

Министерство образования и науки Российской Федерации Федеральное государственное бюджетное образовательное учреждение высшего профессионального образования
«Хабаровская государственная академия экономики и права»
В. Л. Бутуханов, Н. Н. Минаева
Химия
Учебное пособие
для студентов направлений 38.03.07 «Товароведение» и 19.03.04 «Технология продукции и организация общественного питания»
Хабаровск 2014
ББК В 3
Х 12
Химия : учеб. пособие / В. Л. Бутуханов, Н. Н. Минаева. – Хабаровск : РИЦ ХГАЭП, 2014. – 124 с.
В учебном пособии изложены общие принципы неорганической, физической, коллоидной и органической химии. Пособие предназначено для студентов 1, 2, 3-го курсов направлений 38.03.07 «Товароведение» и 19.03.04 «Технология продукции и организация общественного питания» всех форм обучения.
Рецензенты – завкафедрой химии Тихоокеанского государственного университета, канд. хим. наук, доцент Т. Б. Панасюк; доцент кафедры химии Дальневосточного
государственного университета Т. В. Хекало
Утверждено издательско-библиотечным советом академии в качестве учебного пособия
© Хабаровская государственная академия экономики и права, 2014
|
СОДЕРЖАНИЕ |
Введение |
.........................................................................................................................5 |
Глава 1. Периодическая система элементов как классификация элементов по строению электронных оболочек атомов………………………7
1.1. Развитие периодического закона. Закон Мозли. |
|
Современная трактовка периодического закона……………………............. |
7 |
1.2.Структура периодической системы элементов Д. И. Менделеева……........7
1.2.1.Порядковый номер химического элемента………………………........8
1.2.2.Период………..…………………………………………...…………......8
1.2.3.Группа…....………..……………………………………………...……...8
1.2.4.Подгруппа………………………………………………………………..9
1.2.5.Лантаноиды и актиноиды…………………………...……………….....9
1.3.Электронное строение атома………………………………………………...10
1.3.1.Электронная конфигурация атома……………………………………10
1.3.2.Энергетическая диаграмма уровней и подуровней………………….10
1.4.Периодичность свойств атомов химических элементов………...………...11
1.4.1.Электронное строение последних энергетических уровней………..12
1.4.2.Размеры атомов………………………………………………………...12
1.4.3.Энергетическая характеристика атома……………………………….12
1.5.Периодические изменения свойств простых веществ……………...……...13
Глава 2. Дисперсные системы………………...…………………………………..14
2.1.Классификация дисперсных систем…………………...…………………14
2.2.Газовое состояние и газовые законы………………………………………..16
2.3.Жидкое состояние. Концентрация растворов и способы её выражения……………………………………………………..19
2.4.Свойства разбавленных растворов. Закон Рауля.
Криоскопия, эбуллиоскопия………………………………………………....21
2.5.Осмос, осмотическое давление. Закон Вант-Гоффа.
Роль осмоса в биологических процессах……………………...……………24
Глава 3. Элементы химической термодинамики……………...……………….31
3.1.Предмет и метод химической термодинамики.
Некоторые понятия и определения………………………………………...31
3.2.Законы термодинамики. Энтропия………………………………………….33
3.3.Энергия Гельмгольца и энергия Гиббса.
Критерии направления самопроизвольно протекающих процессов……...37
3.4.Экзо- и эндотермические процессы. Тепловой эффект реакции.
3
Закон Гесса и Лаувазье-Лапласса…………………...……………………...40
Глава 4. Химическая кинетика и катализ……………………..………………..42
4.1.Средняя и истинная скорости реакции……………………………………...42
4.2.Влияние концентраций на скорость, порядок и молекулярность реакций…………………………………………43
4.3.Понятие об активных молекулах. Энергия активации……………...……..47
4.4.Влияние температуры на скорость реакции……………...………………...48
4.5.Влияние катализаторов на скорость реакций.
Катализ в живой природе…………………………………………………….50
4.6.Понятие о теориях катализа…………………………...…………………….53
4.7.Роль диффузии………………………………………………………………..55
4.8.Механизм химических реакций…………………………………...………...56
Глава 5.Химическое равновесие………………………………………...…………61
5.1.Закон действующих масс, принцип ле Шателье………………...…………61
5.2.Равновесие в водных растворах. Электролитическая диссоциация. Закон разбавления Оствальда………………………………..63
Глава 6.Теоретические основы органической химии…………………………...66
6.1.Классификация и номенклатура органических соединений………………66
6.2.Изомерия органических соединений………………………………………..72
6.2.1.Структура изомерия………………...…………………………………73
6.2.2.Пространственная изометрия (стереоизомерия)………...…………..74
6.3.Химическая связь и строение органических соединений………………...74
6.3.1.Строение атома углерода и его особенности………………………...74
6.3.2.Типы химических связей…………………...…………………………78
6.3.3.Строение молекул этана, этилена, ацетилена………………………..81
6.4.Сопряжение в органических соединениях…………………………………….84
6.4.1.Открытые сопряжённые системы………………...…………………..85
6.4.2.Замкнутые сопряжённые системы……………………………………87
6.5.Электронные эффекты органических соединений……………………………90
6.5.1.Индуктивный эффект заместителей……………...…………………..90
6.5.2.Мезомерный эффект заместителей…………………………………...92
Заключение.……………………………………...………………………….105
Библиографический список………………………………………………………..107
4
Введение
История химии началась ещё в древности. Химия как совокупность практических знаний и умений, синтеза полезных для человека веществ известна давно. Однако формирование химии как науки началось лишь в конце XVIII в.
Современная химия характеризуется огромными успехами в познании структуры веществ и механизма химических процессов. Современная химия позволяет создавать новые вещества, не встречающиеся в природе, снабжает человека эффективными фармацевтическими препаратами против различных заболеваний. Подсчёты показали, что 25% медикаментов – это природные препараты (т.е. растительного и животного происхождения), 75% – синтезированные.
В своём развитии химия опирается на достижения мировой науки, в развитие которой большой вклад внесли отечественные учёные – М.В. Ломоносов, Д.И. Менделеев, А.М. Бутлеров.
Многие учёные начинали свою творческую деятельность ещё в 19 веке. Это основатель геохимии академик В.И. Вернадский, создатель школы органического синтеза и органического катализа академик Н.Д. Зелинский, основатель школы физико-химического анализа академик Н.С. Курнаков, основатель советской школы биохимиков А. Н. Бах. В годы Советской власти сформировались такие виднейшие химики, как академик Н.Н. Семенов; Герой Социалистического Труда, лауреат Нобелевской премии (созданная им теория цепных и разветвлённых реакций – крупнейший вклад в мировую науку, его школа химической кинетики – одна из ведущих в мире); академик А.Н. Фрумкин
– создатель теории электрохимических процессов, глава советской школы электрохимиков; академик А.Н. Несмеянов – основатель советской школы элементорганики, академик К.А. Андрианов – создатель нового раздела химии – кремнийорганические соединения.
В своём развитии химия опирается также на практический опыт и потребности всех отраслей народного хозяйства, которые связаны с химической наукой и химической промышленностью.
При химическом взаимодействии между атомами, как правило, взаимодействуют внешние (валентные), наиболее удалённые от ядер электроны, в результате чего при условии антипараллельности спинов электронов возникает химическая связь.
При этом электроны, движущиеся вначале в единичных атомах, после образования молекулы Н2 движутся в едином поле молекулы, что приводит к
5
качественному изменению организации вещества. Это и означает появление химической формы движения.
Следует подчеркнуть, что в химических процессах кроме атомов и молекул участвуют такие самостоятельные, качественно особые дискретные частицы, как ионы, свободные радикалы, макромолекулы. Подчиняясь общим закономерностям протекания химических реакций, этим частицам свойственны и свои, особые закономерности.
Характерный для новейшей химии, как и для всей науки ХХI в., процесс глубокой внутренней дифференциации в значительной степени связан с открытием качественного многообразия химических частиц. Их строение, превращения и свойства стали предметом изучения специальных разделов химии: неорганической химии, электрохимии, коллоидной химии, физической химии, химии высокомолекулярных соединений, биохимии т.д.
Самостоятельной отраслью химии является наука о методах определения качественного и количественного состава вещества — аналитическая химия.
Без современных методов анализа был бы невозможным синтез новых химических соединений, контроль за технологическими процессами и качеством продукции. Важную роль играет анализ в научно-техническом прогрессе, способствуя развитию естественных наук, таких как геохимия, биохимия, биология, физика, медицина и т. д.
На грани соприкосновения физики и химии возникла физическая химия. Предметом её изучения стали общетеоретические вопросы, касающиеся строения и свойств молекул химических веществ и происходящих при этом физических явлений. B центре внимания физико-химиков стоят проблемы физико-химии полупроводников, кинетики каталитических реакций, зависимость между условиями протекания реакций и свойствами продуктов этих реакций (полимеры), проблемы структуры молекул, подбора катализаторов, биокатализа.
Большое развитие получила новейшая отрасль физической химии — радиационная химия. Достижения радиационной химии и химии изотопов широко используются в медицине (исследование обменных процессов в костной ткани, лечение опухолей, диагностика болезней).
6
ГЛАВА I. ПЕРИОДИЧЕСКАЯ СИСТЕМА ЭЛЕМЕНТОВ КАК КЛАССИФИКАЦИЯ ЭЛЕМЕНТОВ ПО СТРОЕНИЮ ЭЛЕКТРОННЫХ ОБОЛОЧЕК АТОМОВ
1.1. Развитие периодического закона. Закон Мозли. Современная трактовка периодического закона
Периодический закон и периодическая система элементов Д.И. Менделеева существует почти 150 лет. Период развития периодического закона характеризуется двумя основными этапами – химическим и физическим. Химический период характерен тем, что в основу классификации элементов была положена атомная масса. Периодичность пытались подтвердить на основе химических и физико-химических свойств. Этому же периоду отвечает Менделеевская формулировка закона: «Свойства простых тел, а также формы и свойства соединений элементов находятся в периодической зависимости от величины атомных весов элементов».
Следует отметить, что Д.И. Менделеев, открыв всеобщий закон природы (1869), не объяснил причины периодического изменения свойств элементов. На химическом этапе невозможно было раскрыть физическую величину периодичности, так как не было данных об электронной структуре атомов. Раскрыть же причину периодичности свойств можно только на основе строения атомов, структура которых получила теоретическое объяснение в квантовой механике.
Более глубокие знания о строении атома позволили сформулировать периодический закон Д.И. Менделеева так: «Свойства элементов находятся в периодической зависимости от электронной структуры атомов». В такой формулировке закона отражена идея, что периодическая повторяемость свойств связана со строением электронной оболочки атомов и каждый раз появление нового типа электронов с иными квантовыми числами обусловливает новые свойства элементов. Повторяемость же подобных свойств связана с тем, что периодически повторяются однотипные электронные конфигурации атомов. В этом и заключается физический смысл периодичности.
1.2. Структура периодической системы элементов Д.И. Менделеева (короткая форма таблицы)
В табличном варианте периодической системы элементов Д.И. Менделеева каждый элемент имеет свой порядковый номер и занимает одну клетку таблицы,
7
химические элементы расположены в направлении увеличения их порядкового номера по периодам, группам и подгруппам.
1.2.1. Порядковый номер химического элемента
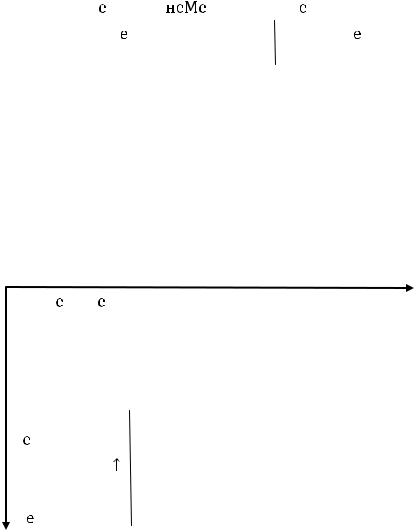
В каждой клетке таблицы, кроме порядкового номера, названия и символа химического элемента, указаны его атомная масса, распределение электронов по энергетическим уровням и электронная конфигурация валентных электронов.
Порядковый номер химического элемента в периодической системе – это важная характеристика атома:
N |
= |
Zя |
= |
Np |
= Ne = |
A |
= Nn |
|||||||
порядковый |
|
|
заряд |
|
|
число |
|
|
число |
|
|
молярная |
|
число |
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|||||||||
номер |
|
|
ядра |
|
|
протонов в |
|
|
электронов |
|
|
масса |
|
нейтронов в |
элемента |
|
|
атома |
|
|
ядре атома |
|
|
в атоме |
|
|
атома |
|
ядре атома |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1.2.2. Период
Период – горизонтальный ряд химических элементов, расположенных в порядке увеличения заряда яда и числа электронов в атоме.
Атомы одного периода имеют одинаковое число энергетических уровней, которое равно номеру периода, обозначенному арабской цифрой. В таблице
имеется 7 периодов. |
|
|
|
||
N (периода) = |
N (энерг. ур.) |
= |
n |
||
|
|
|
|
|
|
номер |
|
число энергетических |
|
|
главное квантовое число |
|
|
|
|||
периода |
|
уровней в атоме |
|
|
последнего энергетического уровня |
|
|
|
|
|
|
1.2.3. Группа
Группа – вертикальный ряд химических элементов, атомы которых имеют одинаковое число валентных электронов, но разное число энергетических уровней. Номера групп обозначены римскими цифрами. В таблице имеется 8 групп.
|
|
||
N (группы) = |
В |
|
|
номер |
число валентных |
||
группы |
электронов |
||
8

1.2.4. Подгруппа
Подгруппа – часть группы, в состав которой входят химические элементы с одинаковым распределением валентных электронов в атоме.
В результате смещения химических элементов влево и вправо каждая группа делится на две подгруппы – главную А и побочную Б.
Главная подгруппа А
В находится:
находится:
-на последнем n-м энергетическом
уровне;
-ns- и npподуровнях.
Включает:
s-элементы: ns1 или ns2;
р-элементы: ns2 npx, где х = 1,2…6.
1.2.5. Лантаноиды и актиноиды.
Побочная подгруппа Б
В находится:
находится:
-на последнем n-м и предпоследнем
(n-1)-м энергетических уровнях;
-ns- и (n-1)d-подуровнях.
Включает:
d-элементы: (n-1)dx ns2, где х=1,2…10.
На ns-подуровне некоторых d-элементов может быть один электрон или не одного.
Лантаноиды и актиноиды – два семейства f-элементов, которые в виде двух отдельных рядов вынесены за пределы таблицы. В последние годы в эти ряды часто включают лантан и актиний.
|
|
Лантаноиды |
|
|
Актиноиды |
Порядковые номера от 57 до 71. |
Порядковые номера от 89 до 103 |
||||
N (периода) = 6 |
N (периода) = 7 |
||||
N (группы) = III |
N (группы) = III |
||||
Подгруппа – побочная Б |
Подгруппа – побочная Б |
||||
|
|
|
|
||
В |
|
находятся: |
В |
|
находятся: |
на трёх последних энергетических |
на трёх последних энергетических |
||||
уровнях; |
уровнях; |
||||
|
|
|
|
|
|
9

подуровнях 4fx5dy6s2, где х=1,2…14 |
подуровнях 5fx6dy7s2, где х=0,1,2…14 |
у=0,1,2; |
у=0,1,2; |
Включает: |
Включает: |
f-элементы |
f-элементы |
Число валентных электронов у атомов этих химических элементов может быть больше или меньше трёх за счёт провала электронов с f-подуровня на d- подуровень или наоборот.
1.3. Электронное строение атома
Электроны в атоме располагаются по энергетическим уровням и подуровням, что изображается чаще всего двумя способами – в виде электронной конфигурации атома или энергетической диаграммой расположения электронов по уровням и подуровням в атоме.
1.3.1. Электронная конфигурация атома
Электронная конфигурация атома отображает расположение электронов в атоме по уровням и подуровням.
При этом следует помнить, что в соответствии с принципом Паули максимально может быть на s-подуровне любого уровня 2 электрона, на р- подуровне – 6 электронов, на d-подуровне – 10 электронов, на f-подуровне – 14 электронов.
В соответствии с правилом наименьшей энергии (правилом Клечковского) уровни и подуровни заполняются в следующей последовательности:
1s 2s 2p 3p 3s 4s 3d 4p 5s 4d 5p 6s 4f 5d 6p 7s 5f 6d 7p.
Электронную конфигурацию атома принято изображать в виде электронной формулы, которая записывается в порядке возрастания номера энергетического уровня, т.е. без учёта последовательности заполнения подуровней электронами.
1.3.2. Энергетическая диаграмма уровней и подуровней
Энергетическая диаграмма уровней и подуровней – это наглядная схема электронной структуры атома, которая в общепринятом, сжатом по вертикали изображении в отсутствии электронов имеет вид
10
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Энергетические уровни |
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5 |
|
|
|
|
|
|
|
|
|
|
|
|
f |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6 |
|
|
|
|
|
|
d |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7 |
|
|
p |
|||||||||||||
|
|
|
|
|
|||||||||||||
|
|
s |
|||||||||||||||
подуровни
На диаграмме обозначаются:
- атомная орбиталь или энергетическая ячейка
-электроны в ячейке или
или 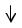 ;
;
-спаренные электроны 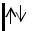
 ;
;
-неспаренные электроны или
Согласно принципу Паули, на одной атомной орбитали (в одной энергетической ячейке) не может находиться больше двух электронов. Следовательно, на s-подуровне (одна атомная орбиталь) может находиться 2 электрона, на р-подуровне (три орбитали) – 6 электронов, на d-подуровне (5 атомных орбиталей) – 10 электронов, на f-подуровне (7 атомных орбиталей) – 14 электронов.
Согласно правилу Гунда, электроны стремятся занять наибольшее число ячеек в подуровне.
1.4. Периодичность свойств атомов химических элементов
Периодическая система элементов Д.И. Менделеева отражает периодичность конфигураций валентных электронов и позволяет на их основе объяснить закономерное изменение физических и химических свойств элементов.
11
1.4.1. Электронное строение последних энергетических уровней
На последних энергетических уровнях атомов распределяются валентные электроны В , которые при протекании химических реакций переходят от атома к атому. Если атом отдаст все В
, которые при протекании химических реакций переходят от атома к атому. Если атом отдаст все В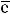 , то элемент приобретает высшую положительную степень окисления (в. ст. ок.). Число валентных электронов, их электронные конфигурации и положительные степени окисления элементов периодически повторяются при непрерывном увеличении заряда ядра атома.
, то элемент приобретает высшую положительную степень окисления (в. ст. ок.). Число валентных электронов, их электронные конфигурации и положительные степени окисления элементов периодически повторяются при непрерывном увеличении заряда ядра атома.
В больших периодах (4,5,6,7) наблюдается периодическое изменение числа валентных электронов и высших положительных степеней окисления дважды – в одном и другом рядах.
Некоторые d-элементы (Cu, Ag, Au) и f-элементы имеют аномально высокие положительные степени окисления за счёт d-электронов предпоследнего и f- электронов третьего от конца уровней.
1.4.2. Размеры атомов
Радиусы атомов с увеличением заряда ядра в группах сверху вниз увеличиваются в связи с ростом числа энергетических уровней, в периодах от начала к концу – уменьшаются, так как при одинаковом числе энергетических уровней усиливается притяжение валентных электронов к ядру атома.
В атомах d- и f-элементов действие возрастающего заряда ядра на радиус атома более слабое, потому в периодах и группах радиусы изменяются в меньшей степени, а у актиноидов они почти постоянны.
1.4.3. Энергетическая характеристика атома
Способность атома удерживать электроны количественно оценивается энергиями ионизации и сродства к электрону или их суммарной величиной, называемой электроотрицательность.
Энергия ионизации Еи – это энергия, необходимая для отрыва от одного моля атомов одного или более молей валентных электронов. Энергия ионизации атомов химических элементов периодически изменяется: повышается от начала периода к его концу, а в главных подгруппах – снизу вверх.
Энергия сродства к электрону Еср. – это энергия присоединения молей электронов к одному молю нейтральных атомов.
Энергия сродства атома к электрону также повышается в периоде от начала к концу, в группе – снизу вверх.
12

Электроотрицательность ЭО характеризует способность атомов в соединениях притягивать к себе электроны и рассчитывается по формуле
ЭО=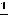 (Еи+Еср).
(Еи+Еср).
Относительная электроотрицательность ОЭО – предложена Полингом, который составил таблицу ОЭО почти всех химических элементов периодической системы. ОЭО рассчитывается по формуле
ОЭО=
 > 1, или < 1, или =1.
> 1, или < 1, или =1.
ОЭО выражена небольшими числами, позволяющими легко сравнивать способность атомов отдавать или принимать электроны, оценивать длину и полярность химической связи во многих соединениях элементов. ОЭО также изменяется периодически, т.е. растёт в периодах от начала к концу и в группах снизу вверх.
Наиболее электроотрицательные элементы – фтор, кислород, хлор, азот.
1.5. Периодичность химических свойств простых веществ
Металлы – s, d, f-элементы и некоторые р-элементы (III группы главной подгруппы А, кроме бора В, и элементы Sn,Pb, Bi, Sb, Po из главных подгрупп 4,5,6,7 групп). Атомы металлов имеют больший, чем у неметаллов, радиус, низкую электроотрицательность и 1,2 или 3 валентных электрона на последнем энергетическом уровне, которые они легко отдают при химическом взаимодействии:
Ме |
– |
ne |
→ |
Меn+ |
Восста- |
|
Отдача |
Процесс |
Окисли- |
новитель |
|
электронов окисления |
тель |
|
Металлы легко окисляются, т.е. они проявляют восстановительные свойства, образуют при окислении положительно заряженные ионы. Металлы называют электроположительными элементами. Величина степени окисления металлов, находящихся в IV, V, VI, VII группах, переменная: от +2 до высшей положительной степени окисления, равной номеру группы.
Неметаллы – это р-элементы, находящиеся в конце периодов в IV,V, VI, VII, VIII группах и бор в III группе. Атомы неметаллов имеют более 3 валентных
электронов |
на |
последнем |
энергетическом |
уровне, |
высокую |
|
|
|
13 |
|
|
электроотрицательность, малые радиусы; они способны как отдавать валентные электроны, так и принимать на последний уровень электроны до 8, т.е. до образования электронной оболочки инертного газа.
неМе-х ← х |
|
+ |
|
|
|
– n |
|
→ неМе+n |
|||||
|
|
|
|||||||||||
Приём |
|
|
|
окислитель или |
Отдача |
|
|
||||||
|
|
|
|
|
|||||||||
Восстановление |
|
восстановитель |
Окисление |
||||||||||
|
|||||||||||||
Неметаллы обладают окислительными и восстановительными свойствами, могут взаимодействовать как с металлами, так и с другими неметаллами.
Периодическое изменение металлических и неметаллических, восстановительных и окислительных свойств простых веществ определяется периодичностью электронного строения соответствующих атомов.
Схема изменения основных характеристик атомов и свойств простых
веществ в периодах и группах. |
|
|
|
|||||||||
|
|
|
|
|
Изменения в периодах |
|||||||
|
|
|
|
|
|
|
|
|
|
|||
|
Zя↑, N |
|
↑, B |
|
↑, ОЭО ↑ |
|
усиливаются неметаллические |
|||||
|
|
|
|
|||||||||
|
r (атома) ↓ |
|
|
и окислительные свойства |
||||||||
|
N (энерг. ур.) = const |
|
простых веществ |
|||||||||
|
|
|||||||||||
Изменения |
Zя↑ |
усиливаются металлические |
||||||||||
|
|
|
|
|
|
|||||||
в главных |
N |
|
|
|
↑ |
и восстановительные |
||||||
подгруппах |
N (энерг. ур.) |
свойства простых веществ |
||||||||||
|
ОЭО↓ |
|
|
|
||||||||
|
|
|
|
|
||||||||
|
B |
|
= const |
|
|
|
||||||
Глава I I. ДИСПЕРСНЫЕ СИСТЕМЫ
2.1. Классификация дисперсных систем
Микрогетерогенные системы, состоящие из вещества, взятого в избытке (среда) и распределённого в нём в виде мелких частиц другого вещества (дисперсная фаза), называются дисперсными. В целом все вещества, образующие систему, называются компонентами системы. Компоненты систем могут существовать в разных фазах. Фазой называется однородная часть системы, отделённая от другой части системы поверхностью раздела. Фазы, представлены жидкостями или твёрдыми веществами, называются конденсированными. Дисперсные системы классифицируются по размерам
14
частиц дисперсной фазы, по агрегатному состоянию компонентов, по интенсивности взаимодействия частиц дисперсной фазы с дисперсионной средой.
По размерам частиц дисперсные системы классифицируются на микрогетерогенные, или грубодисперсные, с размером частиц дисперсной фазы более 1 мкм (1мкм = 10-6м, к таким системам относятся суспензии и эмульсии), ультрамикрогетерогенные, или тонкодисперсные, с размером частиц 0,1 – 0,001 мкм, истинные растворы, где дисперсными частицами являются молекулы, атомы, ионы (размер их порядка одного ангстрема, 1А = 10-10 м).
В зависимости от фазового состояния компонентов различают гомогенные и гетерогенные дисперсные системы. В гомогенных системах между компонентами отсутствует поверхность раздела, т.е. вся система представлена одной фазой (раствор ацетона в воде, раствор сахара в воде, и т.д.). Гетерогенные системы – неоднородные системы – состоят из ряда компонентов, которые разделяются поверхностью раздела (кровь, газированная вода, раствор цемента и др.). Между отдельными фазами в системе при определённых условиях может устанавливаться динамическое равновесие, которое называется фазовым.
Количество фаз в системе можно определить с помощью правила фаз Гиббса, которое устанавливает зависимость между числом степеней свободы и количеством фаз и компонентов:
С = К-Ф+2, где С – число степеней свободы; К – количество компонентов, образующих данную систему; Ф – количество фаз в системе.
Под числом степеней свободы (вариантностью) системы понимают число внешних и внутренних факторов (температура, давление, концентрация), влияющих на состояние системы, значение которых можно изменять в определенных пределах без изменения количества фаз. Системы, не имеющие ни одной степени свободы, называются безвариантными. Отсутствие степеней свободы означает, что такие системы могут существовать только при строго определенных условиях. При изменении хотя бы одного из условий равновесие системы нарушается и одна из фаз исчезает. При изменении давления или температуры одна из фаз исчезает.
Системы, имеющие две степени свободы, называются бивариантными.
15
2.2. Газовое состояние и газовые законы
Газовое состояние возникает при слабом взаимодействии между молекулами смешиваемых веществ. При высоком давлении и низких температурах силы взаимодействия возрастают, в результате чего вещество из газообразного может перейти в конденсированное состояние (жидкое или твёрдое). В целом состояние газообразных веществ зависит от давления, температуры и объёма, который занимает газ.
При изучении газообразного состояния было введено понятие об идеальном газе. Принято считать, что между молекулами идеального газа отсутствуют силы взаимного притяжения и молекулы при соударениях ведут себя, как абсолютно упругие шарики. Отсутствие этих двух условий характеризует реальные газы, поскольку даже при незначительном взаимодействии между молекулами свойства реальных газов отклоняются от свойств идеальных газов. Многие газовые законы были выведены без учёта того, что молекулы газов имеют свой собственный объём и что они на определённом расстоянии могут взаимодействовать. Известно, что чем больше расстояние между молекулами газов, тем слабее это взаимодействие, поэтому реальные газы при высоких температурах и низких давлениях по свойству приближаются к идеальным газам.
Если между газовыми смесями нет химического взаимодействия, они подчиняются общим газовым законам.
Закон Дальтона выражает зависимость между общим давлением смеси газов и парциальным давлением её компонентов. Под парциальным давлением газа понимают то давление, которое имел бы газ, если бы он один занимал объём, который занимает вся смесь, и находился бы при той температуре, что и смесь. Согласно закону Дальтона, давление смеси газов равно сумме парциальных давлений её компонентов. Математически закон Дальтона выражается следующим уравнением:
р = р1+р2+р3+…+рn,
где р – общее давление смеси газов; р1, р2, р3, …; рn – парциальные давления компонентов смеси.
Газы, как правило, незначительно растворяются в жидкостях. Растворимость газов резко изменяется в зависимости от температуры и давления: с увеличением давления растворимость газов возрастает, с повышением температуры – понижается. Эта зависимость имеет большое биологическое значение для организмов, живущих в воде. С повышением температуры дыхание организмов
16
затрудняется, увеличивается потребность в кислороде, тогда как его концентрация в воде уменьшается. Сильное нагревание может привести к гибели организмов от удушья из-за недостатка кислорода. При насыщении воды кислородом в организме становится менее чувствительными к повышению температуры.
Английский химик Генри (1803 г.) при изучении растворимости газов установил закон, который формулируется так: при постоянной температуре растворимость данного газа в определённом объёме жидкости прямо пропорциональна парциальному давлению газа. Математически закон Генри можно выразить уравнением:
С = kp,
где С – весовая концентрация газа в насыщенном растворе; k – коэффициент пропорциональности, или константа Генри (зависит от природы газа и растворителя, от температуры, но не зависит от давления); р – парциальное давление газа.
Дальтон также установил подобную зависимость для смеси газов: растворимость газа в жидкости пропорциональна его парциальному давлению. Закон Дальтона является важным дополнением к закону Генри, и поэтому оба закона часто объединяют в один общий закон Генри – Дальтона, который точно соблюдаются лишь тогда, когда условия приближаются к условиям существования идеальных газов и жидкостей. Во всех остальных случаях, особенно при высоких давлениях и низких температурах, наблюдаются
значительные отклонения от закона. |
|
|
|
||
Закон Генри – Дальтона |
можно использовать при |
расшифровке |
|||
физиологических |
процессов, |
поскольку |
он |
позволяет |
установить |
закономерности растворения газов в крови и в тканях тела. Известно, что газы (кислород и азот) в организм и кровь попадают главным образом из внешней среды, основное же количество углекислоты образуется в тканях организма (в воздухе СО2 содержится 0,03 мас. %).
Количество растворённого газа для крови, как и для других жидкостей, определяется коэффициентом абсорбции газа α, который представляет собой объём газа в миллилитрах (при нормальных условиях), растворённый в миллилитре жидкости при соответствующей температуре и при парциальном давлении газа, равном 101325 Па, и зависит от парциального давления газа в данной пробе крови.
Растворимость газов в крови несколько меньше, чем в воде (таблица 1).
17
Коэффициент абсорбции газов (α) по Бору
|
|
|
|
|
|
Таблица 1. |
|
|
|
|
|
|
|
|
|
|
Газ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
О2 |
|
N2 |
|
CO2 |
|
|
Объект |
|
|
|
|
|
|
|
Температура, 0С |
|
|
|
|
|
||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
15 |
38 |
15 |
38 |
15 |
|
38 |
|
|
|
|
|
|
|
|
Вода |
- |
0,0237 |
- |
0,0122 |
- |
|
0,555 |
0,033 |
0,023 |
0,017 |
0,012 |
0,994 |
|
0,541 |
|
Плазма крови |
|
||||||
0,031 |
0,022 |
0,016 |
0,011 |
0,934 |
|
0,511 |
|
Цельная кровь |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Пользуясь этой таблицей, можно рассчитать количество растворённого в крови газа при любом его парциальном давлении по формуле
m = 
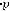



