

Генетика и МГК. Ситуационные задачи с решением
.pdfПри необходимости проводятся хирургические операции для лечения пороков сердца, аномалий желудочно-кишечного тракта, обследование у невролога,
кардиолога, офтальмолога, дефектолога, логопеда, педиатра и др.
Раннее начало лечения позволит максимально развить способности детей с синдромом Дауна и улучшить качество их жизни. Сразу после рождения чрезвычайно важно осуществлять развитие двигательной, познавательной сферы ребенка, его психических функций. Существуют специальные дошкольные и школьные учреждения, где разработана программа для детей с соответствующим уровнем интеллекта.
Профилактика
Профилактики рождения детей с синдромом Дауна не разработано. Людям, входящим в группу риска по рождению ребенка с данным заболеванием, во время планирования беременности рекомендуется пройти генетическое исследование.
Задача№4
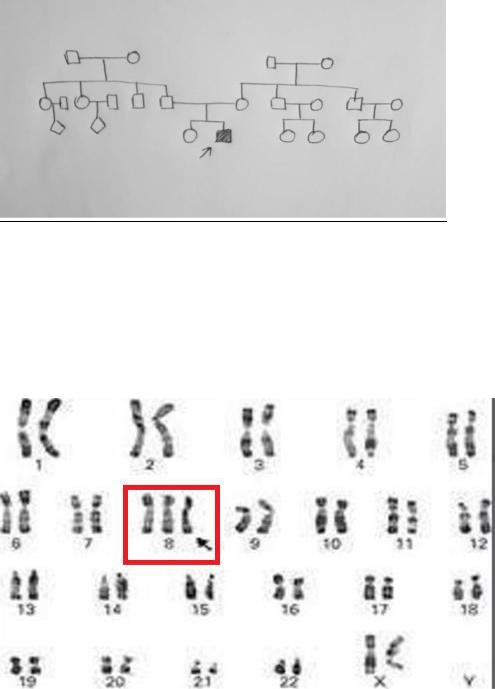
В ККМГ обратились супруги по поводу отставания в психофизическом развитии сына. Мальчик родился доношенным вторым ребёнком в семье. Первой родилась здоровая дочь. Родотвенники со сторонысупруга: родители, брат и две сестры здоровы. У сестёр по одному здоровому ребёнку. Со стороны супруги родители, два брата и их две дочери здоровы.
Кариотипы супругов в норме. Супруге 32 года, супругу – 36 лет.
Данные осмотра ребёнка
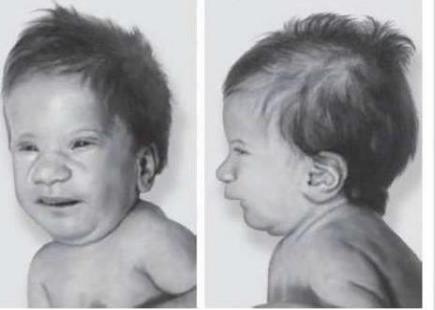
Отмечается умеренная задержка моторного и речевого развития, снижение интеллекта.
Лицо ассиметричное; выступающий лоб; микроцефалия; множественные контрактуры суставов, особенно кистей и стоп. Характерна глубокая борозда между I и II костями
плюсны; большие ушные раковины.
Проведите медико – генетическое консультирование. Изложите решение генетической задачи.
Охарактеризуйте все использованные методы генетики и возможную пренатальную диагностику
1)Первый этап-уточнение диагноза
Отмечается умеренная задержка моторного и речевого развития, снижение интеллекта.
Лицо ассиметричное; выступающий лоб; микроцефалия; множественные контрактуры суставов, особенно кистей и стоп. Характерна глубокая борозда между I и II костями
плюсны; большие ушные раковины.
2) Родословная
Это спонтанная мутация отягающаяся здоровьем родителей
3) Анализ кариограммы
Синдром трисомии 8
47 XY, +8
Заключение
мы видим что в родословной больных не было, все здоровые, значит можно судить о том, что это спонтанная мутация, но явно отягощения возрастом родителей.
Мы предполагаем, что это трисомия по 8 хромосоме
Мы опираемся на Цитогенетический метод, в кариограмме мы увидели, что 3 восьмых хромосомы
Более современный метод, который точно подтвердит нашил предположения, это наш метод фиш.
Мы берём кариотип каждой клетки и наблюдаем в ней по представленной идиограмме 47 +8
В кариограмме мы увидели что 8 хромосом три - опираясь на условие задачи мы
запишем это как 47 ху + 8
Полная трисомия летальна. Ну мы имеем дело с тем, что ребёнок уже родился, то есть было решено его сохранить.
