внимание на следующие недостатки: дислексия, беглое чтение, разговорные отклонения. Очень часто, девочки имеют диспраксию. Также, они могут иметь более высокий рост по сравнению с другими девочками их возраста.
В некоторых случаях, у детей с трисомией по X хромосоме могут иметься умеренные лицевые аномалии: вертикальные складки кожи охватывающие внутренние углы глаз, широко расставленные глаза (гипертелоризм) и маленькая окружность головы. Большинство младенцев также имеют сниженный мышечный тонус (гипотония) и
клинодактилию. Лица с трисомией по X хромосоме могут часто проявлять тревогу, синдром дефицита внимания и гиперактивности (СДВГ). В некоторых случаях, такие нарушения улучшаются с возрастом. Также, некоторые девочки имеют незначительные поведенческие или эмоциональные нарушения в то время как другие имеют более серьезные проблемы, которые могут потребовать кратковременного вмешательства. Раннее выявление и лечение таких нарушений является очень полезными для таких детей.
Конкретные терапевтические стратегии будут зависеть от нескольких факторов, включая возраст девочки, конкретные симптомы, проявления и их тяжесть. Раннее вмешательство рекомендуется для всех младенцев и детей с диагнозом трисомии по X хромосоме. Опыт показывает, что дети с этим синдромом очень хорошо реагируют на раннее вмешательство (логопедия, трудотерапия, физиотерапия и другие методы).
Младенцы и дети с трисомией X также должны пройти обследование почек и сердца, чтобы исключить наличие отклонений в этих органах.
Коррекция симптомов заключается в гормональной терапии. При явлениях
недостаточности функции яичников необходима циклическая заместительная терапия эстрогенами и прегнином или прогестероном.
3) Третий этап – заключение и подтверждение диагноза.
Заключение.
Пациентка с данным диагнозом может иметь детей. Для исключения бесплодия рекомендуется прохождение обследования, в результате которого будет выявлено либо опровергнуто нарушение функции яичников. При наличии каких-либо нарушений следует
прибегнуть к гормональной терапии.
Профилактика синдрома трисомии по Х хромосоме на данный момент не разработана. Пока что у генетиков нет возможности влиять на механизмы деления клеток после зачатия.
Методы для подтверждения диагноза.
Кариотипирование.
Лучшим способом диагностики болезни остается кариотипирование. Оно предусматривает анализ хромосомного набора пациентки с установлением наличия дополнительной X-хромосомы. Для определения кариотипа используют как прямые, так и
непрямые методы исследования. В первом случае материал, взятый из костного мозга, лимфатических узлов или других тканей, изучают сразу же после получения. Однако прямой метод информативен только тогда, когда в материале имеется достаточное количество метафаз митоза, так как только в этой фазе хромосомы приобретают присущие им особенности строения и возможна их точная идентификация.
FISH-диагностика.

Задача №13
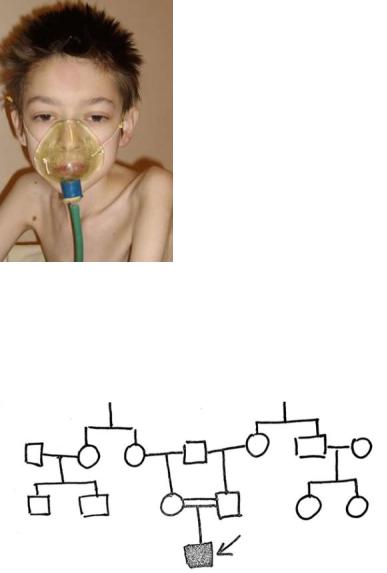
В ККМГ направлен детским неврологом ребёнок с целью уточнения диагноза. У неё периодически отмечались эпилептические припадки. Из данных анамнеза стало известно, что ребёнок (девочка) единственная, родилась недоношенной.
Родители: супруге 34 года; супругу 39 лет; здоровы. В акушерском анамнезе матери ребёнка двое мёртворождённых дочерей. Кариотипы родителей в норме.
Её родители, сестра и брат здоровы, имеют по одному здоровому ребёнку. Родители супруга здоровы; брат здоров и имеет троих здоровых сыновей.
При осмотре ребёнка обнаружено:
адинамия, гипотрофия мышц, судорожное сокращение мышц; задержка психофизического развития; нарушение слуха зрения.
Биохимический анализ: в крови обнаружен высокий уровень содержания молочной кислоты; уменьшение концентрации кальция.