 ,
,
где m – количество растворённого газа, об. %; р – парциальное давление газа в данной пробе крови; α – коэффициент абсорбции газа при данной температуре.
Кровь переносит кислород от лёгких к тканям, а углекислый газ в обратном направлении – от тканей к лёгким. Это чрезвычайно важный процесс, который нарушается при разных патологических состояниях, поэтому информация о количестве этих газов в крови используется для диагностических целей и изучения патогенеза заболевания.
И.М. Сеченов исследовал закономерности процесса поглощения углекислого газа физиологическими жидкостями и солевыми растворами. Он доказал, что растворимость газов в растворителе уменьшается при наличии в нём электролитов и может быть рассчитана по формуле
С = С0е-ра, где С – искомая растворимость газа; С0 – растворимость газа в чистом
растворителе; е – основание натурального логарифма; р – константа, зависящая от температуры, природы газа и электролита, а – концентрация электролита. И.М. Сеченов также установил, что 2/3 всего количества углекислого газа растворяется в плазме крови, а 1/3 связывается с красными кровяными тельцами и что процесс перехода СО2 из крови в лёгкие аналогичен диффузии газа из жидкости в воздух. На основании этих исследований Сеченов определил роль гемоглобина крови в переносе кислорода и углекислого газа.
18
2.3. Жидкое состояние. Концентрация растворов и способы её выражения
По свойствам жидкости занимают промежуточное положение между газами и твёрдыми телами. Подобно газам, жидкости принимают форму того сосуда, в котором они находятся. Жидкости имеют свой собственный объём и, подобно твёрдым телам, малосжимаемы, обладают значительной плотностью. В отличие от твёрдых тел жидкости обладают текучестью, что связано со способностью молекул свободно перемещаться, а также совершать поступательное и вращательное движения. Силы взаимодействия, возникающие между молекулами жидкостей, значительно больше сил между молекулами газов, но меньше сил между молекулами твёрдых тел, чтобы препятствовать перемещению частиц друг относительно друга. Одним из свойств поверхности жидкости является её стремление к сокращению своего объёма, что проявляется в сферической форме капель и в других свойствах поверхностей жидкостей.
Жидкости, так же как и твёрдые тела, обладают определённой структурой, например, жидкая вода имеет структуру, подобную структуре льда. Подобно индивидуальным жидким веществам растворы имеют так называемую внутреннюю структуру ближнего порядка. Это следует понимать так: разбавленные растворы имеют структуру, подобную структуре растворителя, концентрированные – подобную структуре растворённого вещества.
Растворы – это однофазные системы переменного состава, образованные двумя и большим количеством компонентов. Однородность растворов роднит их по свойствам с химическими соединениями, переменность состава обусловливает свойства, характерные для механических смесей. Растворы играют исключительно важную роль в жизни человека, в природе и технике. Значительное количество известных химических реакций протекает в растворах. Воды мирового океана и атмосфера являются растворами. Физиологические жидкости – плазма крови, лимфа, спинномозговая жидкость, желудочный и кишечный соки, пот, моча и другие – также являются растворами. Почти все лекарства оказывают лечебное действие на организм в растворённом состоянии. С растворами связаны почти все физиологические процессы, и управление ими в определённой степени будет зависеть от наших знаний о внутренней структуре и свойствах растворов.
Одной из важнейших характеристик любого вещества является его растворимость, т. e. способность распределяться в том или ином растворителе.
В зависимости от количества растворённого вещества растворы бывают
19
насыщенными, ненасыщенными, перенасыщенными и разбавленными. Концентрацию растворов можно выражать по-разному. На практике наиболее употребительны следующие способы выражения концентрации:
1.Процентная концентрация показывает, сколько граммов данного вещества содержится в 100 г раствора. Например, 10%-ный раствор NaCl — это значит, что в 100 г раствора содержится 10 г NaCl и 90 г воды.
2.Молярная концентрация показывает, сколько молей растворённого вещества содержится в 1 л раствора.
3.Нормальная концентрация показывает, сколько грамм-эквивалентов растворенного вещества содержится в 1 л раствора.
4.Моляльная концентрация показывает; сколько молей растворенного вещества содержится в 1000 г растворителя.
Титр показывает, сколько граммов растворённого вещества содержится в 1 мл раствора. Наряду с перечисленными способами выражения концентрации существуют и другие, применяющиеся для выражения концентрации биологически активных веществ. Например, содержание биологически активных веществ в тканях выражают в микрограммах (1 мкг = 10-6 г), отнесённых к 1 г живой ткани. Концентрацию биологически активных веществ в физиологических жидкостях выражают в граммах на литр, которая показывает сколько граммов вещества содержится в 1000 мл жидкости.
При изготовлении растворов лекарственных препаратов пользуются процентной концентрацией. При этом концентрацию лекарственных препаратов выражают следующим образом:
1.В массовых процентах, показывающих массовое количество вещества, содержащегося в 100 г раствора.
Например:
Rp: Sol Natril bromidi |
2% |
– |
200,0 |
Misce, Da. Signa. |
По |
1 |
столовой ложке 3 раза в день |
Для приготовления такого раствора 4 г NaBr растворяют в 196 г воды.
2. В массо-объёмных процентах, показывающих массовое количество вещества, содержащегося в 100 мл раствора.
Например: |
|
Rp.: Natril bromidi |
5,0 |
Kalii bromidi |
5,0 |
Aq. destill. |
200,0 |
20
Misce. Da. Signa. По 1 столовой ложке 3 раза в день.
Для приготовления такого раствора 5 г NaBr и 5 г KBr растворяют в воде до получения 200 мл раствора.
3. В объёмных процентах, показывающих объёмное содержание растворённого вещества в 100 мл раствора.
Например:
Pp.: Sol. Hуdrogenil peroxydi dilutae 5,0 Spiritus aethylici 950 5,0
Misce. Da. Signa. По 10 капель в ухо.
Для приготовления данной прописи пероксид и спирт берут по указанному объёму.
В современной медицинской практике применяют и другие способы выражения количества лекарственных препаратов в рецептах, однако выражение концентрации лекарственных веществ в процентах наиболее распространено.
2.4. Свойства разбавленных растворов. Закон Рауля. Криоскопия, эбулиоскопия
Между молекулами жидкости действуют силы притяжения, которые образуют внутреннее молекулярное поле сил, жидкости, характеризующиеся одинаковыми межмолекулярными полями, смешиваются неограниченно, при их смешивании не выделяется и не поглощается тепло, общий объём равен сумме объёмов исходных жидкостей и свойства смеси изменяются пропорционально составу. Жидкие смеси, обладающие такими свойствами, называются идеальными растворами. Приготовление же реальных растворов сопровождается либо поглощением (растворение NaC1 в Н2О), либо выделением тепла (растворение Н2SO4 в Н2О), изменением объёма и т. д. Это обусловлено тем, что в природе практически не существует двух жидкостей, молекулярные силовые поля которых были бы совершенно одинаковыми. Однако реальные растворы с предельно малой концентрацией растворённого вещества можно считать идеальными, поскольку взаимодействие между молекулами растворённого вещества в них практически отсутствует.
Рассмотрим некоторые свойства идеальных растворов.
Из опыта известно, что температура кипения растворов выше температуры кипения чистых растворителей. Чем это объясняется? Давление насыщенных паров над какой-либо жидкостью при постоянной температуре является
21
постоянной величиной. При добавлении к жидкости какого-нибудь нелетучего соединения между eгo молекулами и молекулами растворённого вещества возникает взаимодействие, приводящее к образованию гидратов (растворитель – вода) или сольватов (неводный растворитель). Связывание молекул растворителя с молекулами растворённого вещества препятствует переходу молекул растворителя в парообразную фазу. Поэтому при одной и той же температуре упругость паров растворителя над раствором будет меньше, чем над чистым растворителем. Начинают кипеть растворы тогда, когда упругость их паров достигает внешнего давления, а следовательно, растворы закипают при более высокой температуре.
Французский химик Рауль, исследуя зависимость между давлением насыщенного пара и температурами кипения и замерзания растворов, установил закон, который позволяет рассчитать, на сколько повышается температура кипения или на сколько понижается температура замерзания растворов по сравнению с чистыми растворителями. Закон Рауля формулируется так: повышение температуры кипения, как и понижение температуры замерзания разбавленных растворов, пропорционально концентрации растворенного вещества.
Согласно закону Рауля, повышение температуры кипения
tk = E· m,
где m – моляльная концентрация вещества; Е – эбулиоскопическая константа. Если m = 1, то tk = Е, т.е. эбулиоскопическая константа показывает, на
сколько повышается температура кипения раствора, если в 1000 г растворителя содержится 1 моль вещества. В этом и заключается физический смысл эбуллиоскопической константы. Опыт также показывает, что температура замерзания растворов ниже температуры замерзания соответствующих чистых растворителей.
Температура замерзания воды, как и любого водного раствора, называется температура, при которой лёд, жидкая вода и пар находятся в динамическом равновесии. Чем выше концентрация раствора , тем ниже температура его замерзания по сравнению с чистым растворителем. По закону Рауля, понижение температуры замерзания раствора
t3 = mK,
где m – моляльная концентрация раствора; K – криоскопическая константа.
Если m = 1, то t3 = K, т.е. криоскопическая константа показывает, на сколько понижается температура замерзания раствора, если в 1000 г
22
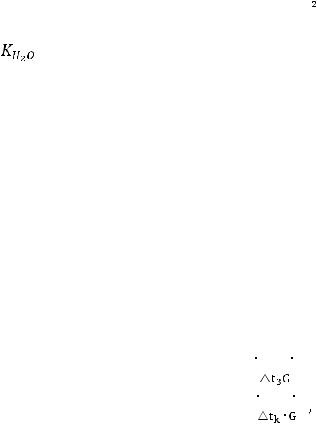
растворителя содержится 1 моль вещества.
Криоскопическая константа так же, как и эбулиоскопическая, не зависит от природы растворённого вещества, а зависит от природы растворителя. Эбулиоскопическая константа для воды 
 = 0,510 С, для бензольных растворов Е = 2,50 С, для хлороформа Е = 3,80 С. Криоскопическая константа для
= 0,510 С, для бензольных растворов Е = 2,50 С, для хлороформа Е = 3,80 С. Криоскопическая константа для
воды |
= 1,860 С. |
Следует отметить, что закон Рауля справедлив для разбавленных растворов неэлектролитов, в растворах электролитов наблюдается отклонение от закона, причём с повышением концентрации вещества отклонение опытных данных от расчётных увеличивается.
Измеряя температуры замерзания или температуры кипения растворов неэлектролитов, по закону Рауля можно рассчитать молекулярную массу растворённого вещества.
Метод определения молекулярной массы вещества, основанной на измерении повышения температуры кипения растворов, называется эбуллиоскопическим, основанным на измерении понижения температуры замерзания растворов – криоскопическим. Расчёт ведут по формулам:
М= 

