

Генетика и МГК. Ситуационные задачи с решением
.pdf1)Первый этап-уточнение диагноза
Клинический осмотр
Исходя из данных осмотра:низкий рост, брахицефалия, прогнатия, на шее крыловидные кожные складки, короткая шея с низкой линией роста волос на затылке, грудная клетка широкая, щитовидная, широко расставленные соски, сандалевидная стопа, инфантильность.Можно сделать вывод-у пациентки синдром Шерешевского-Тернера
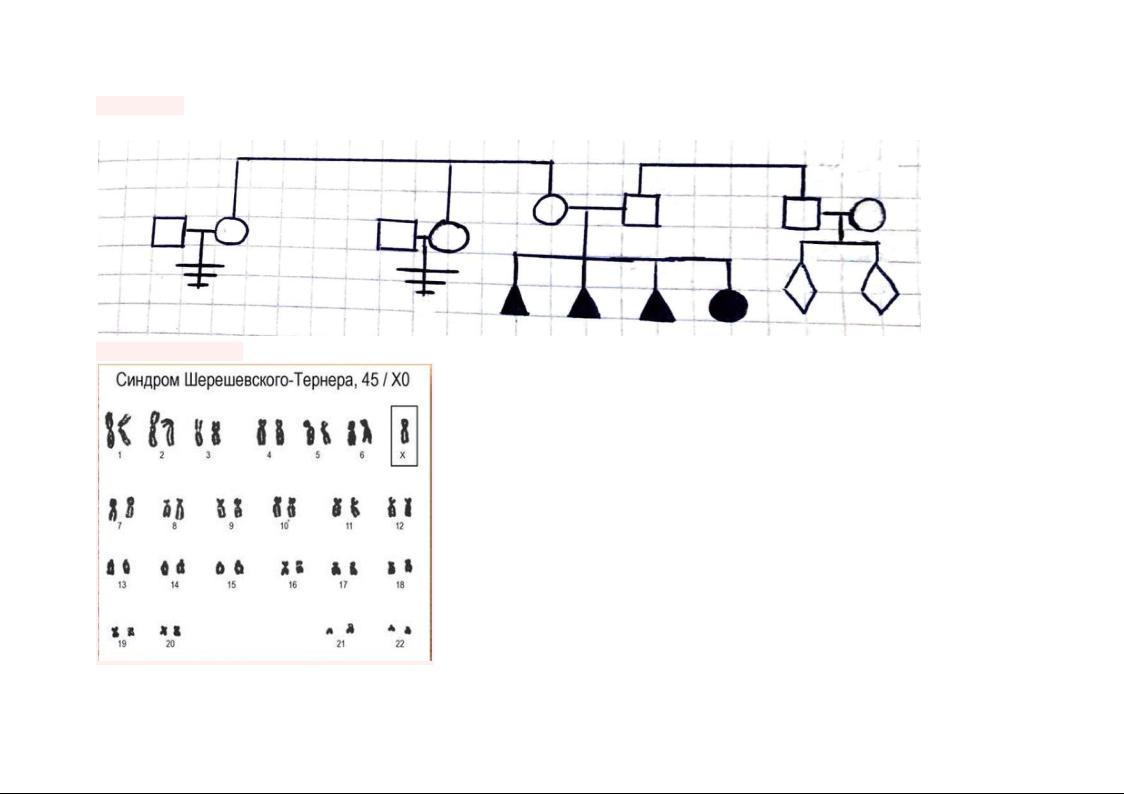
Родословная Исходя из анамнеза, можно составить родословную семьи пациентки
Анализ кариограммы
(45, Х0)

Анализ полового хроматина
Нет телец Барра
Анализ дерматоглифа -Дистальное расположение осевого трирадиуса
-Увеличена частота завитков на пальцах -Высокий гребневый счёт на пальцах
При дерматоглифическом исследовании выявляется изменение кожных узоров пальцев и ладоней. Обычно обнаруживают увеличение частоты ульнарных петель на больших и указательных пальцах. Дистальный осевой трирадиус встречается у 50% больных с синдромом Шерешевского—Тернера. Чаще, чем у здоровых, наблюдается поперечная складка ладони и единственная складка на V пальце. Ладонные узоры очень большие дистальные петли или завитки с большим гребневым счетом.