Биопсия мышц: обнаружен феномен «рваных красных волокон» (разрывы миофибрилл)
1 этап - уточнение диагноза
При осмотре ребёнка обнаружено:
адинамия, гипотрофия мышц, судорожное сокращение мышц; задержка психофизического развития; нарушение слуха зрения.
Биохимический анализ: в крови обнаружен высокий уровень содержания молочной кислоты; уменьшение концентрации кальция.
Биопсия мышц: обнаружен феномен «рваных красных волокон» (разрывы миофибрилл)
Родословная
Исходя из выше перечисленных данных, можно сделать вывод, что ребенок страдает синдромом MELAS - Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes — «митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды».
Основными клиническими критериями диагноза MELAS являются:
•материнский тип наследования; начало до 40 лет;
•нормальное психомоторное развитие до заболевания;
•непереносимость физических нагрузок;
•мигренеподобная головная боль с тошнотой и рвотой; инсультоподобные эпизоды;
•энцефалопатия с эпилептическими приступами и/или деменцией;
•лактат-ацидоз;
•рваные красные волокна в биоптатах скелетных мышц;
•прогрессирующее течение.
2 этап - прогноз потомства
Большая часть детей пациентки будет страдать данным заболеванием, так как синдром MELAS передается от матери всем детям с яйцеклеткой, которая содержит мутантные митохондрии. Дети могут иметь разную выраженность симптомов.
В процессе формирования первичных половых клеток серия последовательных делений приводит к тому, что в образовавшихся клетках соотношение митохондрий с мутантной и нормальной мтДНК будет существенно отличаться от их соотношения в исходной (материнской) клетке, т. е. будет наблюдаться иной уровень гетероплазмии - состояние, при котором в цитоплазме клетки сосуществуют митохондрии с двумя разными вариантами мтДНК (нормальной и мутантной), в отличие от гомоплазмии, при которой мтДНК всех митохондрий клетки представлена одним типом (мутантным или нормальным)
3 этап - заключение и подтверждение диагноза
Заключение : вероятность передачи заболевания потомкам будет зависеть от того, какие митохондрии будут содержаться в яйцеклетке, участвующей в оплодотворении. Выраженность симптомов у больных детей колеблется от легкой до тяжелой формы. Пожтверждение диагноза:
Молекулярно-генетический анализ
1. Получение образцов ДНК:
—выделение митохондриальной ДНК
—рестрикция ДНК — получение отдельных фрагментов ДНК (использование рестриктаз, которые разрывают ДНК по строго определенным последовательностям нуклеотидов из 4-6 пар оснований — сайтах рестрикции).
2. Амплификация — накопление (умножение, клонирование) одинаковых фрагментов ДНК:
—использование классических трудоемких методов: создание рекомбинантных плазмид —> введение их в бактериальную клетку —> размножение клеток —> выделение заданных фрагментов ДНК;
— использование полимеразной цепной реакции: разделение двухцепочечной ДНК на одноцепочечную —> присоединение праймеров (затравка) к одноцепочечным молекулам ДНК (по принципу комплементарности) —> синтез многочисленных полинуклеотидных цепей на одноцепочечных молекулах в границах присоединения праймеров.
3.Электрофорез фрагментов ДНК — разделение фрагментов по молекулярной массе и электрическому заряду на поверхности агарозного геля. Каждый фрагмент имеет определенные размеры и занимает в геле определенное место в виде дискретной полосы.
4.Идентификация конкретных фрагментов ДНК с помощью блот-гибридизации по Саузерну. Фрагменты ДНК переносятся на специальные фильтры, где проводится их гибридизация с радиоактивными синтетическими зондами или клонированными фрагментами ДНК. Зонд выявляет необходимый фрагмент ДНК путем связывания с комплементарными ему нуклеотидными последова-тельностями фрагмента.
Изменение положения и длины фрагмента по сравнению с контролем (зондом), исчезновение или появление нового фрагмента свидетельствует о перестройках последовательности нуклеотидов в исследуемом гене.
Для пренатальной диагностики используется исследование уровня лактата-пирувата в амниотический жидкости, для чего проводится амниоцентез - инвазивная процедура, заключающаяся в пункции амниотической оболочки с целью получения околоплодных вод для последующего лабораторного исследования.
Задача №14
В ККМГ обратились супруги с целью уточнения диагноза заболевания. У ребёнка (мальчика) 6 лет отмечается отставание физического развития; хронический гайморит (воспаление носовых пазух); нарушение пищеварения: рвота, запоры; хронический бронхит; дыхательная недостаточность. Предварительный диагноз – муковисцидоз.