М= 
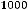
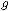
где М – искомая молекулярная масса вещества; К и Е – соответственно криоскопическая и эбуллиоскопическая константы; 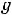 – навеска растворённого вещества, г;
– навеска растворённого вещества, г; 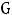 – масса растворителя, г.
– масса растворителя, г.
Эбулиоскопией можно пользоваться тогда, когда растворённое вещество нелетучее и растворитель не разлагаются при кипении раствора. Метод криоскопии более удобен, поскольку при температуре замерзания растворов растворённые вещества не разрушаются. Оба метода иногда применяют для определения молекулярных масс биологически активных веществ. Кроме того, криоскопия так же, как и эбулиоскопия, применяется для определения активности ионов в растворах слабых электролитов. Данные по определению активности можно использовать для расчёта осмотического давления, степени электролитической диссоциации, константы нестойкости, а также для расчёта относительного понижения давления насыщенного пара растворителя.
23
2.5. Осмотическое давление. Закон Вант-Гоффа. Роль осмоса в биологических процессах
Процесс проникновения молекул растворённого вещества между молекулами растворителя, в результате которого концентрация раствора выравнивается, называется диффузией. Это явление обусловлено тепловым движением молекул растворённого вещества. Если же в сосуд А поместить чистый растворитель – воду, а в сосуд В – водный раствор сахара и внизу закрепить полупроницаемую перегородку (проницаемую для молекул воды и непроницаемую для молекул сахара), то вода из сосуда А будет переходить в сосуд В, а молекулы сахара из сосуда В переходить не будут. Таким образом, в описанном опыте наблюдается односторонняя диффузия. Переход молекул растворителя в раствор через полупроницаемую перегородку называется осмосом. Причина этого явления заключается в следующем. Молекулы растворителя могут перемещаться через перегородку в обоих направлениях, но скорость их перемещения будет разной, поскольку растворитель содержит молекулы только растворителя, в растворе же молекулы растворителя сольватационно связаны с растворённым веществом и поэтому их концентрация меньше. Для того чтобы концентрации стали равными, молекулы растворителя (из чистого растворителя) должны с большей скоростью переходить в раствор, чем из раствора в растворитель. Давление, которое необходимо приложить, чтобы скорости обоих процессов стали равными, называется осмотическим. Существует и другое определение: давление, которое необходимо приложить к полупроницаемой перегородке (на 1 см2), чтобы растворитель не проникал через неё, называется осмотическим давлением раствора. Осмотическое давление возникает только тогда, когда раствор отделяется от растворителя полупроницаемой перегородкой. Поэтому осмотическое давление следует рассматривать не как свойство растворённого вещества, растворителя или раствора, а как свойство системы, состоящей из раствора, растворителя и разделяющей их полупроницаемой перегородки (мембраны). В процессе осмоса растворитель перемещается через мембрану от раствора менее концентрированного к раствору более концентрированному.
Расчёты показали, что осмотическое давление в разбавленных растворах подчиняется законам, применяемым к идеальным газам. Эта закономерность, впервые установленная Вант-Гоффом в 1866 г., формулируется следующим образом: осмотическое давление раствора равно давлению, которое производило
24
бы растворённое вещество, находясь при той же температуре в газообразном состоянии, и занимая объём, равный объёму раствора. Математически закон Вант-Гоффа записывается так:
р = СRT,
где р – осмотическое давление; R – универсальная газовая постоянная; С – молярная концентрация; Т – абсолютная температура.
При растворении электролитов количество частиц в растворе увеличивается по сравнению с исходным количеством молекул за счёт диссоциации последних. Поэтому растворы электролитов не подчиняются закону Вант-Гоффа. Для корректирования отклонения от закона был введён изотонический коэффициент i, названный коэффициентом Вант-Гоффа. Физический смысл изотонического коэффициента можно представить так. Пусть растворено N молекул вещества, из них N1 продиссоциировало на n ионов каждая, в результате чего в растворе образовалось N1n ионов, количество непродиссоциировавших молекул равно N – N1, а общее количество частиц в растворе (непродиссоциировавших молекул и ионов) равно (N – N1) + N1n. Изотонический коэффициент равен отношению общего количества частиц в растворе к исходному количеству молекул:
i = |
– |
, |
|
т.е. изотонический коэффициент показывает, во сколько раз увеличилось количество частиц в растворе по сравнению с исходным количеством молекул. Выражается изотонический коэффициент положительным числом. Для неэлектролитов i = 1, для электролитов i 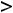 1, а для растворов, в которых происходит полимеризация растворённого вещества, i
1, а для растворов, в которых происходит полимеризация растворённого вещества, i 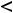 1.
1.
Осмотическое давление в реальных растворах рассчитывают по формуле
р= iСRT.
Втаком виде закон Вант-Гоффа применим к разбавленным растворам неэлектролитов и к очень разбавленным растворам слабых электролитов.
Растворы с одинаковым осмотическим давлением называются изотоническими. Однако поскольку осмотическое давление зависит от количества частиц растворённого вещества, то изотонические растворы одного и того же вещества должны иметь одинаковую концентрацию.
Вклетках роль полупроницаемой перегородки выполняет клеточная оболочка (протопласт), непроницаемая для растворённого вещества. Если клетку поместить в раствор, в котором концентрация вещества выше, чем в клетке, то
начнётся переход растворителя из клетки в раствор, клетка сморщится
25
(плазмолиз). Растворы с более высоким содержанием вещества, чем в клетке, называются гипертоническими, с более низкой концентрацией вещества – гипотоническими. При помещении клетки в гипотонический раствор наблюдается переход растворителя в клетку, это вызывает увеличение объёма клетки вплоть до её разрушения (гемолиз). Изотонические растворы не вызывают изменения объёма клетки, поэтому они широко применяются в медицинской практике для возмещения объёма крови и повышения кровяного давления, а также при обезвоживании организма (например, при ожогах, сильной рвоте), для этого используются 0,85 – 0,90% раствор NaCl и 4,5 – 5,0% раствор глюкозы.
В живых организмах в нормальном состоянии внутриклеточная жидкость находится в изотоническом равновесии с внеклеточной жидкостью, например кровью. Общее нормальное осмотическое давление плазмы крови человека равно 7 · 108 – 8 · 108 Па. Равенство осмотического давления клеточного содержимого и раствора, окружающего клетку, является важнейшим физиологическим условием нормального функционирования клетки. Поэтому осмос и осмотическое давление играет важную роль в процессах осморегуляции живых организмов. Исключительно важна роль осмоса для живых организмов в распределении воды между тканями и клетками.
Осмотическое давление измеряется приборами, называемыми осмометрами. Знание величины осмотического давления необходимо для расчёта осмотической работы, производимой тканевой или клеточной мембраной для создания равной концентрации воды по обе стороны полупроницаемой мембраны. Осмометрия широко применяется для определения молекулярной массы таких биологически важных веществ, как белки, углеводы, нуклеиновые кислоты и др.
Понятие растворитель и растворённое вещество в опредлённой степени условно. Растворителем принято считать то вещество, которого в растворе больше. Иногда за растворитель принимают вещество, которое при охлаждении раствора кристаллизуется первым. Если система гетерогенна, то обычно растворителем считают жидкое вещество,
До конца XIX в. существовало представление, что растворённое вещество и растворитель не взаимодействуют. Это представление было положено в основу так называемой физической теории растворов, основоположником которой был Вант-Гофф. Согласно этой теории, растворитель рассматривался как химически индиферентная среда, в которой равномерно распределялись частицы
26
растворённого вещества (ионы, молекулы). Другими словами, растворы рассматривались как механические смеси. С помощью физической теории удалось объяснить некоторые свойства разбавленных растворов: повышение температуры кипения, понижение температуры замерзания, понижение давления пара над раствором, т. е. те свойства, которые в разбавленных растворах зависят только от количества частиц, находящихся в растворе, но не зависят от их природы. В то же время физическая теория растворов не объясняла свойств концентрированных растворов.
В1887 г. Д И. Менделеевым была предложена химическая теория растворов. Согласно этой теории, частицы растворённого вещества взаимодействуют с молекулами растворителя, в результате чего образуются нестойкие соединения переменного состава, называемые сольватами (неводный растворитель) или гидратами (растворитель — вода). Гидраты и сольваты возникают не за счёт основных химических связей, а главным образом за счёт вандерваальсовских сил или водородной связи. Так как энергия этих связей невелика, то образующиеся соединения непрочные.
Подтверждением теории Менделеева служили факты, что при растворении солей СаСl2, СuSO4 и других в воде при кристаллизации растворов последних выделяются кристалогидраты СаС12· 6H2O, CuSO4· 5Н2О и др., и что при нагревании кристаллогидраты разлагаются с образованием настоящих растворов. Проявление тепловых эффектов при растворении, изменение физических свойств растворов при изменении состава раствора — все это свидетельствовало о том, что растворитель не индифферентен по отношению к растворённому веществу и что молекулы растворителя и растворённого вещества взаимодействуют между собой.
Однако следует отметить, что поскольку растворы в определённой степени проявляют свойства и механических смесей (переменность состава), и химических соединений (однородность, однофазность), то обе теории и физическая, и химическая не исключают, а дополняют друг друга.
В1891 г. И.А. Каблуков развил теорию Д. И. Менделеева и показал, что гидратации, а также сольватации подвергаются не только молекулы, но и ионы
растворённого вещества. Например, диссоциация кислоты проходит по схеме НС1↔H+ + Cl-,
однако, учитывая гидратацию иона Н+, уравнение диссоциации следует писать так:
НС1+H2О↔ H3О+ + Cl-.
27
Таким образом, ион гидроксония H3О+ является гидратированным ионом воды.
Kaк уже отмечалось выше, жидкие вещества имеют определённую структуру. Если повышается концентрация вещества в растворе, то, как правило, усиливается взаимодействие между компонентами раствора и это усложняет в целом его структуру. Структура разбавленных растворов приближается к структуре чистых растворителей, структура концентрированных растворов определяется концентрацией растворённого вещества, а также природой растворителя и растворяемого вещества. Природа растворителя и растворённого вещества в свою очередь определяется, главным образом, структурой их молекул и характером связей, которые возникают в молекулах и между молекулами. Все эти факторы и определяют различную растворимость веществ.
К сожалению, до настоящего времени нет теории, с помощью которой можно было бы предсказать или вычислить растворимость заранее. Однако установлены закономерности, подтверждающие влияние природы растворителя и растворяемого вещества на растворимость. Например, существует эмпирическое правило, что подобное растворяется в подобном: полярные молекулы лучше растворяются в полярных растворителях, неполярные молекулы – в неполярных. Так, хлороводород, который полностью диссоциирует в воде, совсем не диссоциирует в бензоле, в воде его растворимость высокая, а в бензоле практически отсутствует. Бензол хорошо растворяется в гексане (неполярные молекулы) и не растворяется в воде (полярные молекулы). Подобные закономерности наблюдаются и при некоторых биологических процессах. Правило — подобное растворяется в подобном — лежит в основе некоторых теорий клеточной проницаемости. При этом имеется в виду, что оболочки многих клеток содержат неполярные вещества – липиды.
Известно также правило Семенченко: растворимость данного вещества проходит через максимум в ряду растворителей, расположенных по возрастающему значению межмолекулярного взаимодействия в них. Установлено, что максимум отвечает тому растворителю, молекулярное поле сил которого близко к молекулярному полю сил растворённого вещества. Однако следует подчеркнуть, что это правило также не всегда справедливо, особенно для концентрированных твёрдых растворов, а также при наличии химического взаимодействия между компонентами.
Твёрдое состояние – одно из агрегатных состояний, свойственное
28
кристаллическим и аморфным веществам. Для кристаллических веществ характерен так называемый дальний порядок во взаимном расположении частиц, из которых состоит вещество, для аморфных – ближний порядок. Дальний порядок обозначает строгую периодическую повторяемость структурного элемента в бесконечно большом количестве периодов. Для ближнего порядка характерно, что определённый порядок во взаимном расположении можно зафиксировать только для близлежащих частиц; а для отдалённых друг от друга частиц (атомов, ионов, молекул) зависимость в расположении отсутствует.Этот порядок и определяет тот факт, что рентгеноскопические исследования не обнаруживают кристаллической решётки у аморфных тел. Аморфная структура, в свою очередь, определяет общие свойства аморфных веществ: отсутствие определённой температуры плавления, теплопроводности; коэффициент линейного расширения, скорость распространения света и другие параметры не зависят от направления. Такие вещества называются изотропными в отличие от кристаллических тел, в которых многие свойства зависят от направления (анизотропия).
К аморфным телам относятся многие технически важные материалы: стекло, естественные и синтетические смолы, каучук, целлюлоза, белки, а также некоторые вещества, образующие растительные и животные клетки и ткани.
Основным методом исследования дальнего и ближнего порядка в структуре является рентгеноструктурный анализ, основанный на явлении дифракции рентгентовских лучей.
Твёрдые растворы – это однородные кристаллические или аморфные фазы переменного состава, состоящие из двух и более компонентов. Для кристаллических твёрдых растворов характерно, что атомы растворённого компонента замещают атомы растворителя в его кристаллической решётке (растворы замещения), располагаются между ними (растворы внедрения) или образуют особый тип раствора с дефектной кристаллической решёткой (растворы вычитания).
Для кристаллических тел характерны явления изоморфизма и полиморфизма. Явление изоморфизма заключается в том, что из раствора двух кристаллических веществ может кристаллизоваться твёрдая фаза, содержащая оба исходных вещества в неопределённых соотношениях. Например, из растворов алюмокалиевых К2SO4·Al2(SO4)3·24H2O и хромокалиевых K2SO4·Cr2(SO4)3·24H2O квасцов выделяются кристаллы, в состав которых входят хром и алюминий. Изоморфные кристаллические решётки могут возникать изза
29
того, что атомы и ионы одного вещества распределяются в узлах кристаллической решётки другого, при этом тип элементарной ячейки исходных кристаллов должен быть один и тот же, а размеры ячеек близкими. Однако возможно образование твёрдых изомолярных смесей и за счёт диффузии, в результате чего атомы (или ионы) одного вещества внедряются в промежутки кристаллической ячейки другого вещества.
Полиморфизм заключается в том, что одно и то же вещество в зависимости от условий его получения может иметь различную структуру кристаллов. Как известно, углерод существует в двух основных структурных формах: алмаз и графит. Графит имеет гексагональную кристаллическую решётку, а тип связи — ковалентно-металлический. Алмаз имеет кубическую структуру и является типичным ковалентным соединением.
Кристаллическое строение имеют многие биологически важные вещества. Так, с помощью рентгенографии было установлено, что к кристаллическим веществам относятся некоторые вирусы, миоглобин, инсулин, пепсин, гемоглобин, рибонуклеаза и др. Удалось определить также их молекулярную массу, форму, размер и симметрию молекул.
Существуют жидкие вещества, названные жидкими кристаллами. Для жидких кристаллов характерно то, что их структура является промежуточной между структурами жидкости и кристаллического твёрдого тела (такое состояние называют иногда мезоморфизмом). Это выражается в том, что частицы могут свободно перемещаться друг относительно друга, однако их ориентация сохраняется. Поэтому в жидкокристаллическом состоянии могут находится лишь те вещества, молекулы которых имеют удлинённую форму, поэтому его водный или водно-спиртовой раствор обладает свойствами жидкого кристалла. Так, если каплю жидкого мыла подсушить, а затем рассмотреть в поляризационный микроскоп, то станет видна его анизотропность.
Полипептиды, ДНК также при растворении образуют жидкие кристаллы. Жидкокристаллическую структуру имеют и некоторые ткани живых организмов, например миелиновая оболочка нервов. Поэтому пучок нервных волокон в поляризационном свете выглядит ярко светящимся.
Структура жидкого кристалла может изменяться под действием внешних факторов: электрического и магнитного полей, тепла, излучения и др. Изменение структуры приводит к тому, что изменяется длина волны света, отражаемого жидким кристаллом. Это свойство применяется в технике, например, для распознавания внутренних дефектов в деталях. Плёнки на основе жидких
30
кристаллов используются для изучения распределения температуры поверхностей.
Ученые считают также, что жидкие кристаллы можно использовать для диагностики болезней.
Глава III. ЭЛЕМЕНТЫ ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ
3.1. Предмет и метод химической термодинамики. Некоторые понятия и определения
Термодинамика — наука о превращениях энергии, Поскольку все процессы, происходящие внутри и вокруг нас, идут с превращением энергии, то термодинамика описывает огромное количество процессов. Законы термодинамики, используемые для описания химических превращений веществ, составляют предмет химической термодинамики.
Химическая термодинамика позволяет заранее решить вопрос о возможности протекания реакций, определить необходимые для протекания процесса температуру и давление, позволяет предсказать и рассчитать устойчивость вещества при различных условиях. Химическая термодинамика изучает также энергетические эффекты процессов и их зависимость от условий протекания этих процессов. Другими словами, задача химической термодинамики заключается в применении термодинамического метода к изучению химических процессов.
Xимическая термодинамика оперирует целым рядом понятий и определений. Важнейшими являются следующие:
Система — тело или группа тел, мысленно обособленных от окружающей среды; состояние системы — совокупность всех физических и химических свойств системы. Если изменяется хотя бы одно из свойств системы, то это приводит к изменению состояния всей системы в целом.
Различают системы изолированные, закрытые и открытые. Изолированная система не обменивается с окружающей средой массой и энергией (работой), в изолированных системах общий запас энергии остаётся постоянным, энергия может только с одного вида превращаться в другой. Система, которая может обмениваться массой с окружающей средой, называется открытой. Система, которая не может обмениваться массой с окружающей средой, называется закрытой.
Любое свойство системы, являющееся независимым переменным, называется термодинамическим параметром состояния. Если же свойство не
31
является независимым переменным, то оно называется функцией состояния. Постоянное состояние свойственно только изолированным системам. Изменение состояния системы называется термодинамическим процессом. При изменении состояния системы изменяются и параметры. Основными легкоопределяемыми параметрами системы являются температура, давление и объём. Зная эти параметры, можно определить другие, зависящие от них свойства системы.
В связи с существованием различных форм движения материи и различных видов энергии энергия между системами также передаётся в различных формах. Для механической формы движения материи характерна форма обмена энергий, являющаяся механической работой, возникающей при перемещении тела под действием механической силы. Если нет механического движения, то нет и работы. B целом же количество работы А равно количеству энергии, которой обменялись системы в форме механической работы. Поскольку работа является мерой передаваемой энергии, количество её измеряется в тех же единицах, что и энергия. Считается, что работа является формой передачи упорядоченного движения материи, так как при совершении работы частицы тела движутся в определённом направлении. Существует и такая форма обмена энергией, при которой частицы движутся ненаправленно, хаотично, и тогда системы обмениваются энергией в форме тепла.
Любая термодинамическая система обладает определённым запасом энергии. Основным термодинамическим понятием является понятие о внутренней энергией U. Внутренняя энергия представляет собой сумму энергий всех видов движений (поступательного, вращательного, колебательного) частиц, образующих систему, т. е. это энергия молекул, ионов, атомов, ядер и электронов, внутриядерная энергия и энергия межмолекулярного взаимодействия. Не относится ко внутренней энергии кинетическая энергия тела (системы) и потенциальная энергия в поле тяготения. Запас внутренней энергии определяется природой вещества, его массой и состоянием системы. Определить абсолютное значение внутренней энергии невозможно, но поскольку внутренняя энергия является функцией состояния, т. е. изменяется с изменением состояния, то можно определить изменение внутренней энергии U системы. Например, пусть имеется система, характеризующаяся параметрами р1, υ1, T1 (состояние I); при изменении параметров р2, υ2, T2 система перейдёт в состояние II. Каждому состоянию системы отвечает определённый запас внутренней энергии, который в общем виде можно записать:
32
UI = f (р1, υ1, T1);
UII = f (р2, υ2, T2).
Если система из состояния I перешла в состояние II, то
U = UII - UI,
если система из состояния II перейдёт в состояние I (для этого надо восстановить исходные параметры системы), то
U = UI - UII.
Таким образом, изменение внутренней энергии системы равно разности энергий конечного и исходного состоянии, а сама внутренняя энергия не зависит от пути процесса. Если отнести внутреннюю энергию к одному молю вещества, то внутренняя энергия будет зависеть только от состояния системы.
Важным термодинамическим понятием является энтальпия Н. Энтальпия — это теплосодержание системы и одновременно свойство вещества, подобно тому, как свойством вещества является его внутренняя энергия. В общем виде энтальпия определяется уравнением:
Н = U + p ,
,
где U – внутренняя энергия; р – давление; 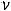 – объём системы.
– объём системы.
Энтальпия, так же как и внутренняя энергия, является функцией состояния системы, т. е. запас теплосодержания системы зависит только от природы веществ и их состояния и не зависит от пути перехода системы от одного состояния в другое.
3.2. Законы термодинамики. Энтропия
Первый закон является частным случаем закона сохранения и превращения энергии:
Q = U +А,
где Q – тепло, поглощаемое или выделяемое системой; U – изменение внутренней энергии системы (U = U2 – U1, U2 – внутренняя энергия конечного состояния, U1 – внутренняя энергия исходного состояния системы); А – работа, совершённая системой (или над нею).
Приведённое уравнение показывает, что сумма изменения внутренней энергии и работы, совершённой системой (или над нею), равна теплоте, сообщённой системе (или выделенной ею).
Несложный анализ уравнения первого закона термодинамики показывает, что если изменение состояния системы сопровождается увеличением внутренней энергии (+U = + Q - A), то приращение внутренней энергии
33
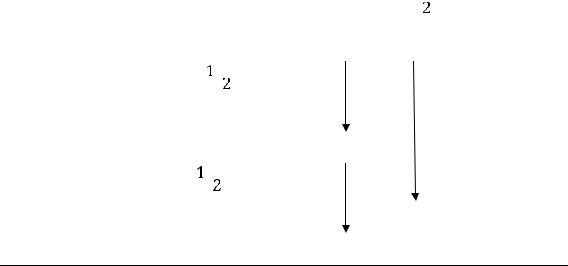
происходит за счёт получения энергии извне (+Q) и работу над системой совершают внешние силы (- А).
Если же в системе уменьшается запас внутренней энергии (-U = - Q + A), то это значит, что количество тепла в системе уменьшается, а работа (+А), совершаемая системой, осуществляется за счёт энергии самой системы.
Если осуществляется процесс, при котором внутренняя энергия не увеличивается (U = 0), тогда Q = A, т.е. тепло, подводимое к системе, полностью расходуется на производство работы над системой и эта работа превращается в тепло, которое выделяется системой. При отсутствии теплового обмена системы с окружающей средой -U = А, т.е. работа самой системы возможна за счёт уменьшения внутренней энергии.
Таким образом, первый закон термодинамики утверждает постоянство внутренней энергии в изолированной системе, доказывает эквивалентность различных форм энергии при их взаимном превращении, устанавливает соотношение, связывающее изменение внутренней энергии с количеством подведённой теплоты и произведённой работы.
Закон Гесса. В 1936 г. Русский академик Г.И. Гесс, измеряя теплоты различных реакций, открыл закон: тепловой эффект реакции (при постоянном давлении) не зависит от пути реакции, т. е. от того, образуется ли продукт непосредственно из исходных веществ или через промежуточные соединения.
Тепловой эффект (энтапия реакции) определяется только природой и состояние исходных веществ и образовавшихся продуктов.
Рассмотрим для примера реакцию горения графита. Она может происходить по двум путям – I и II, как это показано на схеме:
Исходные вещества: С (графит) + 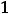 О2 (r)
О2 (r)
|
II путь |
I путь |
|
|
|
|
|
Реакция: С (графит) + |
О2 (r) → |
|
|
→ промежуточное соединение СО(r) |
|
||
∆H2= –110,62 кДж |
|
||
|
|
|
∆Н1= –393,77 кДж |
Реакция: СО (r) + + |
О2 (r) → |
||
→продукт реакции СО2 (r)
∆H3 = –283,15 кДж
34
По первому пути графит сгорает сразу до диоксида углерода (∆Н1), а по второму – сначала образуется оксид углерода СО (∆Н2), который затем дожигается до СО2 (∆Н3).
По закону Гесса энтальпия реакции не зависит от пути процесса. Поэтому ∆Н1 = ∆Н2 + ∆Н3. Величины ∆Н1 и ∆Н2 легко определить путём сжигания графита и СО в калометре. Величину же ∆Н2 таким образом определить нельзя из-за невозможности сжигания графита с образованием только чистой СО без СО2. Таким образом, закон Гесса позволяет находить энтальпии реакций, которые не могут быть непосредственно измерены. В рассматриваемом случае:
∆Н2 = ∆Н1 – ∆Н3 = –393,77 – ( –283,15) = –110,62 кДж/моль Однако первый закон термодинамики не затрагивает вопроса возможности
протекания и направления спонтанных процессов, т. е. процессов, происходящих самопроизвольно без затраты работы извне.
Второй закон термодинамики позволяет установить возможность или невозможность протекания процессов и их направление. Второй закон термодинамики тесно связан с обратимостью и необратимостью процессов. Следует отметить, что термодинамическое понятие обратимости не совпадает с понятием обратимости в химической кинетике. С позиций химической кинетики в обратимых процессах прямая и обратная реакции характеризуются определёнными скоростями, причём величина разности скоростей не ограничивается. Для термодинамической обратимости требуется, чтобы реакция проходила в практически равновесных условиях, когда скорости прямого и обратного процессов различаются на бесконечно малую величину.
Самопроизвольно протекающие процессы характеризуются конечной скоростью, поэтому они являются неравновесными, а следовательно, и необратимыми. Примером необратимых процессов является диффузия газов, процессы, проходящие со взрывом, передача тепла, старение организмов. Абсолютно термодинамически обратимых процессов не существует, поскольку невозможно, чтобы не только система, но и окружающая её среда вернулись точно в первоначальное состояние.
Второй закон термодинамики вводит новую функцию состояния – энтропию S. Энтропия является мерой неупорядоченности состояния системы. Чем более неупорядоченна система, тем больше энтропия. Так, с повышением температуры энтропия возрастает. Если в системе протекают обратимые процессы, то энтропия такой системы остаётся постоянной, следовательно, S = 0(S = S2 – S1, где S2 – энтропия конечного состояния, а S1 – энтропия исходного состояния
35
системы).
Самопроизвольно протекающие процессы в изолированных системах всегда идут с увеличением энтропии, т.е. S  0. Например, если взять ящик с перегородкой, одно отделение наполнить азотом, а другое – водородом, а затем перегородку убрать, то газы смешаются, причём процесс протекает самопроизвольно и необратимо. Смешение газов увеличивает беспорядок в системе (хаотичность), а следовательно, возрастает и энтропия системы.
0. Например, если взять ящик с перегородкой, одно отделение наполнить азотом, а другое – водородом, а затем перегородку убрать, то газы смешаются, причём процесс протекает самопроизвольно и необратимо. Смешение газов увеличивает беспорядок в системе (хаотичность), а следовательно, возрастает и энтропия системы.
Если после смешения газов создать условия для протекания химической реакции (давление, катализатор), то процесс пойдёт с уменьшением энтропии (S ˂ 0), поскольку система из хаотичного перейдёт в более упорядоченное состояние.
Такие процессы, как кристаллизация, конденсация идут с уменьшением энтропии, так как в системе устанавливается больший порядок, уменьшается хаотичность. При плавлении, испарении, растворении энтропия возрастает.
Единой формулировки второго закона термодинамики не существует. Есть много определений этого закона, на первый взгляд не похожих друг на друга. Однако все формулировки имеют общее начало: они характеризуют свойства необратимых процессов, отражают принцип возрастания энтропии. Впервые основной принцип второго закона был сформулирован М.В. Ломоносовым в 1747 г.: «...холодное тeлo В, погруженное в тёплое тело А, не может воспринять большую степень теплоты, чем какую имеет А».
Далее принцип второго начала развивался, что и нашло своё отражение в ряде формулировок:
1. Невозможно создать вечный двигатель (perpetuum mobile) второго рода (Оствальд).
2. Теплота не может сама собой переходить от холодного тела к горячему (Клаузиус).
3. Различные виды энергии стремятся переходить в теплоту, а теплота, в свою очередь, стремится рассеяться, т. е. распределиться между всеми телами наиболее равномерным образом (Томсон).
Состояние любой совокупности частиц можно определить характеристиками макросостояния такими, как температура, давление, масса. Характеристики каждой частицы вещества — положение в пространстве, скорость и направление движения — это характеристики микросостояния вещества. Так как вещества состоят из огромного количества частиц, то определённому макросостоянию отвечает огромное количество различных микросостояний. Количество
36
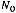
микросостояний, с помощью которых осуществляется данное макросостояние системы, называется вероятностью состояния W.
Больцман впервые дал статистическое толкование второго закона термодинамики и показал, что между энтропией и вероятностью состояния существует зависимость:
S = 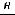 ln W,
ln W,
где R — универсальная газовая постоянная; N0 — число Авогадро.
Если W = 1, т. е. макросостояние системы можно осуществить только одним способом, то S = О. Чем больше значение W, тем больше энтропия, т. е. в системе увеличивается беспорядок.
Связывая понятие об энтропии с состоянием Вселенной, Больцман вывел, что «мир стремится к хаосу». Эта идея перекликалась с формулировкой Клаузиуса: «Энтропия мира стремится к максимуму». Такие представления явились поводом для утверждения теории «тепловой смерти» Вселенной, сторонники этой теории доказывали, что увеличение хаоса (a значит, и энтропии) приведёт полному выравниванию температур во всей Вселенной, а это означало бы возможность протекания любых процессов, а следовательно, вызвало бы «тепловую смерть» Всeлeнной. Эти идеалистические и антинаучные толкования основывались на абсолютно ошибочном представлении о Вселенной как об ограниченной системе. Правильное толкование законов термодинамики применительно к Вселенной возможно только в том случае, если признать, что Вселенная бесконечна, не имеет ни конца, ни начала как во времени, так и в пространстве.
Метафизическое толкование закона критиковалось многими учёными, в том числе и Ф. Энгельсом, который в своей работе «Диалектика природы» подвергнул серьёзной критике теорию «тепловой смерти» Вселенной.
Считается, что сущность второго закона термодинамики наиболее полно выражает следующая формулировка: самопроизвольно могут протекать только те процессы, при которых система из менее вероятного состояния переходит в более вероятное.
3.3. Энергия Гельмгольца и энергия Гиббса. Критерии направления самопроизвольно протекающих процессов
Для характеристики процессов, протекающих в изолированных системах, применяют различные термодинамические функции. В частности, при постоянной температуре и постоянном давлении или объёме наряду с энтропией
37
S используются следующие функции:
1. Изобарно-изотермический потенциал — G (или Z). Его называют свободной энергией Гиббса, свободной энтальпией, или упрощённо изобарным потенциалом. Используется он при рассмотрении процессов, протекающих при постоянных температуре и давлении. B химической термодинамике энергия Гиббса определяется из соотношения:
G = H — TS,
а её изменение вычисляется из соотношения
G = H — TS.
Анализ этого уравнения показывает, что если процесс протекает при низких температурах (величина Т мала), то и произведение TS тоже мало. Если реакция происходит с ощутимым тепловым эффектом, то, как правило, H 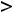 TS. Тогда в выражении G = H — TS можно пренебречь величиной TS, а следовательно, приближённо можно считать, что G = H.
TS. Тогда в выражении G = H — TS можно пренебречь величиной TS, а следовательно, приближённо можно считать, что G = H.
При высоких температура справедливо соотношение H  TS, тогда, пренебрегая значением Н, получим:
TS, тогда, пренебрегая значением Н, получим:
G = — TS.
Приведённые равенства могут быть использованы при решении вопроса о направлении самопроизвольно протекающих процессов. Так, знак изменения энтальпии реакции при низких температурах и знак изменения энтропии при высоких температурах позволяют предварительно судить о направлении процесса. Известно, что при низких температурах самопроизвольно могут протекать экзотермические реакции, а следовательно, Н должно иметь отрицательное значение. При высоких температурах протекают, как правило, эндотермические реакции, сопровождающиеся увеличением энтропии, т. е. величина S имеет положительное значение.
2. Изохорно-изотермический потенциал F (или А) применяется для характеристики процессов, протекающих при постоянных температуре и объёме. Его ещё называют свободной энергией Гельмгольца, или упрощённо изохорным потенциалом. Определяется F из соотношения
F = U — TS.
Изменение энергии Гельмгольца вычисляют по формуле
F = U — TS.
Что выражают эти функции?
1. По изменению G можно судить о принципиальной возможности протекания реакции, по знакуG определяют направление процесса. Если G =
38
0, то происходит равновесная реакция, направление которой определяется только концентрацией отдельных её компонентов. Подобный процесс имеет место при аккумулировании энергии в мышечной ткани.
Если G 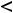 0, то реакция идёт спонтанно с выделением энергии в форме полезной работы. В биохимии такие процессы называются экзэргоническими. Если G
0, то реакция идёт спонтанно с выделением энергии в форме полезной работы. В биохимии такие процессы называются экзэргоническими. Если G  0, то изменение состояния системы происходит только при затрате работы извне эндэргоническими процессами.
0, то изменение состояния системы происходит только при затрате работы извне эндэргоническими процессами.
Все самопроизвольно протекающие процессы должны сопровождаться увеличением отрицательного значения G, причём чем больше по абсолютной величине отрицательное значение G, тем больше действие «движущей силы» реакции в данных условиях.
Следует, однако, подчеркнуть, что изменение в сторону отрицательных значении изобарного потенциала реакции указывает только на принципиальную возможность прохождения этой реакции. Может быть, что скорость реакции незначительна, и тогда, несмотря на то, что по расчёту G  0, взаимодействие практически не происходит. И только при значительном повышении температуры или при применении катализатора реакция может пройти до конца.
0, взаимодействие практически не происходит. И только при значительном повышении температуры или при применении катализатора реакция может пройти до конца.
2. Изменение свободной энергии Гиббса является количественной мерой (максимальной) энергии, которая может быть получена в форме полезной работы, например, в процессе обмена веществ.
3.Возможность расчёта свободной энергии Гиббса (изобарного потенциала)
исвободной энергии Гельмгольца (изохорного потенциала) позволяет количественно определить энергию химических превращений при обмене веществ, что имеет большое значение прежде всего для биохимии, поскольку благодаря такому расчёту химические реакции, протекающие в живой клетке, можно оценивать не только с точки зрения превращения материальных субстратов, но и энергетически. Удобным примером может служить синтез сахарозы (приведённые ниже значения изобарного потенциала вычислены применительно к физиологическим условиям):
Глюкоза + Фруктоза↔ Сахароза + Н2 О (G = + 21 кДж/моль).
Прямая реакция является эндэргонической и поэтому не может протекать самопроизвольно. Для её осуществления необходимо, чтобы она сопрягалась с экзэргической реакцией, сопровождающейся выделением большого количества свободной энергии. Такой реакцией является реакция гидролиза АТФ, в которой в качестве продуктов реакции образуются аденозиндифосфорная (АДФ) и фосфорная (Ф) кислоты:
39
АТФ+Н2О↔ АДФ+ Ф (G = 50,3 кДж/моль).
Сопряжение реакций достигается образованием в качестве промежуточного продукта глюкозо-1-фосфата:
АТФ + Глюкоза →Глюкоза-1-Ф+ АДФ (G = - 29,4 кДж/моль);
Глюкоза-1-Ф + Фруктоза → Сахароза + Ф (G = 0).
Поскольку изменение свободной энергии сложной системы является величиной аддитивной, суммарно процесс можно записать так:
АТФ + Глюкоза + Фруктоза → Сахароза + АДФ + Ф (G = — 29,4 кДж/моль). Этот пример сопряжения типичен для многих биологических процессов, в
которых экзэргический характер одной из реакций обеспечивает протекание до конца другой эндэргической реакции.
3.4. Экзо- и эндотермические процессы. Тепловой эффект реакции. Закон Гесса и Лавуазье — Лапласа
Раздел химической термодинамики, изучающий тепловые эффекты реакций, называется термохимией. Уравнения реакций, для которых указывается, их тепловой эффект, называются термохимическими.
Тепловые эффекты химических реакций обусловлены тем, что их протекание связано с разрывом одних химических связей и образованием других. Разница в энергиях разорвавшихся и образующихся связей и проявляется в виде результирующего теплового эффекта химической реакции.
Химические реакции, сопровождающиеся выделением тепла, называются экзотермическими, а сопровождающиеся поглощением тепла — эндотермическими. Тепловые эффекты в уравнениях реакций записываются с соответствующими знаками: «+» для экзотермических и « — » для эндотермических и выражаются в джоулях (Дж) или килоджоулях (кДж):
1.Н2 + ½ 02 = Н2О (п) + 242 кДж;
2.Н2 + ½ 02 = Н2О (ж) + 286 кДж (68,4 ккал);
3.N2 + ½ 02 = N2О - 81,6 кДж (19,5 ккал).
Тепловой эффект, сопровождающий образование 1 моля соединения из простых веществ, называется теплотой образования данного соединения. Приведённые выше тепловые эффекты являются в то же время теплотами образования воды и оксида азота (I).
Процессы растворения сопровождаются выделением или поглощением тепла и зависят от природы растворителя и растворяемого вещества. Для характеристики этих процесcов вводится понятие теплоты растворения. Теплота
40
растворения – это то количество тепла, которое выделяется или поглощается при растворении 1 моля вещества в таком количестве растворителя, что дальнейшее прибавление последнего уже не вызывает дополнительного теплового эффекта.
Известно, что процесс растворения веществ протекает в несколько стадий: а) разрушение кристаллической решётки и
б) диффузия (сопровождаются поглощением тепла соответственно – Q1 и
Q2);
в) гидратация (сольватация) сопровождается выделением тепла +Q3.
Теплота растворения представляет собой алгебраическую сумму тепловых эффектов всех стадий:
Qp = (+ Q3) + (- Q1) + (- Q2), Q1 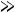 Q2.
Q2.
Следует отметить, что затрата энергии на диффузию значительно ниже, чем значение Q1 и Q3. Поэтому, если на разрушение кристаллической решётки затрачивается больше энергии, чем выделяется при сольватации, то в целом пpoцесс растворения сопровождается поглощением тепла. Если же энергия, выделившаяся при гидратации (сольватации), больше энергии, затраченной на разрушение кристаллической решётки, то процесс растворения сопровождается выделением тепла.
Большое значение для развития химии имело определение теплот, нейтрализации. Гессом было установлено, что теплота нейтрализации граммэквивалента кислоты щелочью является постоянной величиной:
NaOH + ½ Н2SО4 → ½ Na2SO4 + Н2О + Q; NaOH + HCl → NaCl + Н2О + Q;
KОН + НNО3→ KNО3 + Н2О + Q; Q = const.
Причина постоянства теплового эффекта Q разных реакций во времена Гесса являлась совершенно непонятной. Позднее её объяснила теория электролитической диссоциации С. Аррениуса. Согласно этой теории, любая реакция нейтрализации сводится к одному и тому же процессу:
Н+ + ОН- → Н2О + Qн.
Показатели теплоты нейтрализации слабых кислот и оснований отличаются от показателей теплоты нейтрализации сильных кислот и оснований и зависят от природы взятых веществ, поскольку процесс распада слабых электролитов на ионы затруднён.
В основу термохимических расчетов положены законы которые практически являются следствием закона сохранения энергии. Первый закон термохимии,
41