Дети при трисомии не сразу погибают, прогноз такой, что проживают обычно до года. То есть некоторое время он конечно проживет.
Теперь, если супруги планируют ребёнка в будущем, мы должны рассчитать риск рождения ребёнка с таким же синдромом - так как это случайная мутация и все
родственники здоровы, то риск при трисомийном синдроме до 4%, риск небольшой и можно пренебречь, прогноз благоприятный. Общая частота встречаемости синдрома Варкани это 1:50000
НО супруги немолодые, супругам уже за 30, возраст такой пограничный, и мы бы посоветовали обязательную консультацию.
И вот сейчас здесь распишем: из неинвазивных это - узи (можно делать на протяжении всей беременности); а из инвазивных - амниоцентез ( взятие
околоплодной жидкости); метод кариотипирования (точно поможет определить норм кариотип или нет)
Вот именно мать должна быть под контролем врача генетика.
Вообще всегда при пограничном возрасте супруги должны наблюдаться у специалиста
Данные задачи:
В ККМГ обратились супруги П – вы с целью прогноза потомства. Четыре месяца тому назад у них родился ребёнок с множественными врождёнными пороками развития. Предварительно ему был поставлен диагноз хромосомного синдрома. Из анамнеза стало известно, что матери ребёнка 42 года, отцу44. Наследственных болезней в роду у них нет. Их родственники: родители, братья и сестра здоровы; имеют по одному здоровому ребёнку. Кариотипы родителей в норме.
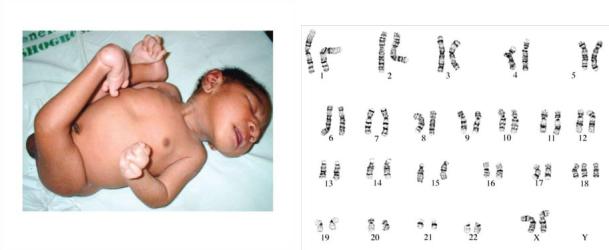
Данные осмотра ребёнка: понижен вес, гипоплазия мышц, тугоподвижность суставов, долихоцефалический череп с выступающим затылком, избыточная кожа на затылке, короткая шея, флексорное сгибание кисти с наложением указательного пальца на III, а V на IV; «стопа –
качалка», врождённые пороки сердца.
Дерматоглиф:
•значительно повышена частота дуговых узоров пальцев
•радиальные петли часто встречаются на 1-ом, 3-ем, 4 или 5 пальцах
•радиальное окончание главных ладонных линий
•высокий осевой трирадиус (дистальный трирадиус)
•диссоциация гребеней
•единственная сгибательная складка ладони(ЧПБ)
•единственная сгибательная складка мизинца.
1)Первый этап-уточнение диагноза
Клинический осмотр:
Данные осмотра ребёнка: понижен вес, гипоплазия мышц, тугоподвижность суставов, долихоцефалический череп с выступающим затылком, избыточная кожа на затылке, короткая

шея, флексорное сгибание кисти с наложением указательного пальца на III, а V на IV; «стопа –
качалка», врождённые пороки сердца. Можно сделать вывод у ребенка Синдром Эдвардса
Родословная:
Исходя из анамнеза, можно составить родословную семьи
Анализ кариограммы
47 , ХХ, +18 Синдром Эдвардса
Анализ дерматоглифа
При многих хромосомных аномалиях у новорожденных имеются характерные дерматоглифические изменения (аномальные узоры и складки на коже ладоней). При синдроме Эдвардса некоторые признаки можно обнаружить почти в 60% случаев. Они
имеют значение в основном для предварительной диагностики при мозаичной или частичной форме болезни. При полной трисомии 18 к дерматоглифике не прибегают, так как для подозрения синдрома Эдвардса хватает других, более заметных аномалий развития.
Основными дерматоглифическими признаками синдрома Эдвардса являются:
дуги на подушечках пальцев располагаются с большей частотой, нежели у здоровых людей;
кожная складка между последней (ногтевой) и предпоследней (срединной) фалангами пальцев отсутствует;
у 30% новорожденных на ладони имеется так называемая поперечная борозда (обезьянья линия, линия Симиан).
2)Второй этап -прогноз потомства
Дети с трисомией в 18 хромосоме чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21и 13. Для женщин
старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков. Выживание после года жизни составляет около 5–10%
Во всех случаях прогноз при синдроме Эдвардса крайне неблагоприятный: в среднем мальчики живут 2-3 месяца, девочки – 10 месяцев. До 1 года доживает лишь 10% больных, до
10 лет – не более 1%. Относительно благоприятные шансы в отношении выживания имеют дети с мозаичной формой синдрома Эдвардса.