Возраст родителей: мать – 28 лет; отец – 29 лет. Из анамнеза стало известно, что родители являются полусибсами. Мать супруги здорова и имеет здоровую сестру, у которой двое здоровых сыновей. Мать супруга здорова ; у неё есть брат, у которого двое здоровых дочерей. Бабушки по линии родителей ребёнка проживали в одной местности.
Данные осмотра пациента: астеническое телосложение: значительное понижение веса; бледные кожные покровы; постоянные приступы кашля с густой, гнойной мокротой. В лёгких жёсткое дыхание, хрипы, Грудная клетка деформирована.
1 этап - уточнение диагноза
Данные осмотра пациента: астеническое телосложение: значительное понижение веса; бледные кожные покровы; постоянные приступы кашля с густой, гнойной мокротой. В лёгких жёсткое дыхание, хрипы, Грудная клетка деформирована.
Родословная
Исходя из данных осмотра и родословной, можно сделать вывод, что ребенок страдает муковисцидозом в легочной(респираторной) форме.

2 этап - прогноз потомства
Муковисцидоз имеет аутосомно-рецессивный тип наследования.В России в среднем частота болезни 1:10000 новорождённых. По условиям задачи супруги являются полусибсами - сводными братом и сестрой, что повышает риск передачи заболевания потомству.
В целях ранней диагностики муковисцидоз входит в программу обследования новорождённых на наследственные и врождённые заболевания. Исследуют уровень иммунореактивного трипсина в сухом пятне крови. При положительном результате тест повторяют на 21—28 день жизни. При повторном положительном результате назначают потовый тест.
3 этап - заключение и подтверждение диагноза