открытый Лавуазье и Лапласом в 1784г., формулируется так: при образовании соединения поглощается (или выделяется) такое количество тепла, которое выделяется (или поглощается) при его разложении на исходные составные части:
3О2 = 2О3 — 299 кДж; 2О3 = 3О2 + 299 кДж.
Важнейшим законом, лежащим в основе многих термохимических вычислений, является закон суммы тепловых эффектов, открытый и экспериментально подтвержденный Г. И. Гессом в 1840 г. Закон Гесса утверждает, что тепловой эффект реакции не зависит от пути её прохождения (т. е. не зависит от промежуточных стадий реакции), а зависит лишь от её начального и конечного состояний:
С + ½ О2 = СО + 110,6 кДж; СО+ ½ О2 = СО2 +283,2 кДж; С + О2 = СО2 + 393,8 кДж .
Очевидно, что если известны общий тепловой эффект реакции и тепловой эффект одной из двух стадий, то на основании закона Гесса можно вычислить тепловой эффект другой промежуточной стадии.
Глава IV. ХИМИЧЕСКАЯ КИНЕТИКА И КАТАЛИЗ
4.1. Средняя и истинная скорости реакции
Учение о скоростях и механизмах протекания химических реакций называется химической кинетикой.
Главной задачей химической кинетики является исследование скоростей химических реакций и факторов, влияющих на них. Скорость химической реакции – это число элементарных актов реакции, происходящих за единицу времени в единице объёма (для гомогенных реакций) или а единице поверхности раздела фаз (для гетерогенных реакций). Скорость химической реакции измеряется изменением концентрации реагирующих веществ в единицу времени. В кинетике принято при записи вместо слова «концентрация» использовать квадратные скобки; чаще всего пользуются молярной концентрацией.
Различают среднюю и истинную скорости реакции. Рассмотрим это на примере разложения азотной кислоты: 4НNO3 4NO2 + O2 + 2H2O.
42

Началу реакции разложения азотной кислоты соответствует время t1, концу – t2, т.е. длительность реакции составляет t2 - t1 = t. В начале реакции концентрация азотной кислоты равна С1, к концу реакции концентрация кислоты уменьшается до С2, т.е. за время t концентрация кислоты изменяется на С = С1- С2. Тогда средняя скорость реакции разложения азотной кислоты будет равна:
υср = 

 =
= 
Поскольку в ходе реакции концентрации реагирующих веществ непрерывно изменяются, то для характеристики скорости реакции в каждый данный момент времени введено понятие истинной (или мгновенной) скорости. Чем меньше t, тем больше средняя скорость реакции приближается к истинной, что выражается следующим дифференциалом:
υист. = ± ,
где  – бесконечно малый промежуток времени;
– бесконечно малый промежуток времени; 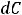 – бесконечно малое уменьшение концентрации любого реагента. Следует учитывать, что концентрация исходных веществ убывает, а продуктов реакции – возрастает. Поэтому для выражения концентрации исходных веществ этот дифференциал необходимо брать со знаком «-», а для продуктов реакции – со знаком «+».
– бесконечно малое уменьшение концентрации любого реагента. Следует учитывать, что концентрация исходных веществ убывает, а продуктов реакции – возрастает. Поэтому для выражения концентрации исходных веществ этот дифференциал необходимо брать со знаком «-», а для продуктов реакции – со знаком «+».
4.2. Влияние концентрации на скорость реакций. Порядок и молекулярность реакций
Зависимость скорости реакции от концентрации выражается следующим законом: скорость гомогенных реакций пропорциональна произведению концентраций реагирующих веществ. Записывается скорость реакций в виде произведения молярных концентраций реагирующих веществ, возведенных в степени, показатели которых равны стехиометрическим коэффициентам соответствующих веществ. Так, скорость реакции
αA + bB AaBb
в общем виде запишется так:
υ = К [A]a [B]b,
где К – константа скорости реакции.
Если [A] = [B] = 1 моль/л, то υ = К, т.е. константа скорости реакции и есть скорость реакции при условии, что исходные концентрации реагирующих веществ равны 1 моль/л, таков физический смысл константы скорости. К зависит от тех же факторов, что и скорость реакции, за исключением концентрации.
43
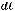
Для любой химической реакции можно записать выражение её скорости в общем виде так:
Н2 + 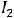 2НI2; υ = K1 [H2] [I2].
2НI2; υ = K1 [H2] [I2].
В гетерогенных системах столкновение частиц происходит на границе раздела фаз, т.е. масса твёрдой фазы не влияет на скорость реакции, а потому и не учитывается в выражении для скорости реакции:
S + O2 SO2; υ = K[O2].
Если реакция протекает в несколько стадий, то для каждой стадии можно определить скорость. Однако в целом скорость реакции определяется скоростью наиболее медленно протекающей стадии.
Кинетические исследования позволяют определить порядок и молекулярность реакций. Число молекул, которые одновременно взаимодействуют и обуславливают акт химического превращения, определяет молекулярность реакции. При этом число молекул образующихся веществ не имеет значения, поскольку молекулярность реакции определяется только числом молекул реагирующих веществ.
Различают следующие реакции: 1) мономолекулярные, в которых претерпевают превращение только один вид молекул или частиц (например, процесс радиоактивного распада); 2) биомолекулярные, в которых элементарный акт осуществляется при столкновении двух молекул одного вещества или двух разных молекул; 3) тримолекулярные, в которых одновременно сталкиваются и принимают участие в элементарном химическом акте три молекулы.
Вероятность одновременного столкновения нескольких молекул очень мала, поэтому тримолекулярные реакции встречаются крайне редко, вероятность столкновения большего числа частиц практически равна нулю.
Порядок химической реакции по данному веществу – это число, равное степени, в которой концентрация данного вещества входит в уравнение скорости этой реакции. Сумма показателей степеней концентрации исходных веществ, входящих в уравнение скорости определяет порядок реакции в целом.
Для реакции первого порядка характерно, что скорость её зависит от концентрации только одного вещества, концентрации только одного вещества, концентрация которого непрерывно уменьшается. Например, скорость реакции
Н22Н
определяется:
- 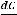 = K [H2],
= K [H2],
44

если [H2] = C моль/л, то -  = K [C], или
= K [C], или
- 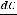 = K dt
= K dt
После интегрирования последнего уравнения в пределах концентраций от С0 (начальная) до С (конечная) и времени от 0 до t получим:
К= 
 log
log  ,
,
К= 
 log
log 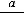 ,
,
где α – начальная концентрация вещества; x — изменение концентрации за время τ, (α — х) —конечная концентрация вещества . Реакции, константы скорости которых выражаются уравнениями, называются реакциями I порядка. К реакциям I порядка относятся конечные стадии ферментативных процессов, взаимодействие антигенов с антителами, реакции изомерного превращения и др. Известны реакции I порядка, в которых за равные промежутки времени реагирует одинаковая часть вещества. Время, необходимое для того, чтобы прореагировала половина исходного количества вещества (или половина того, это имеется в данный момент времени), называется полупериодом реакции ( ).
).
Реакцию II порядка рассмотрим на примере взаимодействия молекулярных водорода и иода:
|
|
|
|
|
|
|
|
|
|
. |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
||||||
Обозначим |
концентрации исходных |
веществ соответственно [ |
|
] = |
|
|||||||||||
|
|
|||||||||||||||
моль/л и [ |
|
] = |
|
моль/л. Тогда скорость этой реакции в общем виде будет |
|
|||||||||||
|
|
|
||||||||||||||
|
|
|
- |
|
[ |
|
][ |
|
] . |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|||||||
Из уравнения скорости видно, что скорость реакции зависит от концентраций двух участвующих в реакции веществ и сумма показателей
степеней равна двум. Если [ |
|
] = [ |
|
|
], то уравнение (VI,7) принимает вид |
|||||
|
|
|||||||||
- |
|
|
|
|
|
|||||
|
|
|
|
|
|
|
. |
|||
|
|
|
|
|||||||
|
|
|
|
|
||||||
После интегрирования и соответствующей замены концентраций получим
.
По двум предыдущим уравнениям можно рассчитать скорости реакций II порядка. Следует отметить, что среди биохимических процессов реакции более высокого, чем II порядка не встречаются.
45

Реакция взаимодействия хлорида железа (III) с хлоридом олова (II) является реакцией III порядка:
2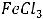 + Sn
+ Sn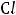
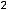
 Sn
Sn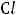
 + 2FеС12,
+ 2FеС12,
если
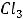


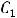
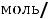
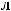 , [Sn
, [Sn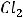 ] =
] =  моль/л ,
моль/л ,
то
.
Если реагирующее вещество присутствует в избытке так, что его концентрация в ходе реакции остаётся практически постоянной, то скорость реакции не изменяется, а количество образующегося продукта зависит только от продолжительности реакций. Иными словами, скорость таких реакций не зависит от концентрации исходных веществ. Эти реакции относятся к реакциям нулевого порядка. Пример реакций нулевого порядка — реакции, катализируемые ферментами:
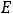
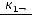

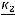




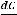

 ,
,
где Е — фермент (ограниченное и определённое количество); S — субстрат (избыток); ЕS — промежуточный комплекс, который, в свою очередь, распадается на фермент и новый продукт P.
Большинство химических процессов осуществляется через промежуточные стадии. Вследствие сложности процессов порядок реакции, как правило, не совпадает с суммой показателей степеней, входящих в кинетическое уравнение. Таким образом, порядок реакции, определённый из уравнения, характеризует формально кинетическую зависимость скорости реакции от концентрации реагирующих веществ. Молекулярность характеризует простейший механизм отдельных стадии сложного процесса. Если процесс происходит в несколько стадий, а также тогда, когда существует большая разница в концентрациях реагентов, порядок реакции не совпадает с её молекулярностью. Примером может служить реакция инверсии тростникового сахaра:
|
|
|
|
. |
|
|
|
|
|
Сахароза |
|
Глюкоза |
|
Фруктоза |
|
|
|
|
|
B общем виде:
.
Реакция идёт до конца, по существу бимолекулярна, но протекает, как реaкция I порядка, поскольку вследствие большой разницы между молекулярными массами сахара воды массовое количество воды, вступающей в
46
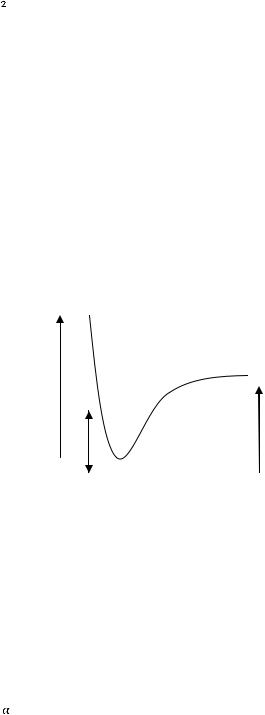
реакцию, малo, что позволяет пренебречь незначительным уменьшением её концентрации и считать последнюю постоянной в условиях разбавленного раствора 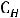
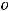

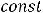 , поэтому она может быть объединена с константой скорости в одну постоянную v = KCсахара.
, поэтому она может быть объединена с константой скорости в одну постоянную v = KCсахара.
4.З. Понятие об активных молекулах. Энергия активации
Молекулярно-кинетическая теория позволяет рассчитать количество столкновений в реагирующей системе. Так, в реакции: H2 + I2 → 2HI
при температуре порядка 300° С и концентрации [H2] = [I2] = 1 моль/л в 1 с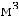 за 1 с происходит
за 1 с происходит  столкновений, а реагирует
столкновений, а реагирует  молекул, т. e. не все молекулы реакционноспособны и не всякое столкновение молекул приводит к их взаимодействию. Молекулы или частицы, имеющие запас энергии, достаточный для того, чтобы при столкновении провзаимодействовать, называются активными.
молекул, т. e. не все молекулы реакционноспособны и не всякое столкновение молекул приводит к их взаимодействию. Молекулы или частицы, имеющие запас энергии, достаточный для того, чтобы при столкновении провзаимодействовать, называются активными.
E
R
Eo |
|
_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ |
||
|
|
|
||
|
|
|
Ea |
Eq |
Изменение потенциальной энергии системы в процессе образования молекулы водорода.
Очевидно, что при повышении температуры процент активных молекул возрастает. Повышая температуру, мы сообщаем дополнительную энергию молекулам. Дополнительная энергия, которая затрачивается на то, чтобы молекулу из неактивного состояния перевести в активное, называется энергией активации 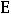 . На что же затрачивается энергия активации? Для тогo чтобы ответить на этот вопрос, рассмотрим, как изменяется потенциальная энергия системы в процессе образования молекулы водород. Предположим, что имеется два атома водорода, далеко расположенных друг от друга. И потому они не взаимодействуют. При достаточном сближении становится возможным перекрывание электронных облаков, образуется химическая связь и выделяется
. На что же затрачивается энергия активации? Для тогo чтобы ответить на этот вопрос, рассмотрим, как изменяется потенциальная энергия системы в процессе образования молекулы водород. Предположим, что имеется два атома водорода, далеко расположенных друг от друга. И потому они не взаимодействуют. При достаточном сближении становится возможным перекрывание электронных облаков, образуется химическая связь и выделяется
47

энергия. При образовании стойкой молекулы водорода выделяется максимальное количество энергии, этому энергетически стойкому состоянию системы отвечает минимум на кривой её потенциальной энергии. Если молекула выводится из энергетически стойкого состояния, то для этого затрачивается дополнительная энергия, что приводит к разрыхлению связей. Таким образом, энергия, которая затрачивается на разрыхление связей, и есть энергия активации. Энергия, которая приводит к разрыву связей между атомами в молекуле, называется энергией диссоциации 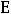 .
.
Как показывает опыт, для того чтобы прошла химическая реакция, не обязателен полный разрыв связей, достаточно их разрыхлить. Частицы с разрыхлёнными связями способны вступать во взаимодействие с образованием так называемых активных (промежуточных) комплексов. Например, реакция образования иодоводорода протекает так: активные молекулы водорода и иода при столкновении образуют промежуточный комплекс, в котором связи Н — I образуются одновременно с разрыхлением связей Н — Н и I—I:
H – H |
|
H . . . H |
H |
H |
||
|
|
. . |
|
|
|
|
|
|
|
|
|
|
|
+ |
→ |
. . |
→ |
|
+ |
|
|
|
. . |
|
|
|
|
|
|
|
|
|
|
|
I – I |
|
I . . . I |
I |
I |
||
Промежуточный активный комплекс |
||||||
Расчёт показал, что энергия активации этой реакции равна 168 кДж, что меньше энергии диссоциации (571 кДж). Таким образом, реaкции, протекающие с образованием промежуточного комплекса, энергетически более выгодны, чем реакции, протекающие с полным разрывом связей между молекулами, которые вступают во взаимодействие. Реакции характеризуются различными величинами энергии активации  . Для биохимических процессов численные значения
. Для биохимических процессов численные значения 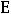 лежат в пределах 21 – 210 кДж/моль, что на 1 – 2 порядка ниже значений
лежат в пределах 21 – 210 кДж/моль, что на 1 – 2 порядка ниже значений 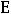 химичeских процeссов, проходящих вне живых организмов. Это можно объяснить тем, что абсолютное большинство биохимических процессов протекает с участием ферментов с образованием промежуточных активных комплексов.
химичeских процeссов, проходящих вне живых организмов. Это можно объяснить тем, что абсолютное большинство биохимических процессов протекает с участием ферментов с образованием промежуточных активных комплексов.
4.4. Влияние температуры на скорость реакции
Известно очень мало реакций, скорость которых уменьшается при увеличении температуры, например окисление NО в N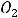 .
.
48

Для абсолютного большинства химических процессов повышение температуры вызывает значительное увеличение скорости реакции. В наиболее простой форме зависимость скорости реакции от температуры представлена эмпирическим правилом Вант-Гоффа, согласно которому при увеличении температуры на 10° С скорость химической реакции увеличивается в среднем в 2 – 4 раза.
B математической форме это можно записать так:
,
где 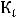 — константа скорости реакции при данной температуре;
— константа скорости реакции при данной температуре; 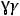 – температурный коэффициент, показывающий, во сколько раз увеличивается скорость химической реакции (следовательно, и константа скорости) при повышении температуры на 10° C. Для приближённых расчётов скорости используют формулу
– температурный коэффициент, показывающий, во сколько раз увеличивается скорость химической реакции (следовательно, и константа скорости) при повышении температуры на 10° C. Для приближённых расчётов скорости используют формулу
где  — скорость реакции при 0° C;
— скорость реакции при 0° C; 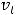 – скорость реакции при заданной температуре (t =
– скорость реакции при заданной температуре (t =  -
-  ,
,  — исходная температура;
— исходная температура;  — конечная температурa). Если принять
— конечная температурa). Если принять  = 2, то при повышении температуры 0 до 10°С скорость реакции увеличится в
= 2, то при повышении температуры 0 до 10°С скорость реакции увеличится в 
 раз.
раз.
Для большинства химических реакций  колеблется в пределах 2 – 3. Ферментативные процессы характеризуются более высокими значениями
колеблется в пределах 2 – 3. Ферментативные процессы характеризуются более высокими значениями  (7 и больше). Особенно большие значения температурного коэффициента характерны для процесса денатурации белков (100 и более). Для большого интервала температур
(7 и больше). Особенно большие значения температурного коэффициента характерны для процесса денатурации белков (100 и более). Для большого интервала температур  не остaётся постоянным и, как правило, уменьшается с повышением температуры.
не остaётся постоянным и, как правило, уменьшается с повышением температуры.
Как было отмечено выше, правило Вант-Гоффа применимо для приблизительных расчётов. Для более точных расчётов используют уравнение Аррениуса — Вант-Гоффа:
,
где К — константа скорости реакции; Т — абсолютная температура; R — универсальная газовая постоянная; 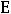 — энергия, активации.
— энергия, активации.
После интегрирования это уравнение принимает вид
49
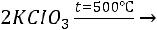
где е — основание натурального логарифма; A — предэкспоненциальный множитель, физический смысл которого, согласно тeории соударений, состоит в том, что он равен общему числу соударений между молекулами за 1 с в 1 с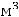 (фактор соударений).
(фактор соударений).
Таким образом, выражение для скорости реакции с учётом энеpгии активации и температуры имеет вид:
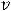

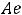

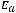
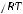




 .
.
Анализ уравнения показывает, что скорость реакции c увеличением температуры изменяется в геометрической прогрессии.
Уравнение Аррениуса – Вант-Гоффа хорошо описывает большинство ферментативных реакций, а также такие сложные биологические процессы, как дыхание растений, брожение, деление клеток в пределах «физиологических температур».
Скорость ферментативных реaкций, как и обычных химических, с повышением температуры увеличивается. Однако после достижения определённого температурного пределa скорость ферментативного процесса в связи с инактивацией фермента начинает уменьшаться. В целом же повышение температуры процесса увеличивает число соударений и количество активных молекул. Kак покaзали расчёты молекулярно-кинетической теории, скорость реакции за счёт увеличения столкновении при нагревании на 10° С максимально возрастает на 2%. Следовательно, резкое увеличение скорости реакции при увеличении температуры определяется, главным образом, значительным увеличением числа активных частиц.
4.5. Влияние катализаторов на скорость реакций. Катализ в живой природе
B химической практике для ускорения химических реакций широко используют катализаторы. Катализаторы – это вещества, которые изменяют скорость реакции, вступая во взаимодействие с исходными веществами и образуя промежуточные продукты реакций. В конце реакции катализаторы восстанавливают свои первоначальный химический состав и массу. Действие катализатора рассмотрим на примере термического разложения бертолетовой соли:
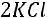
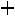