2)Второй этап -прогноз потомства
Прогноз потомства
Пациентка является бесплодной, так как нет необходимого числа Х-хромосом для образования гамет. На первом этапе терапия заключается в стимуляции роста тела анаболическими стероидами и другими анаболическими препаратами. Лечение следует проводить минимальными эффективными дозами анаболических стероидов с перерывами при регулярном гинекологическом контроле. Главным видом терапии больных является эстрогенизация (назначение женских половых гормонов), которую следует проводить с 14—16 лет. Лечение приводит к феминизации телосложения, развитию женских вторичных половых признаков, улучшает трофику (питание) половых путей, уменьшает повышенную активность гипоталамо-гипофизарной системы. Лечение следует проводить в течение всего детородного возраста больных. Если с помощью гормональной терапии удаётся вырастить до нормальных размеров матку, то беременность у таких больных возможна с помощью ЭКО с донорской яйцеклеткой. Случаи, где сохранились свои яйцеклетки, единичны. В последнее время для увеличения показателей окончательного роста проводится терапия соматотропином.
Газеты матери
ХХ, 0
Лечение
Осложнения при мозаичной форме синдрома Шерешевского-Тернера в основном связаны с наличием аномалий развития внутренних органов. Пороки сердца могут привести к ранним проблемам с кровообращением. Недоразвитие почек является причиной почечной недостаточности. Расщепление позвоночника приводит к неврологической симптоматике и тому подобное. Все зависит от исходного состояния организма.
Мозаичная форма болезни в этом плане намного благоприятнее. Опасные осложнения практически не возникают. Патология ограничивается снижением качества жизни, что не слишком влияет на ее продолжительность.
Синдром Шерешевского-Тернера не лечится. Хромосомные аномалии развиваются на уровне молекул ДНК, на которые пока нет прицельного воздействия извне. Поэтому лечение синдрома Шерешевского-Тернера, как мозаичной формы, так и классической, базируется на применении симптоматических лекарств для улучшения качества жизни пациенток и нормализации функции внутренних органов. Чаще всего используют следующие препараты:

●Соматотропин. Гормон роста, который имеет ограниченные показания к применению. Причиной тому является снижение восприимчивости тканей организма к этому гормону. При мозаичной форме болезни он демонстрирует лучшие результаты, чем при классическом варианте заболевания.
●Оксандролон. Применяется в комплексе с соматотропином. Является анаболическим стероидом, способствуя набору мышечной массы и ускорению процесса роста больного ребенка.
●Эстрогены. Группа препаратов для коррекции половой функции пациенток.
Подбор схемы терапии в каждом случае индивидуален. Все зависит от степени выраженности патологии.
3)Третий этап -заключение и подтверждение диагоноза
Заключение
Достижение беременности возможно при помощи вспомогательных репродуктивных технологий. Обычно пациенткам назначают ЭКО с последующим контролем беременности и гормональной поддержкой.
Профилактика синдрома Шерешевского-Тернера на данный момент не разработана. Пока что у генетиков нет возможности влиять на механизмы деления клеток после зачатия.
Методы для подтверждения диагноза
Кариотипирование
Лучшим способом диагностики болезни остается кариотипирование. Оно предусматривает анализ хромосомного набора пациентки с установлением наличия дефектов или отсутствия X-хромосомы.
Для определения кариотипа используют как прямые, так и непрямые методы исследования. В первом случае материал, взятый из костного мозга, лимфатических узлов или других тканей, изучают сразу же после получения. Однако прямой метод информативен только тогда, когда в материале имеется достаточное количество метафаз митоза, так как только в этой фазе хромосомы приобретают присущие им особенности строения и возможна их точная идентификация.