Заключение: у ребенка диагнозтирован муковисцидоз Методы подтверждения диагноза :
Метод FISHфлуоресцентная гибридизация IN SITU - включает следующие этапы:
-создание зондов - однонитевых фрагментов ДНК, к которым присоединяют биотин или дигоксигенин;
-щелочная обработка препаратов IN SITU с целью денатурации хромосомной ДНК за счет
разрыва водородных связей между двумя цепями ДНК;
-гибридизация хромосомной ДНК сзондом путем комплементарного связывания зонда со специфической последовательностью хромосомы;
-обработка препаратов веществами, которые избирательно связываются с биотином и дигоксегенином; для биотина таким веществом является стрептовидин, для дигоксегенина - антидигоксигениновое антитело;
-визуализация хромосом спомощью люминесцентного микроскопа на фоне неокрашенных хромосом.
Пренатальная диагностика: исследование изоэнзимов тонкокишечной щелочной фосфатазы из околоплодных вод, возможно с 18—20 недели беременности. Ложноположительные и ложноотрицательные значения получают в 4 % случаев. (амниоцентез)
1 курс ЛПФ, 4А группа,Хачатрян Елена
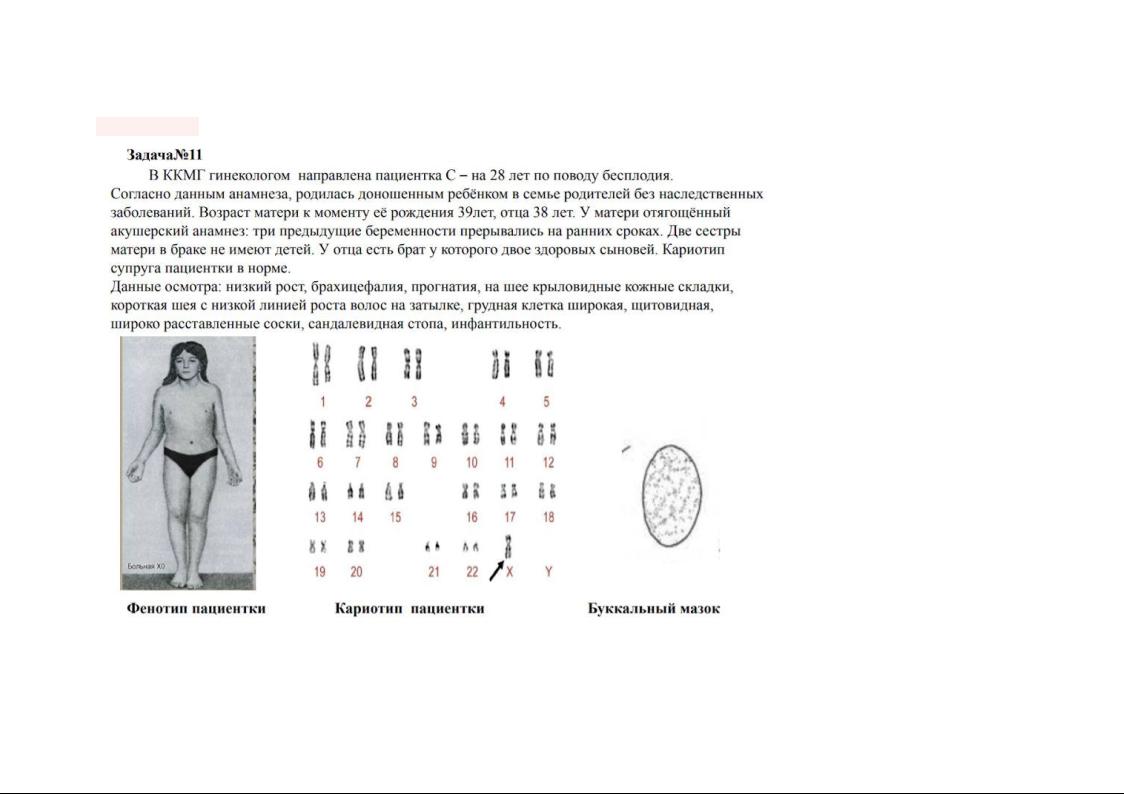
Данные задачи

1)Первый этап-уточнение диагноза
Клинический осмотр
Исходя из данных осмотра:низкий рост, брахицефалия, прогнатия, на шее крыловидные кожные складки, короткая шея с низкой линией роста волос на затылке, грудная клетка широкая, щитовидная, широко расставленные соски, сандалевидная стопа, инфантильность.Можно сделать вывод-у пациентки синдром Шерешевского-Тернера
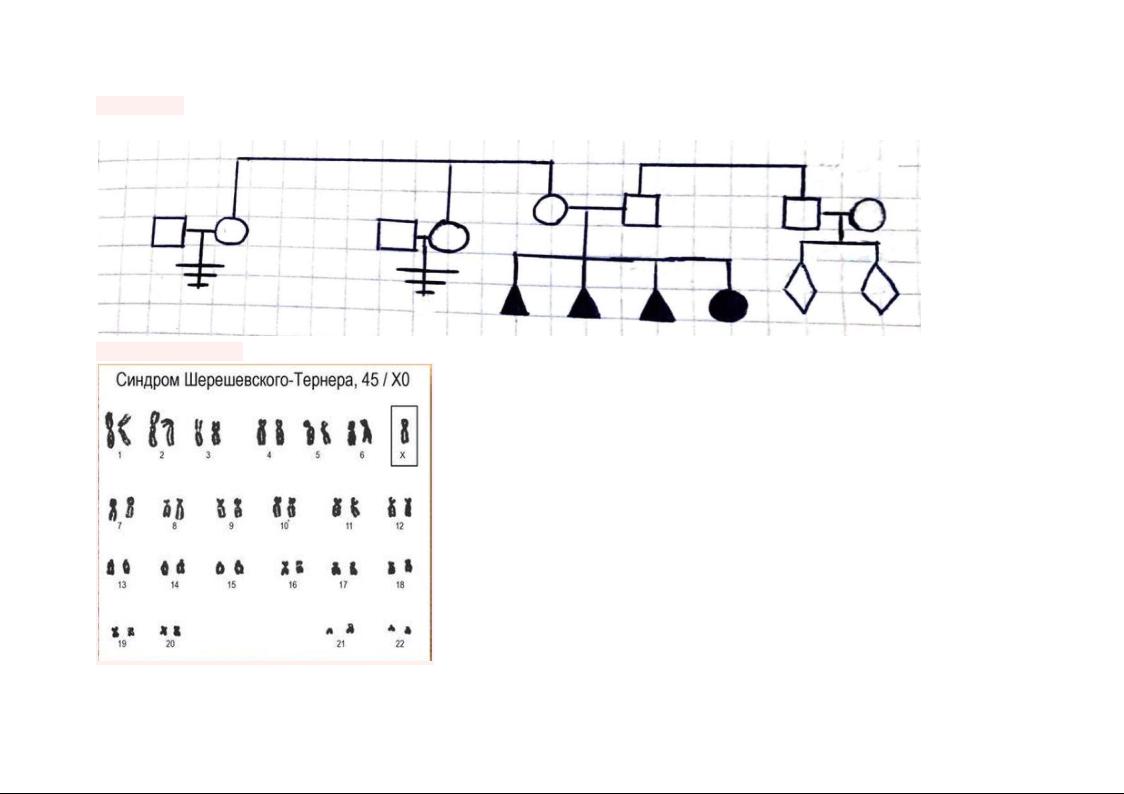
Родословная Исходя из анамнеза, можно составить родословную семьи пациентки
Анализ кариограммы
(45, Х0)