 .
.
50
Однако в присутствии катализаторов (Mn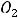 ,
, 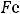
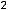
 ,
, 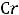
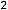
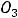 или
или 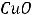 ) гипохлорат калия легко и полностью разлагается при более низкой температуре, процесс протекает через промежуточные стадии:
) гипохлорат калия легко и полностью разлагается при более низкой температуре, процесс протекает через промежуточные стадии:
.
Действие катализаторов проявляется в том, что они вступают во взаимодействие с исходными веществами и образуют нестойкие промежуточные соединения, энергия активации которых меньше энергии активации некатализируемой реакции. Иными словами, катализатор изменяет механизм реакции и позволяет вести процесс энергетически более выгодным путём. Изменение скорости химической реакции под действием катализаторов называется катализом. Различают катализ 1) положительный, при котором скорость реакции увеличивается; 2) отрицательный — скорость реакции уменьшается; 3) автокатализ – катализатором является один из продуктов реакции.
К последнему типу катализа относятся многие процессы горения и некоторые биохимические реакции.
B зависимости от агрегатного состояния реагирующих веществ различают катализ гомогенный и гетерогенный. Если реагирующие вещества и катализатор находятся в одной фазе, катализ называется гомогенным (окисление СО до  в присутствии паров
в присутствии паров 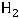 О). Если реагирующие вещества и катализатоp находятся в разных фазах, катализ называется гетерогенным (окислeние S
О). Если реагирующие вещества и катализатоp находятся в разных фазах, катализ называется гетерогенным (окислeние S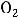 до S
до S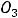 в присутствии твёрдого ванадиевого катализатора).
в присутствии твёрдого ванадиевого катализатора).
Особенностью каталитических процессов является следующее:
1.Одно из исходныx веществ (катализатор) сохраняет в конце реакции неизменным свои состав и количество.
2.Взаимодействие катализаторов с исходными веществами происxодит не в стехиометрических соотношениях. Так, одна массовая часть катализатора вызывает превращение миллиона массовых частей аммиака в процессе окисления его в азотную кислоту. Эта диспропорция особенно велика для таких активных катализатоpов, как ферменты, которые являются ускорителями биологических процессов: одна молекула каталазы за 1 с способна разложить миллион молекул пероксида водорода.
3.В обратимых реакциях катализаторы не смещают равновесия.
51
4. Катализаторы чувствительны к наличию посторонних веществ. Вещества, которые увеличивают каталитическую активность катализатора, называются промоторами, вещества, которые уменьшают или совсем подавляют его действие ядами или ингибиторами. Катализ играет важную роль не только в химико-технологических процессах, но и в живой природе. Вся сложная система управления жизненными процессами в организмах основана на катализе. Такие жизненно важные процессы, как дыхание, пищеварение, синтез белков, превращение химической энергии в механическую и многие другие, осуществляются соучастием ферментов. Ферменты – это биологические катализаторы, которые имеют белковую природу и находятся в организмах в коллоидном или высокомолекулярном состоянии. Механизм ферментативных реакций очень сложен и мало изучен. Доказано, что ферменты характеризуются признаками как гомогенных, так и гетерогенных катализаторов. Ферменты проявляют своё действие в водных растворах, в связи с чем могут быть отнесены
кгомогенным катализаторaм. Кроме того, ферменты — это агрегаты белковых молекул с молекулярной массой, равной сотням тысяч. Это даёт основание считать, что в растворе ферментов существует поверхность раздела и отнести их
кгетерогенным катализаторам. Доказано, что каталитическая активность ферментов во многих случаях определяется наличием в их молекулах особых участков (активных центров), которые обладают специфической pеакционной способностью (гетерогенный катализ). Например, ферменты, участвующие в окислительно-восстановительных реакциях, в биосинтезе белка, проявляют свою каталитическую активность, будучи «вмонтированными» в сравнительно жёсткие структурные кoмпоненты клетки, которые имеют макроповерхность раздела (митохондрии, рибосомы и др.).
По подсчётам биохимиков, в организме человека происходит свыше 10 000 химических превращений. Жизнь организмов требует, чтобы реакции протекали быстро. Однако вещества, которые получают организмы из внешней среды, отличаются высокой химической устойчивостью, неактивностью. Природа в ходе многовековой эволюции создала избирательные и высокоэффективные катализаторы, которые способствуют тому, что при низких температурах и низких давлениях в нейтральной среде малоактивные вещества взаимодействуют в организмах со скоростями, в миллионы раз превышающими скорости этих реакций в обычной химической среде без участия ферментов.
Преимущества ферментативных процессов заключаются в следующем: a) огромная скорость реакций, обусловливающая нормальную жизнедеятельность
52
организмов; 6) высокая избирательность ферментов; в) ферменты подавляют пробочные реакции; г) процессы осуществляются в мягких условиях (низкая температура и давление, нейтральная среда). Центральной проблемой ферментологии является выяснение механизма каталитического действия ферментов и установление связи между молекулярной структурой фермента и его каталитической активностью. Всё это требует подробного изучения кинетики реакций, катализируемых ферментами. В последние годы свойства ферментов и механизм их действия привлекают внимание многих исследователей. Ферменты успешно используются в клиническом и химическом анализах в пищевой и легкой промышленности (в виноделии, изготовление сыров, табака, обработка кожи и т.д.). Особенно широко применяются лечебные ферменты: пепсин, трипсин, химотрипсин, содержащиеся в пищеварительных соках. Гиалуронидаза – фермент, разрушающий межклеточное вещество, используется для ускорения всасывания лекарственных препаратов. Опасное для организма заболевание – тромбоз – лечат с помощью фермента плазмина.
Создана и развивается молодая наука бионика. Задачи бионики – применение биологических идей в науке, технике, промышленности для решения сложных инженерных задач. В этом плане применение ферментов в химической практике представляется блестящим будущим, когда громоздкие, трудоёмкие процессы, протекающие в жёстких условиях (высокая температура, давление и т. д.), можно будет заменить ферментативными реакциями, протекающими в мягких условиях, с большими скоростями, в простом аппаратурном оформлении, на маленьких производственных площадях, с большой производительностью.
4.6. Понятие о теориях катализа
Природа взаимодействия реагирующих веществ с катализатором весьма разнообразна. По характеру промежуточного взаимодействия различают: 1. Кислотно-основный катализ, при котором ускорение реакции обусловлено образованием промежуточных продуктов реакции за счёт присоединения или отдачи ионов водорода или гидроксила. Такие реакции протекают в присутствии кислот или оснований. Так, например, гидролиз сложных эфиров осуществляется в присутствии кислот. 2. Окислительно-восстановительный катализ, при котором осуществляется обмен электронов между реагирующими веществами и катализаторами (например, при окислении 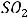 до
до 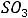 в присутствии ванадиевого катализатора).
в присутствии ванадиевого катализатора).
53
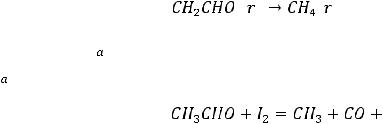
Важнейшими вопросами теории катализа являются исследования механизмов взаимодействия катализаторов с реагирующими веществами, разработка методов подбора катализаторов, которые исключали бы эмпирический путь подбора, требующий огромного числа экспериментов.
Каков же механизм действия катализаторов?
Для гомогенных реакций считают, что катализатор образует в той же фазе промежуточные реакционноспособные продукты, которые позволяют направить процесс по новому энергетически более выгодному пути. Такие процессы протекают в несколько стадий. Например, реакция
 ,
,
для которой  = 191,6 кДж/моль, в присутствии паров йода идёт в две стадии с
= 191,6 кДж/моль, в присутствии паров йода идёт в две стадии с  = 54,5 кДж/моль (ускорение в 1
= 54,5 кДж/моль (ускорение в 1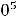
 ):
):
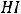 ;
;
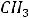
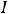
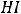

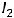 .
.
Хотя промежуточные продукты очень реакционноспособны (это нестойкие соединения с малым периодом жизни), однако для некоторых процессов они выделены, чем доказано их существование.
Более сложный механизм гетерогенного катализа. Поскольку в гетерогенном катализе катализатором, как правило, является твёрдое вещество, то при этом существенную роль играет поглощение поверхностью катализатора реагирующих частиц.
Наиболее широкое признание получили две теории гетерогенного катализа теория мультиплетов (А. Баландин, 1929) и теория активных ансамблей (Н. И. Кобозев, 1938).
В соответствии с теорией мультиплетов необходимым условием гетерогенного катализа является интенсивная адсорбция реагирующего вещества поверхностью катализатора. Так как радиус действия валентно-химических сил невелик, мультиплетная теория утверждает, что в молекуле реагируют только атомы, соприкасающиеся между собой и катализатором. Активные центры на поверхности катализатора расположены закономерно в соответствии со строением кристаллической решётки катализатора. Исходные молекулы должны
54
находиться в монослое, прилегающем к катализатору, и быть ориентированными своими частями к поверхности. Катализ происходит тогда, когда соответствующие связи в реагирующей молекуле ослабляются. Для такого ослабления необходимо удаление друг от друга соседних атомов в молекуле. Когда размеры постоянной решётки кристалла несколько превышают расстояние между атомами в реагирующей молекуле, связи ослабляются и происходит каталитическое ускорение реакции.
Поверхностное соединение, так называемый промежуточный мультиметный комплекс, образуется из одной или нескольких молекул вещества и из нескольких атомов катализатора. В мультиплетном комплексе первоначальные связи деформированы, а реагирующие атомы временно связаны химическими силами с несколькими атомами катализатора мультиплетом. Группа атомов катализатора, вступающая в поверхностное соединение, называется мультиплетом. Обычно эта группа состоит из 2 – 3 атомов. Схематически это можно представить так
Примером гетерогенного катализа являются реакции дегидрогенизации углеводородов, дегидрогенизации и дегидратации спиртов и др. Согласно теории активных ансамблей, активным началом является активный ансамбль, т. е. группа атомов катализатора, адсорбированных поверхностью носителя (обычно асбест, уголь, силикагель). Максимальная каталитическая активность достигается при условии, что ансамбли состоят из 2 – 3 атомов катализатора. Молекулы реагирующих веществ, адсорбируясь и реагируя в ансамбле, занимают фиксированное неподвижное положение.
4.7. Роль диффузии
Диффузия – самопроизвольный процесс переноса вещества, приводящий к установлению равновесного распределения концентраций в результате беспорядочного теплового движения частиц в газах, жидкостях или твёрдых телах.
Влияние диффузии на скорость химических реакций проявляется при условии контакта реагирующих частиц. Известно, что при отсутствии тепловых
55
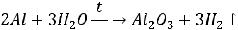
потоков отсутствует и механическое перемешивание, даже газы медленно диффундируют друг в друга. При перемешивании реакции протекают быстрее, поскольку улучшается контакт, а следовательно, увеличивается вероятность столкновения реагирующих частиц. При высоких температурах создаются условия, когда все молекулы активны, и всё-таки не каждое столкновение приводит к химическому взаимодействию. Иногда при достаточно большом количестве активных молекул условия контакта очень затруднены и реакции протекают с малом скоростью или практически не осуществляются. Например, реакция
не идёт до конца, поскольку на алюминии образуется оксидная плёнка, которая мешает контакту молекул алюминия и воды. Несоответствие скорости реакции числу активных молекул объясняется диффузией, при этом считают, что реакция протекает в диффузионной области.
При низких температурах и хороших условиях контакта скорость реакции лимитируется количеством активных молекул, считают, что такие реакции протекают в кинетической области.
Особенно важную роль играют диффузионные факторы в гетерогенных процессах, скорость которых зависит от величины активной поверхности, условии контакта и диффузии.
Диффузионные процессы имеют огромное значение для самых различных областей науки и техники. В биологии диффузионные процессы являются определяющими в явлениях проницаемости тканей, клеточных оболочек, всасывания и поглощения питательных веществ, выделения продуктов обмена. Диффузия определяет механизм и кинетику таких важных процессов, как окисление, сорбция, набухание, кристаллизация и т. д.
4.8. Механизм химических реакций
Механизм химических реакций зависит от характера реагирующих частиц. Простыми называются реакции, которые протекают непосредственно между молекулами, являющимися валентно-насыщенными. Например:
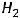
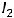

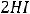 .
.
Доля таких реакций в общем объёме химических превращений невелика, поскольку для их осуществления необходима высокая энергия активации
56
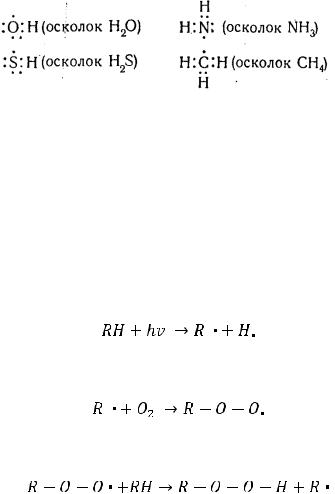
(150 – 450 кДж/моль). Ионные реакции при участии ионов протекают быстро, что свидетельствует о незначительной энергии активации ионных процессов (до 80 кДж/моль). Кроме обычных ионов, возникающих в процессе взаимодействия вещества с растворителем, в ионных реакциях участвуют молекулярные ионы, которые образуются под действием электроразряда, различного рода излучений с высокой энергией. Например:
H2 + γ → H2O+ + e; H2O + e → H2O–.
Реакции с участием молекулярных ионов также протекают с огромной скоростью, что обусловлено малой энергией активации этих частиц.
Радикальные реакции осуществляются при участии свободных радикалов и атомов. Свободный радикал – это молекула или её часть, имеющая неспаренный электрон, вследствие чего радикалы являются валентно-ненасыщенными и очень реакционноспособными
Энергия активации реакций, протекающих с участием радикалов, очень незначительна (порядка 0- 40 кДж/моль).
Многочисленные исследования за последние годы показали, что многие химические реакции протекают с участием свободных радикалов.
Многие биологические реакции, например окислительные, протекают с образованием свободных радикалов. Так, при окислительных процессах под действием лучистой энергии или при других воздействиях органическая молекула RH расщепляется с образованием свободных радикалов по схеме
Далее радикалы взаимодействуют с молекулой кислорода, образуя радикал пероксида
Радикал пероксида взаимодействует с другой молекулой органического вещества, образуя гидропероксид и новый свободный радикал
Две эти реакции могут повторяться много раз, образуя цепь.
57
Исследования парамагнетизма свободных органических радикалов позволили установить существование так называемых «семихинонов», представляющих собой свободнорадикальные промежуточные продукты окисления органических молекул. Семихиноны имеют нечётное число электронов, но, как предполагают исследователи, должны обладать значительно большим периодом жизни, чем известные ранее обычные органические свободные радикалы, отличающиеся высокой активностью. Более высокая стабильность семихинонов объясняется делокализацией электронов в ароматическом ядре. Потенциометрический, спектральный и другие методы исследования дают основания считать, что соединения с нечётным числом электронов, т. е. семихинонные ион-радикалы, должны выполнять роль промежуточных соединений в реакциях биологического окисления, связанных с перемещением электронов.
В последнее время биологи пытаются объяснить биологическую активность важнейших химических компонентов клетки наличием в них неспаренных электронов.
Высказано предположение, что свободные радикалы играют определённую роль в возникновении опухолей. Это предположение опирается частично на тот факт, что многие канцерогенные агенты являются свободными радикалами или содержат последние. Кроме того, получены экспериментальные данные, свидетельствующие о том, что в лейкоцитах при патологии количество свободных радикалов увеличивается в 30 – 40 раз, во многих опухолевых тканях животных и человека в 4 – 5 раз.
Таким образом, свободные радикалы играют важную роль в многообразных биологических процессах, а также в гетерогенном катализе, в реакциях горения и взрыва, в важнейших промышленных процессах пиролиза, крекинга и полимеризации.
Современную теорию цепных и разветвлённых реакций разработали лауреаты Нобелевской премии Н. Н. Семенов и С. Н. Хиншелвуд. В зависимости от механизма различают параллельные, последовательные и сопряжённые реакции.
Для параллельных реакций характерно, что исходные вещества участвуют одновременно в нескольких процессах. Например, при нагревании бертолетовой соли в определ`нных условиях одновременно протекают две реакции:
.
58
Каждая реакция характеризуется своей константой скорости. Суммарный процесс, состоящий из параллельных реакций I порядка, характеризуется константой скорости, которая является суммой констант скоростей всех параллельных реакций. Из двух параллельных реакций преимущественно протекает та, которая характеризуется меньшей энергией активации. Последовательные реакции это реакции с промежуточными стадиями. Опыт показывает, что кинетику таких реакций можно описать уравнением реакции I порядка, если не учитывать начального периода протекания реакции который характеризуется периодом индукции, когда промежуточное вещество в течение некоторого промежутка времени после начала реакции аналитически обнаружить нельзя.
Для сопряжённых реакции характерно одновременное осуществление нескольких реакции, причём протекают они только тогда, когда промежуточные вещества первой реакции являются исходными для второй. Таким образом, промежуточное вещество является связывающим звеном между вторичным и первичным процессами и создаёт условия для протекания обоих процессов. Например, известно, что 
 может окислить
может окислить 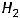
 , но не окисляет непосредственно
, но не окисляет непосредственно 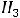
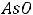
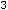 . Если же взять смесь всех кислот, то бромноватая кислота будет окислять и мышьяковистую ортокислоту. По стадиям этот процесс можно представить так:
. Если же взять смесь всех кислот, то бромноватая кислота будет окислять и мышьяковистую ортокислоту. По стадиям этот процесс можно представить так:
HBrO3 + H2SO3 → H2SO4 + HBrO2;
HBrO2 + H2SO3 → H2SO4 + HBrO;
HBrO + H3AsO3 → H3AsO4 + HBr.
Из приведённых реакций видно, что возникающие промежуточные вещества HBrO2 и HBrO окисляют мышьяковистую ортокислоту.
Сопряжённые реакции, так же как и последовательные, широко распространены в природе, особенно в биологических и биохимических процессах. Механизм протекания этих сложных химических реакций исследуется недостаточно из-за сложности определения и изучения промежуточных продуктов реакции.
Для того чтобы реакции протекали с достаточной скоростью, применяют различные методы инициирования (активации). Наиболее распространённым методом активации реагирующих веществ является нагревание, в результате чего молекулы, получив избыточную энергию, переходят в активное состояние: разрыхляются связи или молекулы распадаются на атомы, ионы, радикалы.
59
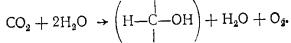
Инициировать реакции также можно действием света (видимого, ультрафиолетового). Реакции, инициируемые светом, называются фотохимическими. Примером таких реакций является синтез углеводородов в растениях, разложение пероксида водорода и нитрата серебра на свету и т. д. В фотохимических процессах реагирующие вещества поглощая кванты света, возбуждаются и переходят в активное состояние.
Фотохимические процессы в сочетании с ферментативными лежат в основе сложных окислительно-восстановительных реакций фотосинтеза. Фотосинтез – основной процесс образования органических веществ на Земле, определяющий кругооборот углерода, кислорода н других элементов, а также основной механизм трансформации солнечной энергии на нашей планете. Конечным результатом фотосинтеза является окисление воды с выделением молекулярного кислорода и восстановление диоксида углерода, этот фотохимический процесс выражается следующим суммарным уравнением:
К фотохимическим реакциям, играющим важную роль для живых организмов, относятся реакции образования озона из молекулярного кислорода под действием ультрафиолетовых лучей Солнца
O2 
 O2* + O2 + O.
O2* + O2 + O.
Значительно ускоряются реакции благодаря инициированию излучением высокой энергии (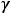 -лучи, нейтроны, рентгеновские лучи и др.). Так, под действием излучений высокой энергии происходит радиолиз воды, состоящий из нескольких стадий
-лучи, нейтроны, рентгеновские лучи и др.). Так, под действием излучений высокой энергии происходит радиолиз воды, состоящий из нескольких стадий
H2O + νγ 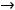 H2O H2O + νγ
H2O H2O + νγ 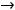 H2O+ + e H2O + e
H2O+ + e H2O + e 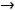 H2O−
H2O−
H2O+ + H2O  H3O+ + OH
H3O+ + OH
H2O− 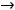 OH + H
OH + H
H• + H• = H2
OH• +OH• = H2O2
H• +OH• = H2O
H2O2 + OH• = H2O + •HO2
HO2 + •HO2 = H2O2 + O2
60
Глава V. ХИМИЧЕСКОЕ РАВНОВЕСИЕ
5.1. Закон действующих масс, принцип ле Шателье
Химические реакции делятся на обратимые и необратимые. При протекании необратимых реакций исходные вещества полностью превращаются в продукты реакции. Необходимым условием необратимых химических реакций является удаление образующихся продуктов из системы.
Обратимыми называются такие реакции, при которых конечные продукты взаимодействуют между собой, образуя исходные вещества. Обратимые реакции идут не до конца, а до известного предела, после чего устанавливается равновесная концентрация веществ в реагирующей системе.
Для обратимых реакций характерно, что скорость прямой реакции υ1 в начальный момент максимальна. По мере того как исходные вещества реагируют, концентрация их уменьшается и соответственно уменьшается скорость прямой реакции. Скорость же обратной реакции υ2 в начале процесса равна нулю, а с течением реакции возрастает, поскольку увеличивается концентрация продуктов реакции. Начиная с некоторого момента времени, скорости прямой и обратной реакций уравниваются, в результате чего устанавливается постоянная концентрация компонентов в реагирующей системе. Состояние системы, при котором скорости прямой и обратной реакции равны, называется химическим равновесием. Химическое равновесие является динамическим, или подвижным.
Схематически обратимую реакцию можно записать так: αА+bB C+dD;
υ1 = K1[A]a [B]b; υ2 = K2[C]c [D]d.
Если система пребывает в химическом равновесии, то υ1= υ2, тогда
K1[A]a [B]b = K2[C]c [D]d,
разделив левую и правую части равенства на величину K2[A]a [B]b, получим:
Отношение констант скоростей называется постоянной величиной и называется константой химического равновесия Кр:
Кр = 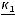 ;
; 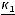



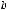 .
.
Уравнение является математическим выражением закона действующих масс, из которого следует, что отношение произведения равновесных концентраций
61

продуктов реакции к произведению равновесных концентраций исходных веществ является величиной постоянной при данной температуре. Величина Кр зависит от температуры и природы реагирующих веществ. Катализаторы не изменяют значения Кр, но сокращают время достижения химического равновесия. Константа равновесия является количественной характеристикой смещения равновесия в системе и изменяется в пределах от 0 до бесконечности. Если Кр = 0, реакция не протекает, если Кр равно бесконечности, то реакция протекает до конца, необратимо. При значении Кр меньше 1 равновесие смещено в сторону образования продуктов реакции.
Все выводы логически вытекают из простейшего математического анализа последнего уравнения: Кр = 0, если числитель равен нулю, а [C]c [D]d = 0, если концентрация одного из продуктов реакции или обоих равны нулю, т.е. продукты реакции не образуются. При делении любого числа на нуль получается бесконечность, а 


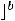

 только тогда, когда исходные вещества полностью превращаются в продукты реакции, т.е. процесс идёт до конца. Если произведение равновесных концентраций продуктов реакции больше произведения равновесных концентраций исходных веществ, то Кр больше одного, и наоборот.
только тогда, когда исходные вещества полностью превращаются в продукты реакции, т.е. процесс идёт до конца. Если произведение равновесных концентраций продуктов реакции больше произведения равновесных концентраций исходных веществ, то Кр больше одного, и наоборот.
Для реакций, протекающих между газами, их молярные концентрации пропорциональны парциальным давлениям (р) соответствующих газовых компонентов. Поэтому в общем виде константу равновесия для газовых систем можно записать так:
Кр = 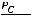
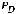 .
.
Закон действующих масс хорошо описывает химическое равновесие в газовых системах, в растворах неэлектролитов и в разбавленных растворах слабых электролитов. Для реакций, протекающих в других системах, вместо концентраций реагирующих веществ следует пользоваться их активностями, тогда в общем виде константа равновесия для реакций всех типов записывается так:
Кр = 

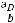 .
.
Большинство химических процессов, протекающих в организме, являются обратимыми. К ним относятся, например, соединение кислорода с гемоглобином, полимеризация и деполимеризация высокомолекулярных соединений, распад и синтез аденозинтрифосфорной кислоты (АТФ),
62
диссоциация электролитов на ионы. Распределение воды в организме также подчиняется законам равновесия; количества воды, распределённые между различными участками организма, должны находиться между собой в динамическом равновесии. Водное равновесие в организме регулируется нервной системой и железами внутренней секреции, а также чисто физическими факторами – гидростатическим и осмотическим давлением.
Таким образом, многие биохимические процессы представляют собой по существу непрерывное смещение разнообразных химических равновесий, которые могут быть описаны законом действующих масс.
Равновесие может смещаться, если изменять условия, которые его определяют. Смещение равновесия в зависимости от концентрации реагирующих веществ, температуры и давления в общем виде определяется правилом ле Шателье, которое формулируется так: если на систему, находящуюся в состоянии динамического равновесия оказывается воздействие, то в системе возникают процессы, направленные против зтого воздействия. Так, например, если в системе увеличивать давление, то равновесие смещается в сторону образования меньших объёмов, понижение давления, наоборот, смещает равновесие в сторону образования больших объёмов. Чтобы увеличить выход продукта реакции:
N2 +202↔ 2N2О,
процесс следует вести при высоком давлении:
FeC13 + ЗNН4SCN↔Fe (SCN)3 + 3NH4 Сl.
В реакции увеличение концентрации исходных солей смещает pавновесие вправо, при добавлении хлорида аммония равновесие сместится влево, раствор обесцветится. В эндотермических реакциях повышение температуры усиливает прямую реакцию, в экзотермических – обратную.
5.2. Равновесие в водных растворах. Электролитическая диссоциация. Закон разбавления Оствальда
Нейтральные молекулы веществ, попадая в раствор, взаимодействуют с молекулами растворителя. Полярные молекулы под действием молекул растворителя распадаются на ионы. Процесс распада молекул вещества на ионы под действием молекул растворителя называется электролитической диссоциацией.
Теория электролитической диссоциации была разработана С. Аррениусом в 1887 г. Её основы сводились к следующим положениям:
63

1.При растворении электролитов в воде образуются ионы двух типов – положительные (катионы) и отрицательные (анионы). Суммарный заряд положительных ионов равен суммарному заряду отрицательных ионов, вследствие чего весь раствор электронейтрален.
2.Химические свойства ионов отличаются от свойств нейтральных атомов и молекул. Например: Na0 – взаимодействует с водой; Na+ – не взаимодействует с водой; Н0 – плохо растворяется в воде; Н+ – содержится в водных растворах в больших количествах и др.
3.При пропускании электрического тока через раствор катионы перемещаются к катоду, а анионы к аноду (отсюда и их название). На электродах ионы разряжаются, превращаясь в нейтральные атомы или группы атомов.
4.С. Аррениус рассматривал электролитическую диссоциацию как обратимую химическую реакцию:
СН3СООН↔СН3СОО- +Н+. 5.Электролитическая диссоциация проходит ступенчато:
а) Н2СО3↔НС
 + Н+, НС
+ Н+, НС
 ↔С
↔С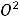
 + Н+,
+ Н+,
б) Н3РО4↔Н2Р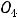
 +Н+, Н2Р
+Н+, Н2Р
 ↔НР
↔НР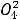
 +Н+, НР
+Н+, НР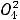
 ↔Р
↔Р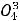
 +Н+.
+Н+.
Как и всякая обратимая реакция, процесс диссоциации характеризуется состоянием равновесия. Равновесие, в свою очередь, характеризуется степенью диссоциации, которая обозначается буквой α и представляет собой отношение числа молекул, распавшихся на ионы N1, к общему числу молекул вещества N, введённых в раствор:
α = 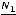 .
.
Значение α изменяется от 0 до 1. Если α = 0, то вещество не диссоциирует, т.е. является неэлектролитом. Вещества, у которых α лежит в пределах 0,3 – 1,0, условно относятся к сильным электролитам (например, кислоты НСlO4,НСl, Н2SO4 (разб.) и др.). Вещества со степенью диссоциации 0,30 – 0,03 относятся к электролитам средней силы (фосфорные кислоты), если α ниже 0,03 – к слабым электролитам (HF, H2O, NH4OH и т.д.). Пределы α указаны для 0,1 н. растворов.
Степень электролитической диссоциации раствора данного вещества определяется по отклонению его свойств от законов Вант-Гоффа и Рауля. Например, осмотическое давление для раствора NaCl, найденное
64