Лечение
Поскольку в большинстве случаев аномалии развития оказываются несовместимыми с жизнью, лечение детей с синдромом Эдвардса сводится к оказанию симптоматической помощи, направленной на поддержание физиологических функций, продление жизни и улучшение ее качества. Хирургическая коррекция врожденных пороков, как правило, является рискованной и неоправданной.
Поскольку дети с синдромом Эдвардса ослаблены и подвержены частой заболеваемости инфекциями мочевыводящих путей, средним отитом, конъюнктивитом, синуситами, пневмониями и пр., они нуждаются в тщательно организованном уходе, полноценном питании, регулярном наблюдении со стороны педиатра.
3)Третий этап -заключение и подтверждение диагоноза
Риск рождения ребенка с синдром Эдвардса теоретически существует в любой супружеской паре; известно, что такая вероятность выше у возрастных родителей (для женщин старше 45 лет – 0,7%). С целью своевременного выявления хромосомной патологии у плода не следует пренебрегать антенатальным скринингом, входящим в программу введения беременности.
Подтверждение диагноза
Важнейшей задачей диагностики служит антенатальное выявление синдрома Эдвардса у плода, поскольку данная патология является медицинским показанием для искусственного прерывания беременности. Заподозрить наличие синдрома Эдвардса можно в процессе УЗИ плода и допплерографии маточно-плацентарного кровотока по косвенным признакам
(множественным аномалиям развития плода, агенезии пупочной артерии, малой величине плаценты, многоводию и пр.).
При оценке степени риска рождения ребенка с синдромом Эдвардса учитываются данные биохимического и ультразвукового скрининга, срок беременности, возраст и масса тела женщины. Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики (биопсии хориона, амниоцентеза, кордоцентеза) с последующим кариотипированием плода.
В случае рождения живого ребенка с синдромом Эдвардса необходимо как можно более раннее всестороннее обследование, направленное на выявление тяжелых пороков развития. Новорожденный с синдромом Эдвардса должен быть осмотрен неонатологом, детским кардиологом, детским неврологом, детским хирургом, детским ортопедом, детским урологом и др. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, служат эхокардиография, УЗИ органов брюшной полости и УЗИ почек.

I этап – уточнение диагноза
1.клинический осмотр:
в ходе осмотра у ребёнка были обнаружены следующие внешние признаки: микроцефалия, уплощённое лицо, монголоидный разрез глаз, эпикантный короткий нос с плоской спинкой, макроглоссия, брахицефалия, низко расположенные деформированные ушные раковины, косоглазие.
2. построение родословной:
3. анализ кариограммы:
47, t 21/21, ХХ
4. дерматоглиф:
• значительно повышена частота дуговых узоров пальцев;
• радиальные петли часто встречваются на 1-ом, 3-ьем, 4-ом или 5-ом пальцах;
• радиальное окончание главных ладонных линий;
• высокий осевой трирадиус(дистальный трирадиус);
• диссоциация гребней;
• единственная сгибательная складка ладони(ЧПБ);
• единственная сгибательная складка мизинца.
5. прогноз потомства:
вероятность рождения ребёнка с таким кариотипом очень мала, но из-за
близкородственного брака возрастает(1/700);
6.Пренатальная диагностика синдрома Дауна включает в себя два этапа. На первом этапе, на сроке беременности 11-13 недель, проводится скрининг, который на
основании специфических признаков узи (толщина воротникового пространства, размер носовой кости плода и д.р.) В комплексе с биохимическим анализом уровне определённых белков крови беременной женщины (свободной b-субъединицы хорионического гормона человека (b-ХГЧ) и ассоциированного с беременностью
плазменного протеина А, с учётом её возраста, позволяет рассчитать для неё риск рождения больного. Однако эти методы не позволяют поставить точный диагноз, в результате проведённого скрининга лишь формируется группа риска беременных с повышенной вероятность рождения больного синдромом дауна. На втором этапе в группе риска проводится инвазивные процедуры для получения плотного материала, необходимого для точного проведения анализа на синдром дауна. Зависимости от срока беременности это может быть биопсия ворсин хориона (8-12 недель), амниоцентез (14-18 недель) или кордоцентез (на более поздних сроках). В полученных
образцах тканей плода проводится определение хромосомного набора.
7.Цитогенетический анализ проводится методом флуоресцентной гибридизации in situ (FISH, от англ. fluorescence in-situ hybridization).