Консультирование
Больные с синдромом Шерешевского-Тёрнера нуждаются в консультации генетика, эндокринолога, кардиолога, кардиохирурга, нефролога, офтальмолога, отоларинголога, лимфолога, гинеколога-эндокринолога (женщины), андролога (мужчины). Для

обнаружения врожденных пороков и сопутствующих заболеваний показано выполнение ЭхоКГ, МРТ сердца, ЭКГ, УЗИ почек, рентгенографии позвоночника, денситометрии, рентгенографии костей стоп и кистей и др. В рамках диагностического обследования женщинам проводится гинекологическое исследование, УЗИ органов малого таза; мужчинам – УЗИ мошонки, исследование андрогенного профиля. В случае аномалиях зубных рядов детям необходима консультация ортодонта. С целью дифференциальной диагностики синдрома Шерешевского-Тёрнера и гипофизарного нанизма необходимо проведение
исследования уровня гормонов гипофиза в крови, рентгенографии турецкого седла, электроэнцефалографии.
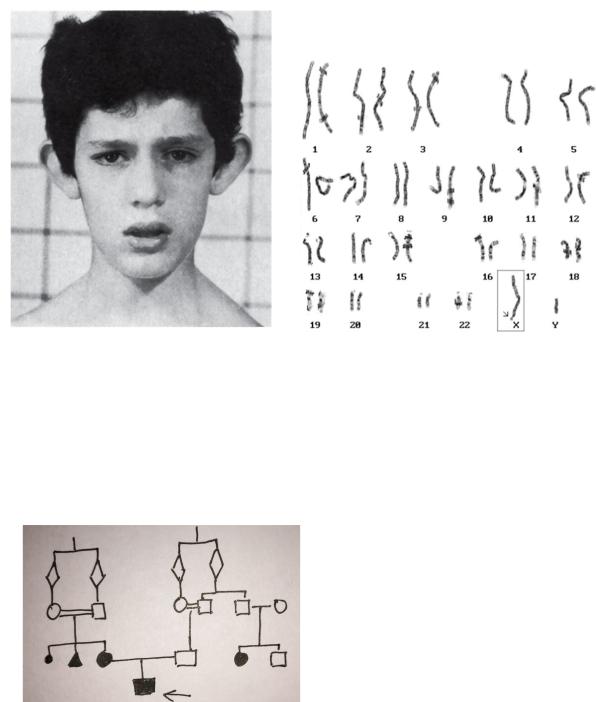
Задача №12
В ККМГ направлен пациент И – в,12 лет по поводу отставания в психическом развитии. Согласно данным опроса родителей, родился доношенным ребёнком. К моменту рождения Матери было 30 лет, отцу 36. Со слов родителей, наследственных заболеваний в роду нет. Родители супругов являются двоюродными братом и сестрой. У матери супруги отягощённый акушерский анамнез: первая беременность закончилась мёртворождением, последующая – рождением больной дочери, третья - спонтанным абортом. У отца супруга есть брат, у которого больная дочь и здоровый сын.
Данные осмотра пациента:
Высокий и широкий лоб, длинное лицо, прогнатизм, большие оттопыренные уши, умственная отсталость, дискоординация движений., мышечная гипотония, быстрая, сбивчивая речь.
Фенотип пациента |
Кариотип пациента |
1 этап - уточнение диагноза
Исходя из данных осмотра: высокий и широкий лоб, длинное лицо, прогнатизм, большие оттопыренные уши, умственная отсталость, дискоординация движений., мышечная гипотония, быстрая, сбивчивая речь, можно сделать вывод, что у пациента синдром хрупкой Х-хромосомы
Родословная семьи
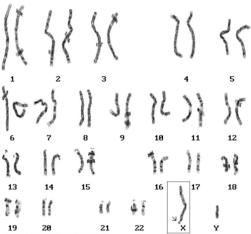
Анализ кариограммы 46, XY ( мутантый ген FMR-1)
Постановка диагноза Синдром хрупкой Х-хромосомы диагностируется путём определения количества ЦГГ-
повторов и их статуса метилирования с помощью эндонуклеазной рестрикции и саузернблоттинга. Наличие более 200 повторов свидетельствует о наличии синдрома хрупкой Х- хромосомы.