экспериментально, в 2 раза больше, чем осмотическое давление, определенное по закону Вант-Гоффа. Причина такого отклонения заключается в том, что каждая молекула NaCl распадается на два иона и количество частиц в растворе увеличивается вдвое, а поскольку осмотическое давление зависит не от природы вещества, а от количества частиц в растворе, то, следовательно, и давление увеличивается вдвое. Та же причина обуславливает повышение температуры кипения и понижение температуры замерзания растворов электролитов:
tкип (опытн.) > tкип (теорет.); tзам (опытн.) > tзам (теорет.).
Как отмечалось выше, электролитическая диссоциация – это обратимый процесс, который характеризуется константой равновесия. Например, диссоциация уксусной кислоты протекает по схеме:
СН3СООН↔СН3СОО- + Н+.
Поскольку это слабый электролит, то, применяя закон действующих масс, можно записать Кр этого процесса:
Кр = 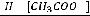 .
.
Константа равновесия в данном случае характеризует диссоциацию уксусной кислоты и называется константой диссоциации (или константой ионизации). В общем виде для слабого электролита, диссоциируеющего по схеме
АВ↔А+ + В-, константа диссоциации запишется:
Кд = 
 Константа диссоциации зависит от природы вещества и от температуры.
Константа диссоциации зависит от природы вещества и от температуры.
Константа диссоциации наиболее полно характеризует способность слабых электролитов распадаться на ионы (электролит тем слабее, чем меньше его константа диссоциации).
Если обозначить исходную молярную концентрацию раствора слабого электролита АВ через С моль/л, степень его диссоциации через α, тогда концентрация ионов будет [А+] = Сα и [В-] = Сα. Концентрация недиссоциированныx молекул будет равна (С — Сα) моль/л. Подставляя соответствующие значения в уравнение для константы диссоциации, получим
Кд = 
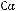

Это уравнение является математическим выражением закона разбавления
65

Оствальда, который, в свою очередь, является частным случаем закона действующих масс в применении к диссоциации слабых электролитов,
Поскольку значение α в растворах слабых электролитов мало, то для приближённых расчётов можно в знаменателе пренебречь величиной α и принять
Кд = Сα2,
или
α = √ 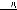 .
.
Из этого выражения видно, что если концентрация вещества увеличится в 100 раз, то степень его диссоциации уменьшится в 10 раз, и наоборот. Таким образом, закон разбавления устанавливает зависимость между α и Кд и позволяет вычислить одну из этих величин, если известна другая.
Степень диссоциации можно определить по данным, характеризующим электропроводность растворов. Известно, что в растворах переносят электричество только ионы, и поэтому электропроводность возрастает пропорционально количеству свободных ионов, т. е. степени электролитической диссоциации. Обозначим через μ молекулярную электропроводность раствора (т. е. электропроводность слоя раствора толщиной 1 см, содержащего 1 моль электролита). При бесконечно большом разбавлении раствора диссоциация становится полной, все ионы принимают участие в проведении электрического тока, и молекулярная электропроводность достигает наибольшего предельного значения μ( ).
).
Отношение молекулярной электропроводности при данном разбавлении μ(γ) к молекулярной электропроводности при бесконечно большом разбавлении равно степени диссоциации α:
α = 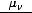
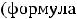
 .
.
ГЛАВА 6. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ОРГАНИЧЕСКОЙ ХИМИИ 6.1. Классификация и номенклатура
органических соединений
Органическая химия – раздел химии, изучающий соединения углерода. Термин был введён шведским химиком Й.Я. Берцелиусом в 1808 году.
Органическая химия изучает свойства органических соединений и методы их получения. Органические соединения – это углеводороды и их производные. Производные углеводородов содержат функциональные группы – атомы или
66
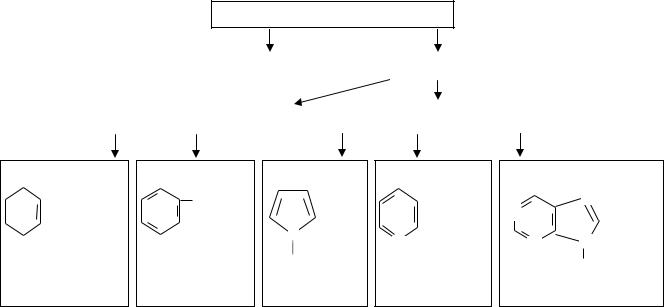
группы атомов, определяющих характерные химические свойства соединения и принадлежность к определённому классу соединений.
Основными признаками классификации органических соединений являются строение углеродного скелета, наличие функциональной группы и степень насыщенности соединений.
По строению углеродного скелета различают ациклические, карбоциклические и гетероциклические соединения. По степени насыщенности подразделяют на насыщенные, ненасыщенные и ароматические соединения.
Ациклические соединения содержат открытую цепь атомов углерода. Карбоциклические соединения содержат замкнутую цепь углеродных атомов и подразделяются на алициклические и ароматические. Гетероциклические соединения содержат циклы, включающие наряду с атомами углерода один или несколько гетероатомов, и также могут быть насыщенными, ненасыщенными и ароматическими, как и карбоциклические соединения.
Классификация органических соединений по строению углеродной цепи:
Органические соединения
ациклические |
|
|
циклические |
|
|
|
|
|
|
|
|
карбоциклические |
|
|
гетероциклические |
|
|
||
|
|
|
|
алициклические ароматические пятичленные шестичленные конденсированные
|
ОН |
|
|
N |
|
|
N |
|
|
|
|
|
|
|
|
N |
N |
|
N |
циклогексен |
фенол |
N |
||
пиридин |
|
|
||
|
H |
|
H |
|
|
пиррол |
|
пурин |
|
По природе функциональных групп органические соединения делятся на классы. Например, свойства карбоновых кислот определяется главным образом наличием в молекуле карбоксильной группы -СООН, алифатических спиртов наличием гидроксильной группы – ОН и т.д. В таблице приведены основные классы органических соединений.
67
ОСНОВНЫЕ КЛАССЫ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Функциональная группа
Отсутствует
F-, Cl-, Br-, I-(Hal) галоген
НО- гидроксильная
RO- алкоксильная
NH2-, -NH-, >N- амино
NO2 - нитро
О =C< карбонильная
карбоксильная
алкоксикарбонильная
карбоксамидная
НS- тиольная
HO3S- сульфо
Класс соединений |
Общая формула |
|
|
|
|
Углеводороды |
R-H |
|
|
|
|
Галогенпроизводные |
R-Hal |
|
|
|
|
Спирты и фенолы |
R-OH, Ar-OH |
|
|
|
|
Простые эфиры |
R-OR |
|
|
|
|
Амины |
RNH2, R2NH, R3N |
|
|
|
|
Нитросоединения |
RNO2 |
|
|
|
|
Альдегиды и кетоны |
|
|
|
Карбоновые кислоты
Сложные эфиры
Амиды карбоновых кислот
Тиолы |
R-SH |
|
|
|
|
Сульфокислоты |
R-SO3H |
|
|
По количеству и однородности функциональных групп органические соединения делятся монофункциональные, полифункциональные и гетерофункциональные. Монофункциональные соединения содержат одну функциональную группу, полифункциональные несколько одинаковых, а гетерофункциональные две и более различных функциональных групп.
68
С2Н5-ОН |
НОСН2 -СНОН-СН2ОН |
СН3-СНОН-СООН |
этанол |
глицерин |
молочная кислота |
монофункциональное |
полифункциональное |
гетерофункциональное |
соединение |
соединение |
соединение |
Соединения определённых классов объединяются в гомологические ряды. Гомологический ряд – это ряд соединений, в котором каждый последующий представитель отличается от предыдущего на группу СН2 , называемой гомологической разностью. Например, гомологический ряд алифатических одноатомных спиртов: СН3-ОН – метиловый,
С2Н5-ОН – этиловый, С3Н7-ОН – пропиловый, С4Н9-ОН – бутиловый и т.д.
Номенклатура органических соединений складывалась на протяжении всего периода развития органической химии. В настоящее время общепринятыми являются три номенклатурные системы: тривиальная, радикало-функциональная и заместительная (систематическая номенклатура), разработанная Международным союзом чистой и прикладной химии (IUPAC).
Тривиальная номенклатура состоит из исторически сложившихся названий, которые не отражают состава и строения вещества. Они являются случайными и отражают природный источник вещества (лимонная и янтарная кислоты, мочевина), характерные свойства (глицерин, гремучая кислота), способ получения (пировиноградная кислота, серный эфир), имя первооткрывателя (кетон Михлера) и т.д. Преимуществом тривиальных названий является их лаконичность, поэтому употребление некоторых из них разрешено правилами
IUPAC.
Радикало-функциональная номенклатура используется для названия простых моно- и бифункциональных соединений, некоторых классов природных соединений: спиртов, кетонов, простых эфиров, аминов и др. Основу названия составляет название данного класса соединений или одного из членов гомологического ряда с указанием заместителей. Названия соединений по радикало-функциональной номенклатуре строятся по следующему алгоритму: 1) перечисляются радикалы; 2) называется функциональная группа соответствующего класса соединений. Например, СН2=СН-ОСН3 винилметиловый эфир; СН3-СО-С6Н5 метилфенилкетон; (СН3)3С-NH-CH(СН3)2 третбутилизопропиламин. В радикало-функциональной номенклатуре наряду с
69
цифрами сохранилось обозначение положения заместителей греческими буквами: α, β,γ и т.д. Например, α,αI-дихлородиметиловый эфир ClСН2-О-СН2Cl; СН3-СН(NH2)-COОН α-аминопропионовая кислота. В симметрично построенных соединениях перед названием соответствующего радикала ставят префикс ди-: СН2 =СН-СН= СН2 дивинил.
Заместительная (систематическая) номенклатура более универсальна.
Название органического соединения состоит из трёх частей – префикса (приставки), корня и суффикса. Корнем названия является название родоначальной структуры. Родоначальной структурой могут быть наиболее
длинная |
углеродная цепь, карбоцикл или |
гетероцикл. Заместители |
подразделяются на два типа: |
|
|
-углеводородные радикалы и характеристические группы, обозначаемые только в префиксе;
-характеристические группы, обозначаемые как в префиксе, так и в суффиксе;
НАЗВАНИЯ АЛКАНОВ И АЛКИЛЬНЫХ РАДИКАЛОВ, ПРИНЯТЫЕ СИСТЕМАТИЧЕСКОЙ НОМЕНКЛАТУРОЙ IUPAC
|
|
|
|
|
|
Алкан |
|
Название |
Алкильный радикал |
|
Название |
|
|
|
|
|
|
|
|
|
|
|
|
CH4 |
|
Метан |
СН3- |
|
Метил |
|
|
|
|
|
|
|
|
|
|
|
|
CH3CH3 |
|
Этан |
CH3CH2- |
|
Этил |
|
|
|
|
|
|
|
|
|
|
|
|
CH3CH2CH3 |
|
Пропан |
CH3CH2CH2- |
|
Пропил |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Изопропил |
|
|
|
|
|
|
|
|
|
|
|
|
CH3CH2СН2CH3 |
|
н-Бутан |
CH3CH2СН2CH2- |
|
н-Бутил |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
втор-Бутил |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Изобутан |
|
|
Изобутил |
|
|
|
|
|
|
|
|
|
|
|
|
70
трет-Бутил
CH3CH2СН2CH2СН3 |
|
н-Пентан |
|
CH3CH2СН2CH2СН2- |
|
н-Пентил |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Изопентан |
|
|
|
Изопентил |
|
|
|
|
|
|
|
Неопентан |
|
|
|
Неопентил |
|
|
|
|
|
СН2=СН2 |
|
Этен |
|
СН2=СН- |
|
Винил |
|
|
(этилен) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
С6Н6 |
|
Бензол |
|
С6Н5- |
|
Фенил |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
С6Н5- СН3 |
|
Толуол |
|
С6Н5- СН2- |
|
Бензил |
|
|
|
|
|
|
|
ХАРАКТЕРИСТИЧЕСКИЕ ГРУППЫ, ОБОЗНАЧАЕМЫЕ ТОЛЬКО В ПРЕФИКСЕ
№ |
Формула |
Название |
№ |
Формула |
Название |
|
|
|
|
|
|
1 |
F- |
Фтор- |
5 |
С6Н5О- |
Фенокси- |
2 |
Cl- |
Хлор- |
6 |
С2Н5О- |
Этокси- |
3 |
I- |
Иод- |
7 |
СН3О- |
Метокси- |
4 |
NO2- |
Нитро- |
8 |
NO- |
Нитрозо- |
НАЗВАНИЯ ХАРАКТЕРИСТИЧЕСКИХ ГРУПП
|
|
|
|
|
Группа |
|
Название |
|
|
(ПЕРЕЧИСЛЕНЫ В |
|
|
|
|
|
|
|
|
|
|
в префиксе |
|
в суффиксе |
|
ПОРЯДКЕ УБЫВАНИЯ |
|
|
||
|
|
|
|
|
СТАРШИНСТВА) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
71
|
|
|
|
|
-(C)OOH* |
|
- |
|
овая кислота |
|
|
|
|
|
|
|
|
|
|
-COOH |
|
карбокси |
|
карбоновая кислота |
|
|
|
|
|
|
|
|
|
|
-SO3H |
|
сульфо |
|
сульфоновая кислота |
|
|
|
|
|
|
|
|
|
|
-(C)HO |
|
оксо |
|
аль |
|
|
|
|
|
|
|
|
|
|
-CHO |
|
формил |
|
карбальдегид |
|
|
|
|
|
|
|
|
|
|
>(C)=O |
|
оксо |
|
он |
|
|
|
|
|
|
|
|
|
|
-ОН |
|
Гидрокси (окси) |
|
ол |
|
|
|
|
|
|
|
|
|
|
-SH |
|
меркапто |
|
тиол |
|
|
|
|
|
|
|
|
|
|
-NH2 |
|
амино |
|
амин |
|
|
|
|
|
|
|
|
|
|
-OR** |
|
алкокси, арокси |
|
- |
|
|
|
|
|
|
|
|
|
|
-F, -Cl, -Br, -I |
|
фтор, хлор, бром, иод |
|
- |
|
|
|
|
|
|
|
|
|
|
-NO2 |
|
нитро |
|
- |
|
|
|
|
|
|
|
|
|
|
*) Атом углерода, заключенный в скобки, входит в состав родоначальной структуры.
**) Алкокси-группы и все следующие за ними перечисляются в префиксе по алфавиту и не имеют порядка старшинства.
6.2. Изометрия органических соединений
Изомерия – это явление существования органических молекул (изомеров) с одинаковой молекулярной формулой, т.е. имеющих одинаковый качественный и количественный состав, но различающиеся последовательностью связывания атомов в молекуле или расположением атомов в пространстве, а вследствие этого различные свойства. Изомерия (от греч. isos — равный)
Впервые термин «изомеры» был введён в 1830 г. шведским химиком Й.Я. Берцелиусом. Теоретическое обоснование изомерии принадлежит русскому химику А.М. Бутлерову, который не только создал теорию строения органических соединений, но и впервые предположил существование изомеров.
72
Различают два вида изомерии – структурную (изомерия строения) и пространственную (стереоизомерия).
Изомеры
Структурные (изомеры строения)
пространственные (стереоизомеры)
конфигурационные конформационные
геометрические оптические (π-диастереомеры)
энантиомеры диастереомеры
6.2.1.Структурная изометрия
Структурные изомеры отличаются друг от друга последовательностью связывания атомов в молекулу. Структурная изомерия подразделяется:
1) на изомерию углеродного скелета |
|
|
а) С4Н8: |
СН3 – СН = СН – СН3 |
СН2 =С(CH3) – СН3 |
|
бутен-2 |
2-метилпропен |
2) изомерию положения функциональной группы или кратной связи в |
||
молекуле |
|
|
а) С2Н4Cl2: |
СН3 – СНСI2 |
CH2CI – CH2CI |
|
1,1-дихлорэтан |
1,2-дихлорэтан |
б) С4Н8 : |
СН2 – СН = СН – СН2 |
CH2 = CH – CH – CH3 |
|
бутен-2 |
бутен-1 |
3) изомерию межклассовую |
|
|
а) С2Н6О: |
С2Н5-ОН |
СН3 -О- СН3 |
|
этанол |
диметиловый эфир |
б) C2H5O2N: |
NH2-СН2 –СООН |
С2Н5-NO2 |
|
глицин (аминоуксусная кисло) |
нитроэтан |
|
73 |
|
6.2.2. Пространственная изометрия (стереоизомерия)
Вещества, имеющие одинаковый состав и порядок связывания атомов в молекуле, но отличающиеся друг от друга их расположением в пространстве, называют пространственными изомерами или стереоизомерами.
Пространственная изомерия подразделяется на три вида – геометрическую изомерию, оптическую изомерию и конфирмационную (или поворотную) изомерию. В основе стереохимии лежат три основополагающих понятия:
конфигурация, конформация, хиральность .
Конфигурация – это определённое пространственное расположение атомов или атомных групп в молекуле относительно хирального центра, плоскости двойной связи или плоскости цикла, без учёта различий, возникающих за счёт вращения вокруг одинарных связей (одной или нескольких).
Конформация – это различное пространственное расположение атомов или атомных групп в молекулах определённой конфигурации, обусловленное вращением вокруг σ-связей. При этом стереохимическая конфигурация молекулы остаётся неизменной.
Хиральность (греч. сheir – рука) – это основное понятие стереохимии, обозначающее свойства объекта быть несовместимым со своим зеркальным отображением и является обязательным условием оптической активности.
6.3. Химическая связь и строение органических соединений
Химические свойства органических соединений обусловлены типом химических связей, природой связываемых атомов и их взаимным влиянием в молекуле. Эти факторы, в свою очередь, определяются электронным строением атомов и взаимодействием их атомных орбиталей.
6.3.1. Строение атома углерода и его особенности
Атом углерода – основа всех органических соединений, и органическая химия своим существованием как самостоятельная наука обязана, прежде всего огромному многообразию соединений углерода.
Углерод – первый элемент IV группы главной подгруппы периодической таблицы химических элементов Д.И. Менделеева. Заряд ядра атома углерода равен +6 и в основном (невозбуждённом) состоянии на внешней оболочке атома имеются только два неспаренных электрона (2s22p2) :
74

С +6 ) |
) |
1s2 2s2 2p2 |
1s2 |
2s2 |
2p2 |
||||
2 |
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
При поглощении дополнительной энергии (676 кДж/моль) один из электронов 2s2 подуровня переходит на свободную орбиталь 2р-подуровня, что приводит к появлению на внешнем электронном уровне атома углерода четырёх неспаренных электронов. Такое состояние называется “возбуждённое состояние
атома” углерода. |
|
|
|
|
|
|
|
|
|
|
|||||||
|
1s2 |
2s2 |
|
2p2 |
1s2 |
2s2 |
|
|
2p2 |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
переход |
возбуждённое состояние атома |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
углерода |
|||||
Четыре неспаренных электрона находятся на разных подуровнях: один на s- подуровне, а три – на р- подуровня, а следовательно, должны различаться по энергии (электроны s – подуровне имеют меньшую энергию, чем на – р- подуровня) и различными свойствами. В действительности соединения
четырёхвалентного углерода |
углеводороды, например, метан СН4 или |
четырёххлористый углерод CCl4, |
имеют четыре энергетически равноценные |
связи в молекулах. Для объяснения данного факта равноценности химических связей Л. Поллингом было предложено понятие – гибридизация. Сущность гибридизации в том, что из нескольких различных по форме, но близких по энергии атомных орбиталей образуется такое же число одинаковых по форме и энергии гибридных орбиталей. Гибридные орбитали могут быть описаны как линейные комбинации s- и р-орбиталей. По форме гибридная орбиталь отличается от исходных s- и р-орбиталей и представляет собой несимметричную объёмную восьмёрку:
s-орбиталь |
p-орбиталь |
|
исходные формы орбиталей |
гибридная орбиталь |
|
Такая гибридная форма орбитали очень выгодна при образовании |
||
химических связей, так как увеличивается |
площадь перекрывания орбиталей |
|
взаимодействующих атомов. Чем больше площадь перекрывания, тем прочнее связь. Для атома углерода характерны три вида гибридизации (три валентных состояния) с участием s- и р- орбиталей: sp3-, sp2- и s-гибридизации. Каждому из этих видов соответствует определённое валентное состояние.
75
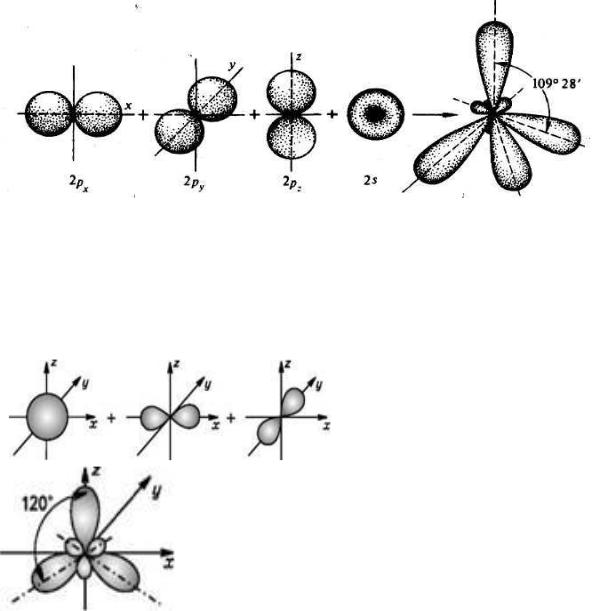
sp3-гибридизация углерода (первое валентное состояние). При смешении одной электронной орбитали 2s-подуровня и трех электронных орбиталей 2рподуровня образуются четыре качественно новые равноценные sp3-гибридные орбитали, направленные в пространстве под углом 109028I (от центра правильного тетраэдра к его вершинам). Поэтому sp3-гибридизацию называют ещё тетраэдрической.
Доля s-облака в гибридных sp3-орбиталях составляет 25 %. В первом валентном состоянии атом углерода образует только простые ковалентные связи.
sp2-гибридизация углерода (второе валентное состояние). Состояние sp2- гибридизации – результат взаимодействия одной s- и двух р-орбиталей:
2s |
2px |
2py |
три sp2-гибридные орбитали |
Образованные три эквивалентные sp2-гибридные орбитали находятся в одной плоскости под углом 120°, поэтому sp2-гибридизация называется
76
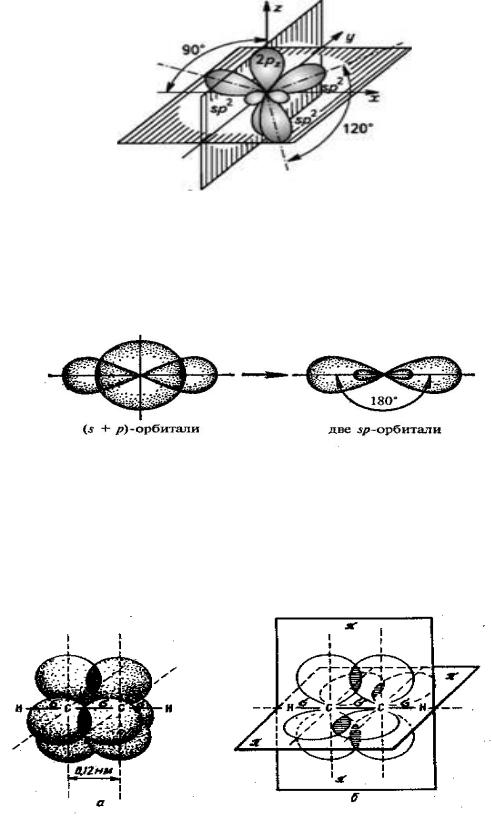
тригональной. Негибридизированная рz-орбиталь расположена в плоскости, перпендикулярной к плоскости гибридных орбиталей:
три sp2-гибридные орбитали + рz-орбиталь
Условная доля s-облака в гибридных sp2-орбиталях составляет 1/3 ( 33%). Такая гибридизация характерна для соединений с двойными связями
sp-гибридизация углерода (третье валентное состояние). Состояние spгибридизации – результат взаимодействия одной s- и одной р-орбиталей:
При этом образуются две sp-гибридные орбитали, расположенные друг к другу под углом 1800 . Отсюда sp-гибридизация называется линейной. Негибридизованные 2py- и 2pz-орбитали расположены в двух взаимно перпендикулярных плоскостях и под прямыми углами к sp-гибридным орбиталям. Условная доля s-облака в гибридных sp-орбиталях составляет 1/2 (50%). В состоянии sp-гибридизации атом углерода образует тройную связь:
77
6.3.2. Типы химических связей
Реакционная способность органических соединений обусловлена прежде всего типом химической связи, природой связываемых атомов и их взаимным влиянием. В зависимости от способа образования связи различают два основных типа – ковалентная связь и ионная связь.
Химическая
связь
Ковалентная Ионная
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ковалентная |
|
|
Ковалентная |
||||
неполярная |
|
|
неполярная |
||||
|
|
|
|
|
|
|
|
КОВАЛЕНТНАЯ СВЯЗЬ является основным типом химической связи в органических соединениях. Ковалентная связь образуется между атомами, равными или близкими по электроотрицательности атомами.
Ковалентная связь – это химическая связь, осуществляемая за счёт общих электронных пар между связываемыми атомами.
Ковалентная связь имеет два механизма образования – обменный и донорноакцепторный.
В обменном механизме общая электронная пара образуется за счёт электронов каждого из атомов:
А• + •B → A:B .
В донорно-акцепторном механизме общая электронная пара образуется за счёт неподелённой электронной пары атома-донора и вакантной орбитали другого атома-акцептора:
A: + B → A : В .
Образовавшаяся ковалентная связь отличается лишь способом образования, но по свойствам идентична ковалентным связям обменного механизма.
78

|
|
донор |
акцептор |
|
|
|
|
|
|
|
|
|
|
||
неподелённая |
|
|
Н |
|
|
|
|
|
Н |
||||||
пара |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
электронов |
R |
|
N : |
+ Н+Сl- |
|
|
R |
|
N+ |
|
H Cl- |
||||
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Н |
|
|
|
|
|
Н |
||||||
|
первичный амин |
|
алкиламмонийхлорид |
||||||||||||
Ковалентная связь характеризуется длиной, направленностью, полярностью, поляризуемостью и энергией.
Энергия связи – это энергия, выделяющаяся при образовании связи или необходимая для разъединения двух связанных атомов. Она служит мерой прочности связи: чем больше энергия, тем прочнее связь.
Длина связи – это расстояние между центрами связанных атомов. Двойная связь короче одинарной, а тройная короче двойной.
Направленность связи обусловливает молекулярное строение органических веществ и геометрическую форму их молекул. Углы между двумя связями называют валентными.
Насыщаемость связи характеризует способность данного атома соединяться с максимальным числом других атомов с образованием ковалентной связи.
Насыщаемость ковалентной связи определяет стехиометрию молекулярных соединений. Если каждую пару связывающих электронов обозначить чёрточкой, то такое изображение связей в молекулах позволит судить о том, в какой степени один атом насыщен другими атомами. Молекулы имеют определённый состав и существуют в виде дискретных частиц с определённой структурой:
Н . . Н или |
Н ─ Н |
|
Н . . С ⁞ |
⁞ С. . Н |
или Н ─ С ≡ С ─Н |
С |
С |
|
Поляризуемость связи выражается в смещении электронов связи под влиянием внешнего электрического поля, в том числе и другой реагирующей частицы. Поляризуемость определяется подвижностью электронов. Электроны тем подвижнее, чем дальше они находятся от ядер атомов. По поляризуемости π-связь значительно превосходит σ-связь, так как максимум электронной плотности π-связи располагается дальше от связываемых ядер. Поляризуемость
79

в значительной мере определяет реакционную способность молекул по отношению к полярным реагентам.
Полярность связи обусловлена неравномерным распределением (поляризацией) электронной плотности. Полярность молекулы количественно оценивают величиной её дипольного момента. Из дипольных моментов молекулы можно вычислить дипольные моменты отдельных связей. Чем больше дипольный момент, тем полярнее связь. Причиной полярности связи служит различие в электроотрицательности связанных атомов.
Электроотрицательность характеризует способность атома в молекуле удерживать валентные электроны. С увеличением электроотрицательности атома возрастает степень смещения в его сторону электронов связи.
Основываясь на значениях энергии связей, американский химик Л. Полинг (1901 – 1994) предложил количественную характеристику относительной электроотрицательности атомов (шкала Полинга). В этой шкале (ряду) типичные элементы-органогены располагаются по относительной электроотрицательности (для сравнения приведены два металла) следующим образом:
Электроотрицательность не является абсолютной константой элемента. Она зависит от эффективного заряда ядра, вида гибридизации атомных орбиталй и влияния заместителей. Атомы, связанные полярной связью, несут частичные заряды, обозначаемые греческой буквой δ (дельта). Смещение электронной плотности полярной σ-связи обозначают прямой стрелкой, полярной кратной связи – изогнутой стрелкой. Ковалентная связь, образованная атомами с одинаковой электроотрицательностью, неполярна, электронная плотность общей электронной пары не смещена ни к одному из атомов.
Например, углерод – углеродные связи в этане, этилене, ацетилене:
|
δ+ δ- |
δ+ δ- |
Н3С – СН3 |
Н3С → Сl |
Н2С = О |
неполярная связь |
полярные связи |
|
Неравномерное распределение электронной плотности в полярной молекуле характеризуется дипольным моментом μ, определяемым экспериментально.
80
Дипольные моменты выражают часто в дебаях D (1D=3,34·10-30Кл·м). Дипольные моменты неполярных связей и неполярных молекул равны нулю. В сложных молекулах полярность связей в большой мере зависит от взаимного влияния атомов и соответствующих электронных эффектов.
Чем больше разность электроотрицательностей атомов, тем выше полярность связи. При разности от 0,5 до 2,0 говорят о сильнополярной связи, если эта разность больше 2,0, связь становится ионной (общая электоронная пара полностью переходит к более электроотрицательному атому).
Ионная связь – химическая связь, образованная за счёт электростатического притяжения противоположно заряженных ионов. Ионная связь образуется между атомами, которые отличаются по электроотрицательности.
Ионную связь называют также электровалентной, гетерополярной. При взаимодействии атомов с резко различной электроотрицательностью (разность более 2) один атом передаёт электроны другому атому, образуются катион и
анион, взаимодействующие электростатически: |
|
||||
|
1ē |
|
|
|
|
A |
+ |
B |
→ |
A+ |
B- |
атом |
|
атом |
|
катион |
анион |
Ионная связь ненасыщаема и не имеет определённого направления в пространстве, электрическое поле ионов одинаково и симметрично во всех направлениях.
Характерные черты веществ с ионными связями – кристаллическое строение, высокие температуры кипения и плавления, растворимость и диссоциация в полярных растворителях. Ионная связь характерна для неорганических соединений и не имеет широкого распространения в органических соединениях
6.3.3. Строение молекул этана, этилена, ацетилена
Строение молекулы этана в зависимости от способа перекрывания атомных орбиталей различают в органических соединениях ковалентные связи двух типов: σ (сигма)-связи и π (пи)-связи. σ-Связи образуются в результате перекрывания двух различных атомных орбиталей – s-, p-, d- и гибридных sp3-, sp2 -, sp- вдоль линии, соединяющей ядра атомов (σ- перекрывание):
81
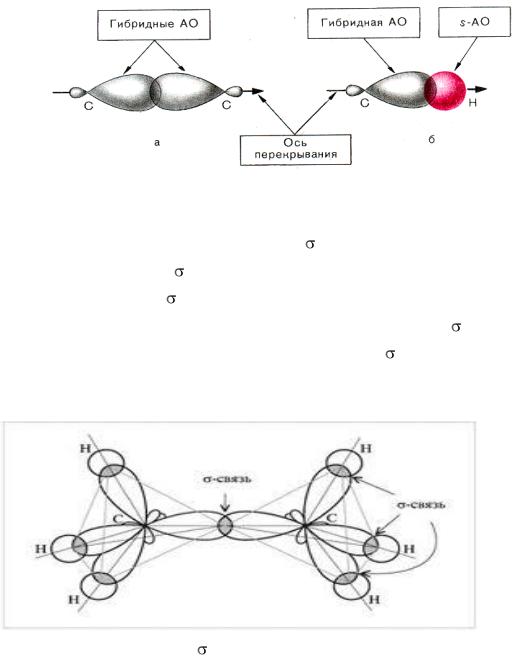
|
|
Образование |
-связи |
|
|
В молекуле |
этана |
-связи |
С-Н |
образуются |
в результате осевого |
s- sp3- перекрывания, а |
-связи С-С за счёт sp - sp3 перекрывания орбиталей. |
||||
Поскольку |
максимальная электронная плотность при - перекрывании |
||||
сосредоточена в пространстве между ядрами атомов, |
- связь обладает большой |
||||
прочностью. Длина углерод – углеродной связи С - |
С равна 0,154 нм, связи |
||||
С – Н - 0,110 нм; валентный угол составляет 109,50. |
|
||||
Образование -связей в молекуле этана Отличительной особенностью молекулы этилена является наличие двойной
углерод – углеродной связи. Атомы углерода в молекуле этилена находятся в sp2-гибридизации. За счёт перекрывания трёх гибридных атомных орбиталей каждого из атомов образуются σ-связи (четыре С—Н и одна С—С); а перекрывание двух негибридизириванных р-орбиталей в плоскости, перпендикулярней плоскости σ-связи (π-перекрывание), приводит к образованию π-связи. Максимальная электронная плотность π-связи сконцентрирована в двух областях — выше и ниже оси, соединяющей центры атомов. π-связь менее прочна, чем σ-; она образуется только между атомами, которые находятся в sp2- или sp-гибридизации.
82
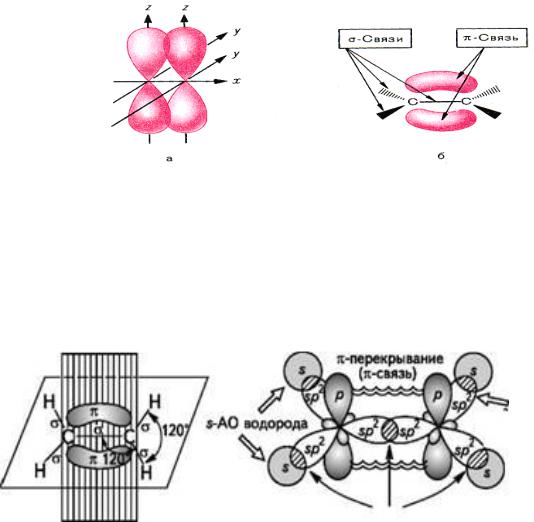
Образование π-связи
Длина углерод – углеродной связи в молекуле этилена составляет 0,134 нм. Углы между σ-связи одной С-С и двух С-Н составляют 1200. В молекуле этилена имеется пять σ-связей и одна π-связь.
Строение молекулы этилена
В молекуле ацетилена атомы углерода находятся в sp-гибридизованном состоянии. В гибридизации участвуют одна s- и одна p-орбитали. Каждый атом углерода имеет две sp-гибридизованные орбитали, расположенные на одной прямой под углом в 1800, и две негибридизованнные p-орбитали, которые расположены перпендикулярно как по отношению к гибридизованным орбиталям, так и по отношению друг к другу.
Две гибридизованные sp-орбитали (по одной от каждого атома) перекрываясь образуют σ-связь С-С (осевое перекрывание), оставшиеся негибридизованнные p-орбитали (2pz- и 2px-) располагаются во взаимноперпендикулярных плоскостях и перекрываются с аналогичными орбиталями другого sp-гибридизованного атома углерода, формируя две π-связи (боковое перекрывание). Каждая из этих π-связей расположена во взаимно-
83
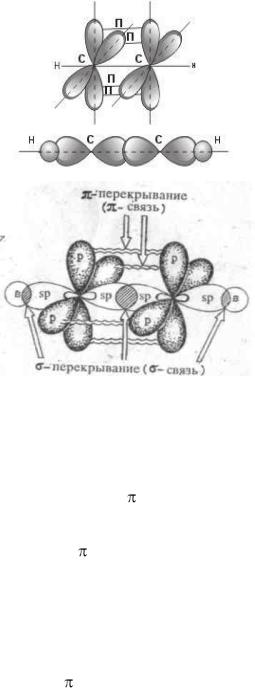
перпендикулярных плоскостях как между собой, так и по отношению к σ-связи, длина углерод – углеродной связи составляет 0,120 нм, валентные углы равны 180 0, молекула ацетилена линейна.
Образование σ- и π-связей в ацетилене
6.4. Сопряжение в органических соединениях
Ковалентная -связь может быть локализованной и делокализованной. Локализованной называется двойная связь, в которой электронная
плотность -связи охватывает только два ядра связываемых атомов. Делокализованная связь характерна для сопряжённых систем. Если в
молекулах присутствуют две двойные связи (или более) разделённые одной одинарной связью или у атома, соседнего с двойной связью, имеется р-орбиталь, то р-орбитали соседних атомов могут перекрываться друг с другом, образуя общую -электронную систему. Такая система называется сопряжённой. Сопряжение (мезомерия, от греч. Mesos – средний) – явление выравнивания, делокализации связей в молекуле, частице. Выравнивание электронной плотности происходит по всей цепи сопряжения, без затухания.
84
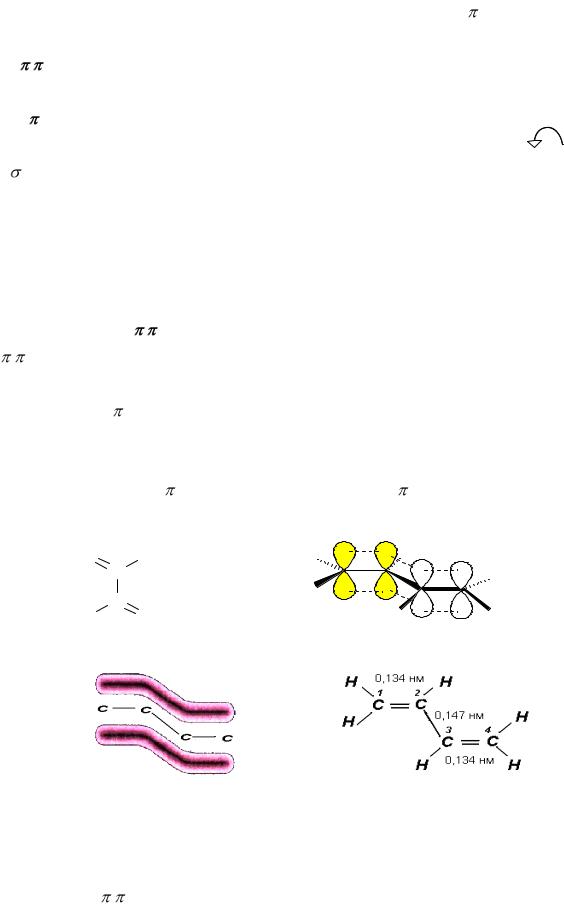
|
Сопряжённые системы в большинстве случаев содержат |
-связь. Известно |
два вида сопряжённых систем: |
|
|
– |
, -сопряжённые систем , состоящие из чередующихся простых и кратных |
|
связей ( = ─ = и т. д.); |
|
|
- |
p, -сопряжённые системы, состоящие из кратной связи, отделенной одной |
|
|
|
● ● |
|
-связью от соседнего атома с неподелённой парой р-электронов ( = ─ Х ). |
|
|
Сопряжение возможно лишь в случае параллельности |
осей симметрии |
взаимодействующих орбиталей. Сопряжённые системы делятся на системы с открытой и замкнутой цепью сопряжения.
6.4.1. Открытые сопряженные системы
Открытые , -сопряжённые системы. Простейшим представителем
, -сопряжённых систем с углеродной цепью является бутадиен-1,3, в молекуле которого атомы углерода находятся в состоянии sp2-гибридизации. Особенность
образования |
-связи в бутадиене-1,3 заключается в |
дополнительном |
||||
перекрывании негибридизированных |
р-орбиталей не только между атомами С1 |
|||||
и С2, С3 и С4 , |
но и атомов С2 и С3, что обеспечивает образование |
единого |
||||
(делокализованного) -электронного |
облака или -молекулярной |
орбитали, |
||||
включающей 4 р-электрона. |
|
|
|
|
||
H2C |
|
H |
H |
H |
|
|
|
|
|
|
|||
C |
|
|
H |
|
||
|
|
H |
|
|
||
|
C |
|
|
|
|
|
H |
CH2 |
|
H |
H |
|
|
|
|
|
||||
|
|
|
|
|
||
а) |
|
|
б) |
|
|
|
в) |
г) |
Образование , -сопряжённой системы в молекуле бутадиена-1,3: а)бутадиен – 1,3;
85
б)перекрывание р-атомных орбиталей в молекуле бутадиена – 1,3; в)делокализованная -молекулярная орбиталь; г) длина С-С связей.
Этот тип сопряжения называется π, π-сопряжением, т. к взаимодействуют орбитали π-связей. В результате делокализации электронов при образовании сопряжённой системы происходит частичное выравнивание длин связей: одинарная становится короче, а двойная – длиннее. Изолированная двойная связь в этилене, алкенах имеет длину 0,137 нм; одинарная связь в алканах имеет длину 0,154 нм. В бутадиене -1,3 две двойные связи сблизились в результате сопряжения и удлинились, стали равны по 0,134 нм, а центральная связь между вторым и третьим углеродными атомами стала короче и составила 0,146 нм.
Цепь сопряжения может включать большое число двойных связей. Чем она длиннее, тем больше делокализация π-электронов и тем устойчивее молекула. В сопряженной системе π-электроны уже не принадлежат определённым связям, они делокализованы т. е равномерно распределены по всей молекуле. Примерами таких природных сопряжённых молекул являются β-каротин и ретинол.
ретинол
β-каротин
Делокализация π-электронов в сопряжённой системе сопровождается выделением энергии, которая называется энергией сопряжения. Такие молекулы более устойчивы, чем системы с изолированными двойными связями.
Система сопряжения может включать и гетероатомы. Примером π, π- сопряженных систем с гетероатомом в цепи могут служить α и β-ненасыщенные
86

карбонильные соединения. Например, в акролеине (пропен-2-аль) цепь сопряжения
СН2 = СН – СН = О
включает три sp2-гибридизованных атома углерода и атом кислорода, каждый из которых вносит в единую π-систему по одному р-электрону.
Открытые p, -сопряжённые системы. Этот тип сопряжения осуществляется путём взаимодействия -орбитали кратной связи с р-орбиталью атома или группы атомов, содержащих неподелённую пару электронов. Примером таких систем являются следующие молекулы:
|
O |
|
H3C - C |
|
|
|
NH2 |
● ● |
|
|
|
|
|
СН2 = СН – О – СН3 |
ацетамид |
|
винилметиловый эфир |
6.4.2. Замкнутые сопряжённые системы
Замкнутые , -, и р, -сопряжённые системы. Циклические сопряжённые системы представляют большой интерес как группа соединений с повышенной термодинамической устойчивостью по сравнению с сопряжёнными открытыми системами. Эти соединения обладают и другими особыми свойствами, совокупность которых объединяют общим понятием ароматические соединения. Причиной выделения этих веществ в особый класс послужило то, что эти вещества в соответствии со своей молекулярной формулой обладают очень высокой степенью ненасыщенности, однако для них более характерны реакции замещения, а не присоединения, кроме того они устойчивы к действию окислителей и температуры. Многие ароматические соединения содержатся в нефти и на протяжении долгих лет сохраняют свои свойства. Наибольший интерес представляют ароматические углеводороды (арены и их производные) и гетероциклические соединения с , - или p, -сопряжёнными системами. Простейшим представителем этой группы веществ является бензол. В 1866 г. А. Кекуле предложил знаменитую формулу бензола:
87

Впоследствии изменился лишь способ её написания. Однако ни формула Кекуле, ни другие её варианты не смогли объяснить химические свойства бензола. Причина особого поведения ароматических соединений оставалась загадкой вплоть до создания современной теории химической связи. Все атомы углерода в молекуле бензола находятся в sp2-гибридизации. Шесть sp2-гибридных облака образуют каркас бензола. Все связи (С─С и С─Н) лежат в одной плоскости. Шесть негибридизованных р-орбиталей расположены перпендикулярно плоскости молекулы и параллельно друг другу. Каждая р-орбиталь в равной степени может перекрываться с двумя соседними р-орбиталями. В результате такого перекрывания возникает единая делокализованная π-система, наибольшая электронная плотность в которой находится над и под плоскостью скелета и охватывает все атомы углерода цикла. π-Электронная плотность равномерно распределена по всей циклической системе. Все связи между атомами углерода имеют одинаковую длину (0,139 нм), промежуточную между длинами одинарной и двойной связей.
Схема образования -связей (а) и , -сопряжённой системы (б) молекуле бензола
а)
б)
88
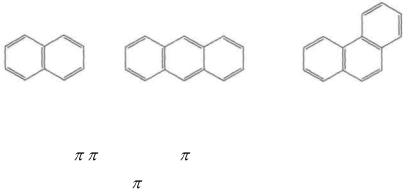
Бензол – плоский правильный шестиугольник с валентными углами 120°, все связи С─С равноценны (0.139 нм) и все атомы углерода sp2-гибридизованы; все связи С─С и С─Н лежат в одной плоскости. Термодинамическая стабильность бензола (его энергия cопряжения или энергия делокализации) – 150
кДж/моль. Энергия делокализации – это количество энергии, которое нужно затратить, чтобы нарушить ароматическую систему бензола. Энергия сопряжения бензола была рассчитана с помощью следующих экспериментальных данных: гидpиpование двойной связи циклогексена, где отсутствует сопряжение, сопровождается тепловым эффектом 121 кДж/моль. Пpи каталитическом гидpиpовании бензола должно было бы выделиться (если считать, что сопряжения нет) 121*3 = 363 кДж/моль. В действительности энергии выделяется значительно меньше 210 кДж/моль. Молекула бензола на 153 кДж/моль беднее энергией по сравнению с гипотетическим циклогексатриеном; эта величина – энергия сопряжения бензола. Она примерно в 10 раз превышает энергию сопряжения бутадиена – 1,3. Такое электронное строение объясняет устойчивость бензола.
В 1931 году немецкий физик Э. Хюккель обосновал критерии (правило)
ароматичности: соединение ароматично, если
1)оно имеет плоский цикл (все атомы цикла находятсяв sp2-гибридизации);
2)сопряжённая π-электронная система содержит (4n + 2) р-электрона (n = 0, 1,
2, 3 и т.д.;
3)характерны реакции электрофильного замещения (SE).
Для плоских конденсированных систем, в которых все атомы углерода находятся в состоянии sр2-гибридизации, число π-электронов сопряжённых ароматических систем также подчиняется правилу Хюккеля: для нафталина – 10 π-электронов; антрацена, фенантрена – 14:
нафталин |
антрацен |
фенантрен |
В гетероциклических ароматических соединениях гетероатом может |
||
участвовать как в |
, -, так и в |
,р-сопряжении: изогнутой стрелкой показана |
делокализация электронов -связи и р-атомной орбитали гетероатома, примером таких молекул могут быть пиррол (б), пиридин (в). В бензоле (а), в отличие от
89
гетероциклических соединений, электронная плотность делокализованной - молекулярной орбитали распределена равномерно между всеми атомами цикла. а) б) в)
бензол пиррол пиридин Число электронов сопряжённых систем во всех трёх соединениях
подчиняется правилу Хюккеля и равно шести.
6.5. Электронные эффекты органических соединений
Идея о взаимном влиянии атомов на реакционную способность органических соединений впервые высказана Бутлеровым в теории химического строения. Одно из основных положений теории химического строения органических молекул является положение о взаимном влиянии атомов или групп атомов в молекулах органических соединений на реакционную способность веществ. Дальнейшее развитие она получила в теории электронных смещений (электронных эффектов). Согласно современным представлениям, реакционная способность органических соединений определяется характером распределения электронной плотности в молекуле и подвижностью (поляризуемостью) электронов ковалентных связей.
В органических соединениях различают два вида электронных смещений – смещение электронной плотности по цепи σ-связей — индуктивный эффект; смещение по системе π-связей — мезомерный эффект.
6.5.1. Индуктивный эффект заместителей
При образовании ковалентной связи между двумя атомами с одинаковой электроотрицательностью электронная пара в равной мере принадлежит связанным атомам (неполярная связь). Если же атомы имеют разную электроотрицательность, электронная плотность ковалентной связи смещается к более электроотрицательному атому (полярная связь).
этан |
хлорметан |
|
ковалентная неполярная связь |
ковалентная полярная связь |
|
Полярную ковалентную связь |
изображают в виде |
стрелки (→), |
направленной к более электроотрицательному атому. Буквой δ (греч. «дельта»)
90
условно обозначают дробные (частичные) заряды на атомах. Символ δ+ отражает пониженную, а δ– – повышенную электронную плотность.
В сложных органических молекулах полярная ковалентная связь оказывает влияние на соседние связи, вызывая их поляризацию за счёт смещения электронной плотности σ-связи в сторону наиболее электроотрицательного элемента:
+ |
+ |
+ |
- |
СН3 СН2 |
СН2 |
СН2 |
Сl |
1-хлорбутан |
- I Cl |
+> + > + |
|
Поляризация распространяется на ближайшие две – три σ-связи и вёдет к возникновению частичных зарядов на соседних атомах. Такое влияние заместителей называется индуктивным эффектом (позднелат. «inductivus» – от лат. «inductio» – наведение, побуждение). σ-Связь поляризуется слабо , а поэтому индуктивный эффект затухает через 3 – 4 σ-связи («быстро гаснет»). Действие индуктивного эффекта заметно только на двух ближайших к заместителю атомах углерода.
Индуктивный эффект обозначается символом I. Графически индуктивный эффект изображается прямой стрелкой (→), совпадающей с -связью, а острие её направлено к более электроотрицательному атому.
В зависимости от направления электронного влияния заместителя различают положительный (+І) и отрицательный (–І) индуктивные эффекты. В качестве стандарта для оценки индуктивного влияния заместителя принят индуктивный эффект атома водорода, который из-за небольшого дипольного момента связи С─Н принято считать равным нулю. Заместители, притягивающие к себе электроны σ-связи в большей степени, чем атом водорода, проявляют отрицательный индуктивный эффект (–І ), а заместители, отталкивающие от себя электроны σ-связи сильнее атома водорода, проявляют положительный индуктивный эффект (+І ). Под притяжением и отталкиванием имеют в виду разницу в положении электронов связи, обусловленную различной электроотрицательностью атома водорода и заместителей. Электроотрицательность (э.о.) атомов зависит от вида гибридизации атомов и падает в ряду sp C > sp2 C > sp3C, что объясняется большим вкладом s- орбитали в гибридизацию.
91