Наследование Синдром ломкой Х-хромосомы — сцепленное с полом доминантное заболевание с
редуцированной пенетрантностью.
Мужчины имеют одну Х-хромосому, соответственно, если она содержит мутантный аллель, у носителя развивается заболевание. Женщины несут две Х-хромосомы, таким образом, их шанс получить нормальный аллель удваивается. Женщина с мутантным геном FMR1 может иметь симптомы болезни или быть здоровой. Несмотря на то, что вторая Х-хромосома может служить резервной копией, только одна Х-хромосома активна в каждой отдельной клетке, вследствие инактивации второй.
Мужчина с ломкой Х-хромосомой не может передать её ни одному из сыновей, только всем дочерям. Женщина с одной мутантной хромосомой имеет одинаковые шансы передать её как дочерям, так и сыновьям с вероятностью 50 %. Наследование синдрома ломкой Х-хромосомы обычно увеличивается с каждым новым поколением, это явление получило название парадокса Шермана.
2 этап - прогноз потомства
Пациентка имеет одну мутантную хромосому и страдает синдромом хрупкой Х-хромосомы, вероятность передачи данного заболевания и мальчикам, и девочкам составляет 50%, так как имеет вторую нормальную хромосому.
В настоящее время симптомы можно облегчить с помощью когнитивно-поведенческой терапии, специфического обучения, медикаментов и, при необходимости, лечения физических аномалий.
3 этап - заключение
Пациентка может иметь детей, но из-за наличия мутации в Х-хромосоме, как минимум половина ее детей будут страдать этим же заболеванием. Передача данного синдрома увеличивается из поколения в поколение.
Подтверждение диагноза Единственным методом, позволяющим однозначно диагностировать заболевание, является
молекулярногенетическое исследование. Однако генетическое исследование является дорогостоящим, занимает достаточно длительное время и требует высокой мотивации и аргументации для направления на него. В международной практике для первичного включения ребенка в «группу риска» по наличию FXS основанием могут служить высокие результаты шкальных анкет, составленных на основе фенотипических особенностей, характерных для синдрома. Оценка включает 14 клинических признаков, экспрессивность каждого из которых оценивается в баллах. Отсутствию клинического признака соответствует оценка – 0 баллов, а наибольшей степени выраженности симптома соответствует 3 балла. Тяжесть клинических проявлений оценивается суммой всех баллов. Проведенные многолетние исследования с помощью ранговой шкалы позволили выделить пороговое значение – 21 балл – как критерий для причисления ребенка к группе риска по синдрому
FXS.
FXS - синдром хрупкой Х-хромосомы, синдром Мартина-Белл

Задача №13
В ККМГ направлен детским неврологом ребёнок с целью уточнения диагноза. У неё периодически отмечались эпилептические припадки. Из данных анамнеза стало известно, что ребёнок (девочка) единственная, родилась недоношенной.
Родители: супруге 34 года; супругу 39 лет; здоровы. В акушерском анамнезе матери ребёнка двое мёртворождённых дочерей. Кариотипы родителей в норме.
Её родители, сестра и брат здоровы, имеют по одному здоровому ребёнку. Родители супруга здоровы; брат здоров и имеет троих здоровых сыновей.
При осмотре ребёнка обнаружено:
адинамия, гипотрофия мышц, судорожное сокращение мышц; задержка психофизического развития; нарушение слуха зрения.
Биохимический анализ: в крови обнаружен высокий уровень содержания молочной кислоты; уменьшение концентрации кальция.
Биопсия мышц: обнаружен феномен «рваных красных волокон» (разрывы миофибрилл)
1 этап - уточнение диагноза
При осмотре ребёнка обнаружено:
адинамия, гипотрофия мышц, судорожное сокращение мышц; задержка психофизического развития; нарушение слуха зрения.
Биохимический анализ: в крови обнаружен высокий уровень содержания молочной кислоты; уменьшение концентрации кальция.
Биопсия мышц: обнаружен феномен «рваных красных волокон» (разрывы миофибрилл)
Родословная