По индуктивному эффекту может поляризоваться и -связь. В этом случае
+ |
+ |
- |
||
его обозначают как I -эффект: |
CН3 |
СН |
|
СН2 . |
|
|
|||
|
|
|||
Поляризация -связи обозначается изогнутой стрелкой. СН3-группа выступает в роли электронодонорного заместителя. Направление (знак) I-эффекта заместителя качественно оценивается путём сравнения со стандартом – атомом водорода, индуктивный эффект которого принят за ноль (шкала Поллинга):
F > О > N, Cl > Br > Csp > |
Csp2 > Csp3 |
S > H |
||||||
4,0 |
3,5 |
3,0 |
2,8 |
2,78 |
2,69 |
2,5 |
2,5 |
2,2 |
–I-эффект проявляют заместители, которые содержат более электроотрица- |
||||||||
тельные атомы, чем атом углерода: -F, -Cl, -Br, -OH и OR-, -NH2, |
||||||||
NHR- и NR2-группы, -NO2, |
>C=O, -COOH, -С≡N, –SO3H и др. |
|||||||
+I-эффект проявляют заместители, содержащие атомы с низкой электроот- |
||||||||
рицательностью (или имеющие отрицательный заряд): |
-Mg-, -Li; - |
|||||||
NR─, -O─, алифатические углеводородные радикалы (-CH3, -C2H5)
ит.п.
6.5.2.Мезомерный эффект заместителей
В молекулах, имеющих только -связи, взаимное влияние атомов в случае их различной электроотрицательности осуществляется через индуктивный эффект. В молекулах, представляющих собою сопряжённые системы, проявляется действие другого эффекта – мезомерного, или эффекта сопряжения. Влияние заместителя, передающееся по сопряжённой системе π- связей, называется мезомерным эффектом (М). Эффект сопряжения проявляется в молекулах, которые имеют кратные связи, разделённые только одной одинарной связью. При этом происходит перекрывание π-электронов этих связей, ведущее к образованию электронных орбиталей, охватывающих более двух атомов углерода. В результате избыток электронной плотности распределяется по всей системе сопряжённых связей. В сопряжении также могут принимать участие р-орбитали, если они отделены от кратной связи только одной -связью. Графически смещение электронной плотности по мезомерному эффекту обозначается изогнутыми стрелками. При этом заместитель сам является участником сопряженной системы, за счёт включения в систему сопряжения дополнительно -связи (например, группы -СН=О, -СООН и др.), неподелённую пару электронов гетероатома (галогена или групп -ОН, -NН2 и
92
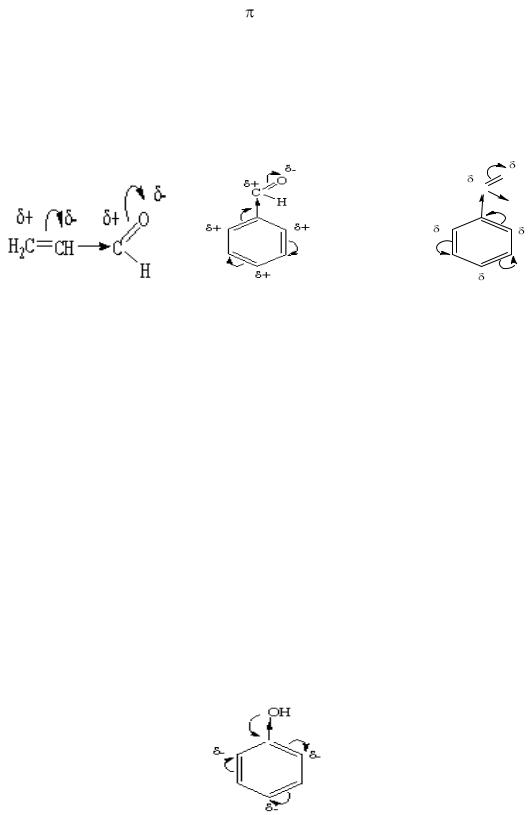
др.), а также вакантную или заполненную р-атомную орбиталь. В отличие от индуктивного, мезомерный эффект передается по системе сопряжённых связей на всю цепь сопряжения, так как -связь более подвижна, легче поляризуется.
Заместители, понижающие электронную плотность в сопряжённой системе,
проявляют отрицательный мезомерный эффект (-М). Эти заместители являются электроноакцепторными. Смещение электронной плотности показано
на следующих молекулах: |
|
|
акролеин |
бензальдегид |
бензойная кислота |
|
|
- |
|
+ |
O |
|
|
|
|
С |
|
|
|
OH |
+ |
|
+ |
|
|
|
|
+ |
|
В акролеине, бензальдегиде альдегидная группа проявляет отрицательный мезомерный ( -МСОН) и отрицательный индуктивный( -JСОН) эффекты и является электроноакцепторным заместителем, так же как и карбоксильная группа в бензойной кислоте, проявляющая -МСООН и -JСООН. Благодаря этому в электронная плотность ароматического кольца бензальдегида и бензойной кислоте ниже, чем в бензоле и особенно в положениях 2,4,6. Заместители, понижающие электронную плотность в сопряжённой системе (смещающие её на себя), проявляют отрицательный мезомерный эффект (-М). К ним относятся заместители, содержащие кратные связи с кислородом (имеющие карбонильную группу). Заместители, повышающие электронную плотность в сопряжённой системе, проявляют положительный мезомерный эффект (+М). Эти заместители являются электронодонорными. Положительным мезомерным эффектом (+М-эффектом) обладают заместители, увеличивающие цепь сопряжения. К ним относятся группы, которые, как правило, связаны с сопряжённой системой через одну σ-связь, обладающие р-орбиталью с неподелённой парой электронов (-OH, -NH2, -OCH3, -O-, -F, -Cl, -Br, -I и др.) или
с одним электроном (-CH2●).
В молекуле фенола заместитель (ОНгруппа) проявляет -IОН и +MОН электронные эффекты. При оценке влияния заместителей по распределению электронной
93
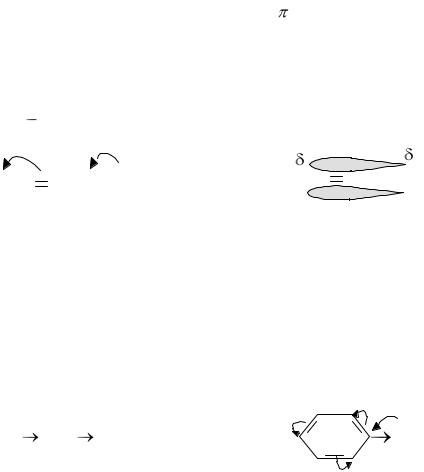
плотности в молекуле необходимо учитывать результирующее действие индуктивного и мезомерного эффектов. Если заместитель имеет р-орбиталь с неподелённой парой электронов (р, -сопряжение), то электронная плотность сопряжённой системы смещается в сторону р-орбитали с меньшим количеством электронов на ней. Электронная плотность на заместителе уменьшается (+М), например:
р- сопряжение
сопряжение
.. |
- |
- |
+ |
|
|||
СН2 CH — NН2 |
СН2 |
CH — NН2 |
|
|
|
|
|
виниламин |
|
+М NН2 |
|
В молекулах органических соединений электронные эффекты заместителей могут действовать либо согласованно, либо в противоположных направлениях. При оценке влияния заместителя на распределение электронной плотности в молекуле необходимо учитывать результирующее действие электронных эффектов. Один и тот же заместитель может быть электроноакцептором (ЭА) или электронодонором (ЭД). Например:
. . |
|
|
. . |
|
|
|
|
СН3 СН2 NH2 |
-INH |
2 |
NН2 |
-INH |
2 |
< +МNH |
2 |
|
|
|
|
|
|||
|
ЭА |
|
|
ЭД |
|
|
|
этиламин |
|
|
анилин |
|
|
|
|
|
|
|
|
|
|
|
6.6. Кислотность и основность органических соединений
Для оценки кислотности и основности органических соединений в современной органической химии используют две теории – протонную
(протолитическую) теорию Брёнстеда и электронную теорию Льюиса.
Согласно теории Бренстеда-Лоури, кислотой называют любое вещество, способное отдавать протон (донор протона), а основанием – вещество, способное присоединять протон (акцептор протона). Отсюда теория называется «протонной» или «протолитической». Для взаимодействия с протоном основание должно иметь неподелённую пару электронов или π-молекулярную орбиталь. Кислотность и основность являются относительными свойствами вещества. Кислотный характер может проявляться лишь в присутствии основания, и наоборот, основный характер – только в присутствии кислоты. В
94
целом кислотно-основный процесс состоит в переносе протона от кислоты к основанию и может быть представлен следующей схемой:
А- Н |
+ |
B → А– |
+ |
В+ -Н |
кислота |
основание cопряжённое |
|
сопряжённая |
|
|
|
основание |
|
кислота |
Кислота А-Н, отдав протон, превращается в основание А–, которое называют |
||||
сопряжённым основанием данной кислоты. Основание В, присоединив протон, переходит в сопряжённую кислоту В+-Н. Кислота А-Н и основание А–, а также основание В и кислота В+-Н являются сопряжёнными кислотно-основными парами.
Многие органические соединения могут одновременно обладать свойствами и основания, и кислоты. Такие вещества называют амфотерными (амфолитами). Мерой силы кислоты А-Н является константа кислотности Kа (a от англ. Acid – кислота), которая обычно определяется по отношению к стандартному основанию – воде: А–Н + Н2О →А– + Н3О+. Мерой кислотности является константа равновесия, называемая константой кислотности (Ka):
Ka=[A-] [H3O+]/[AH].
Чем больше значение Kа, тем сильнее кислота. Как правило, константы кислотности очень малы. Например, для уксусной кислоты Kа при 25 °С частицы вещества, рассматриваемые в рамках протонной теории как кислоты, могут представлять собой нейтральные молекулы (например циановодород), катионы (катион аммония, аквакомплексы металлов, такие как [Al(H2O)6]3+) или анионы
(например гидросульфат-ион HSO4–): |
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
HCN + H2O CN– + H3O+ |
|
|
|
|
|
|||||||
|
|
|
NH4+ + 2 H2O NH3.H2O + H3O+ |
|
|
|
|||||||||||
|
|
|
[Al(H |
O) ] + + H |
O [Al(H |
O) |
OH] + |
+ H O+ |
|
|
|
||||||
|
|
|
2 |
6 3 |
2 |
|
2 |
5 |
|
2 |
3 |
|
|
|
|||
|
|
|
|
|
HSO |
– + H |
O SO 2– + H |
O+ |
|
|
|
|
|
||||
|
|
|
|
4 |
|
2 |
4 |
|
3 |
|
|
|
|
|
|
||
Условно кислоты делят на "сильные" pKa < 0 и "слабые" pKa 2-15.7 |
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Кислота |
HI |
HBr |
|
H SO |
|
HF |
CH COOH |
|
H O |
ROH |
|
NH |
|||||
|
|
|
|
|
2 |
4 |
|
|
|
3 |
|
|
|
2 |
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
рK |
a |
-11 |
-9 |
|
|
-9 |
|
3.4 |
|
|
4.75 |
|
15,7 |
16 – 18 |
|
30 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
В роли оснований протонная теория рассматривает нейтральные частицы (например гидрат аммиака), анионы (цианид-ион, карбонат-ион, ацетат-ион); катионы-основания (например катион гидразиния):
95
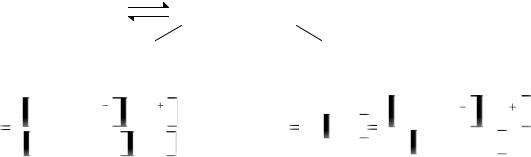
NH3.H2O + H2O NH4+ + OH– + H2O
CN– + H2O HCN + OH–
N2H5+ + H2O N2H62+ + OH– |
|
Особое место занимают протолиты с двойственными свойствами – |
|
амфотерные вещества (амфолиты): |
|
HCO3– + H2O CO32– + H3O+ |
|
Кислота |
Основание |
HCO3– + H2O H2CO3 + OH– |
|
Основание |
Кислота |
Большинство органических соединений можно рассматривать как кислоты, |
|
поскольку в них содержатся |
поляризованные связи атома водорода с |
различными элементами (О, N, S, С).
Органические кислоты классифицируют по природе кислотного центра: ОН-кислоты: спирты, фенолы, карбоновые кислоты, сульфокислоты, гидроксикислоты, аминокислоты;
SH-кислоты: тиоспирты, SH-содержащие аминокислоты и др. соединения; NH-кислоты: амины, имины, гетероциклические соединения с атомом азота; СН-кислоты: углеводороды, радикалы гетерофункциональных соединений.
Сравнительная характеристика кислотных свойств органических соединений
Органические кислоты — слабые электролиты, и процесс диссоциации идет до установления обратимого равновесия, которое характеризуется константой
диссоциации (К). Например, диссоциация уксусной кислоты: |
||
CH3COOH + H2O |
CH3COO- + H3O+ |
|
анион кислоты |
катион гидроксония |
|
(сопряжённое основание) |
(сопряжённая кислота) |
|
|
CH3COO |
H3O |
|
|
|
CH COO |
H O |
|
K |
|
|
|
; |
Ka |
K H2O |
3 |
3 |
CH |
COOH H O |
CH COOH |
||||||
3 |
|
2 |
|
|
|
3 |
|
|
Ка – константа кислотности. Для многих органических соединений Ка очень малы (например, для уксусной кислоты Ка = 1,76 · 10-5), поэтому чаще используется для количественной характеристики кислотных свойств используется величина рКа:
pKa = -lg Ka.
Чем меньше рКа, тем больше кислотность по Бренстеду.
96

Сила кислот определяется устойчивостью образующихся после отщепления протона сопряжённых оснований (анионов). Чем устойчивее сопряжённое основание, тем сильнее кислота. Устойчивость же аниона обусловлена степенью делокализации отрицательного заряда и зависит от ряда факторов:
природы кислотного центра; характера заместителя, связанного с кислотным центром; природы растворителя.
При равных других факторах устойчивость анионов, а следовательно, и кислотность возрастают с увеличением электроотрицательности и поляризуемости атомов кислотного центра. Поскольку в пределах периода периодической системы электроотрицательность атомов возрастает слева направо (поляризуемость не изменяется), то ОН-кислоты сильнее соответствующих NН-кислот, а те, в свою очередь, сильнее СН-кислот.
С–Н |
N–H |
О–Н |
S–H |
О–Н кислоты |
|
кислота |
кислота |
кислота |
кислота |
|
|
|
|
|
|
|
|
С2Н5СH2–Н |
С2Н5NH–H |
С2Н5О–Н |
С2Н5S–H |
С6Н5О–Н |
СН3СОО–Н |
пропан |
этиламин |
этанол |
этантиол |
фенол |
уксусная кислота |
|
|
|
|
|
|
рКа = 50 |
рКа = 30 |
рКа = 16 |
рКа = 10,6 |
рКа = 10 |
рКа = 4,8 |
В пределах группы периодической системы электроотрицательность атомов уменьшается сверху вниз, но увеличивается их объём, а следовательно, возрастает поляризуемость, то есть возможность делокализации внешнего электронного облака. Это способствует повышению стабильности аниона и приводит к возрастанию кислотности. Поэтому SН-кислоты обладают большей кислотностью, чем ОН-кислоты:
CH3—CH2—OH |
CH3—CH2—SH |
этанол |
этантиол |
рKа = 18,0 |
рKа = 10,5 |
Органические кислоты с одинаковыми радикалами в зависимости от природы кислотного центра можно расположить по возрастанию кислотности:
СН-кислоты < NН-кислоты < ОН-кислоты < SН-кислоты
Кислотность органических кислот.
В пределах отдельного типа кислот кислотность зависит от строения радикала, связанного с кислотным центром. Алкильные радикалы, благодаря +I- эффекту, увеличивают электронную плотность в кислотном центре и тем самым
97

дестабилизируют анион, что приводит к уменьшению кислотности. Ароматические радикалы, наоборот, повышают устойчивость аниона за счёт делокализации отрицательного заряда и способствуют увеличению кислотных свойств. Заместители, введённые в алифатические и ароматические радикалы, оказывают влияние на кислотность вследствие проявления ими электронных индуктивного и мезомерного эффектов. Электронодонорные заместители (+І; +М-эффект) понижают кислотность, а электроноакцепторные (-І; -М-эффект)
— увеличивают её. Так, введение в молекулу фенола электроноакцепторных групп приводит к повышению кислотности, введение же электронодонорных групп — понижает кислотность. При этом главную роль играют электронные эффекты заместителей и образование системы сопряжённых связей. Сравним кислотность этанола и фенола:
CH3 — CH2 — OH + H2O |
H3O+ + CH3 — CH2 O- |
этоксид-анион + IС2Н5
 — OH + H2O
— OH + H2O  H3O+ +
H3O+ + 

 — O- :
— O- :
феноксид-анион
Этоксид-анион неустойчив, так как содержит электронодонорную этильную группу. В феноксид-анионе отрицательный заряд делокализуется по сопряжённой системе -связей ароматического кольца, что стабилизирует анион и увеличивает кислотные свойства фенола по сравнению с этанолом.
Высокая кислотность карбоновых кислот обусловлена образованием в карбоксилат-анионе резонансной р- сопряжённой системы, где отрицательный заряд распределяется поровну между атомами кислорода:
|
O |
|
|
O- |
|
O- 1/2 |
R — C |
O- |
|
R — C |
O |
R — C |
O- 1/2 |
|
||||||
|
|
|
|
В алифатическом ряду наиболее сильное влияние на кислотность оказывают заместители, расположенные к кислотному центру ближе.
Cl CH2—COOH |
ClCH2—CH2—COOH |
ClCH2—CH2—CH2—COOH |
хлоруксусная кислота 3-хлорпропановая кислота |
4-хлорбутановая кислота |
|
pKa = 2,86 |
pKa = 4,1 |
pKa = 4,5 |
Наряду с природой кислотного центра и строением радикала значительное влияние на проявление кислотных свойств оказывает растворитель.
98

Как правило, лучше гидратируются (сольватируются) небольшие по размеру ионы с низкой степенью делокализации заряда. Например, в ряду карбоновых кислот с увеличением длины алифатического (гидрофобного) радикала
кислотность уменьшается. |
|
|
|
НСООН |
СН3СООН |
СН3–СН2–СООН |
С15Н31–СООН |
муравьиная |
уксусная |
пропановая |
пальмитиновая |
рКа 3,7 |
4,7 |
4,8 |
4,9 |
Влияние растворителя определяют его диэлектрической проницаемостью и способностью сольватировать растворённые частицы. Чем выше диэлектри-
ческая проницаемость растворителя и сольватационный эффект, тем стабильнее ионы в растворе. При равных прочих условиях сольватация аниона протекает тем сильнее, чем меньше его размер и менее делокализован в нём заряд. Таким образом, влияние сольватационного эффекта растворителя и влияние заместителей на кислотность противоположны друг другу. Наиболее эффективным растворителем является вода, обладающая высокой диэлектрической проницаемостью (ε = 80 при 20 °С) и способностью к сольватации растворённых частиц.
ТИПЫ ОРГАНИЧЕСКИХ ОСНОВАНИЙ Согласно протонной теории, в роли основания может выступать любое
вещество, способное присоединять протон. Для образования химической связи с протоном основание должно иметь неподеленную пару электронов или π- молекулярную орбиталь.
Органические основания в зависимости от природы основного центра (атом с неподеленной парой электронов или электроны π-связи) подразделяются на р- основания и π-основания.
В основаниях центром основности является атом с неподелённой парой электронов. По природе центра основности р-основания классифицируют на следующие типы:
аммониевые (центр основности N): амины, азометины (R-CH=NR), нитрилы (R- C≡N), азотсодержащие гетероциклы;
оксониевые (центр основности O): спирты, простые эфиры, альдегиды, кетоны, сложные эфиры, амиды кислот и др.;
сульфониевые (центр основности S): тиоспирты (R-SH), тиоэфиры (R-S-R). Органические основания, чтобы присоединить протон, должны иметь либо
неподелённую пару электронов у гетероатома – р-основания (аммониевые, оксониевые, тиониевые), либо быть анионами:
99
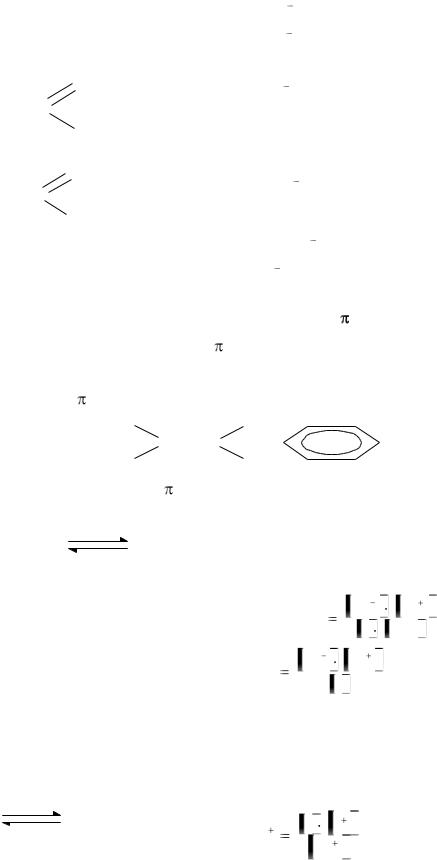
р-основания: |
анионы |
R–ОН — спирты |
НО — гидроксид-ион |
R–O–R — простые эфиры |
RO — алкоксид-ион |
|
O |
альдегиды |
RS — алкил-тиолят-ион |
|
|
||
R — C |
|
|
|
|
H (R) кетоны |
|
|
|
O |
карбоновые |
H2N — амид-ион |
|
|
||
R — C |
|
кислоты |
|
|
|
||
|
OH |
|
|
R–SH — тиолы |
|
RCOO — ацилат-ион |
|
R–NH2 — амины |
Н — гидрид ион |
||
Существует ещё одна группа оснований — -основания, где центром основности являются электроны -связи или ароматической системы. Это слабые основания, которые с протоном образуют не ковалентные связи, а короткоживущие -комплексы:
C  С
С
-основания Реакция основания В: с водой описывается уравнением:
В : + Н2О |
ВН+ + ОН- |
Поскольку органические основания, как и кислоты, слабые электролиты, то
константа равновесия этой реакции будет равна: K |
OH |
BH |
|
|
|
||
B |
H 2 O |
||
|
и соответственно, константа основности: Kв |
OH BH |
|
|
||
B |
||
|
Так как органическая кислота отщепляет протон, а основание его присоединяет, то кислотные и основные свойства можно количественно характеризовать одной величиной – сродством к иону водорода. Это позволяет применять константы кислотной ионизации как для кислот, так и для оснований:
ВН+ |
В + Н+ |
|
B H |
|
|
Kвн |
|
|
|
|
|
|
|
BH |
|
|
|
|
100
Как и для кислот пользуются значениями рКв = -lg Кв. Зная величину рКв, можно перейти к значениям рКвн+. Для этого из отрицательного логарифма ионного произведения воды (рКн2о) при данной температуре надо вычесть значение рКв. Чем сильнее основание, тем выше его рКвн+.
Значение рКвн+ некоторых оснований
Гидроксид натрия |
14 |
Этиламин |
11 |
Аммиак |
9 |
Хинин |
8 |
Анилин, пиридин |
5 |
п-Нитроанилин |
1 |
По аналогии с кислотами сила оснований зависит от ряда факторов:
природы основного центра; характера заместителя, связанного с основным центром.
При равных других факторах с увеличением электроотрицательности атома основного центра, в пределах одного и того же периода, неподелённая пара электронов удерживается прочнее, а следовательно, основность соединения уменьшается. Поэтому оксониевые основания слабее аммониевых. В пределах группы периодической системы с увеличением поляризуемости атома основного центра усиливается делокализация неподелённой электронной пары и соответственно уменьшается основность соединения. Поэтому сульфониевые основания слабее оксониевых. Еще более слабыми основными свойствами обладают π-основания, в которых электронная пара, присоединяющая протон, не является свободной. Таким образом, органические основания в зависимости от природы основного центра можно расположить по возрастанию основности:
Основность органических оснований
π-основания < сульфониевые основания< оксониевые основания< аммониевые основания.
Если |
сравнивать |
основность |
соединений с |
одинаковыми |
радикалами, |
. . |
|
. . |
то оксониевое |
основание |
будет слабее |
СН3 О |
СН3 и СН3 |
NH CH3 , |
аммониевого. В тиониевых основаниях, например,СН3 – S – СН3 электронная плотность атома серы рассредоточена в большем объёме (поляризуемость атома серы больше, чем кислорода), и плотность заряда значительно меньше. Поэтому тиониевые основания слабее оксониевых и не способны образовывать
101
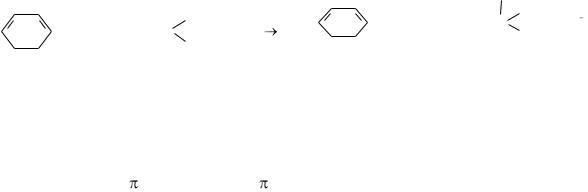
прочные связи с протоном. В целом, сила р-оснований с одинаковыми радикалами уменьшается в ряду: N > O > S. Наибольшую основность среди органических соединений проявляют амины.
Электронодонорные заместители повышают основные свойства, так как увеличивают электронную плотность на атоме основного центра, а электроноакцепторные заместители понижают электронную плотность и выраженность основных свойств. Обсудим влияние этих заместителей на примере новокаина – сложного эфира n-аминобензойной кислоты и диэтиламиноэтанола, который применяется в хирургической практике для местной анестезии. Новокаин плохо растворим в воде, лучше растворимы его соли, например, новокаина гидрохлорид:
|
|
|
O |
|
|
|
|
|
O |
H |
|
|
+ |
|||
.. |
|
|
.. C2H5 |
|
|
|
|
|
|
|
|
C2H5 |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
H2N — |
|
— C — O — (CH2)2 — N |
Cl |
||||||||
|
|
|
|
|
|
|
|
|||||||||
H2N— |
|
—C—O—(CH2)2 |
—N |
+ HCl |
|
|
|
|
|
|
|
C2H5 |
|
|||
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
C2H5 |
|
|
|
|
новокаина гидрохлорид |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
В новокаине два наиболее выраженных основных центра: 1) атом азота, связанный с бензольным ядром, 2) атом азота, связанный с алифатическими радикалами. Неподелённая пара электронов атома азота в первом основном центре вступает в р, -сопряжение с -системой ароматического кольца, что приводит к уменьшению основных свойств. Алкильные радикалы, являясь электронодонорами, увеличивают электронную плотность на атоме азота второго основного центра и его основные свойства. Протон от кислоты присоединяется ко второму центру.
Влияние растворителя на основность определяется главным образом эффектом сольватации. Как и в случае с кислотами, сольватационный эффект растворителя и электронные эффекты заместителей оказывают на основность противоположное воздействиего центра и его основные свойства. Протон от кислоты присоединяется ко второму центру.
Во вторичных и третичных алифатических аминах с атомом азота связаны, соответственно, два и три алифатических радикала, обладающих электронодонорными свойствами. Поэтому можно было бы ожидать, что основность увеличивается в ряду: NH3 < первичный < вторичный < третичный амин.
Есть, однако, ещё один важный фактор, от которого зависит основность – сольватация алкиламмониевого иона молекулами воды. Способность к образованию водородных связей с молекулами воды растёт в ряду: третичный
102

амин  вторичный амин
вторичный амин  первичный амин
первичный амин  NH3. Комбинация этих двух факторов приводит к следующему ряду основности аминов: NH3 третичный
NH3. Комбинация этих двух факторов приводит к следующему ряду основности аминов: NH3 третичный  первичный вторичный амин.
первичный вторичный амин.
Американский учёный Г. Н. Льюис в 1923 году предложил электронную теорию кислот и оснований, которая не противоречит теории Бренстеда-Лоури, но является более общей. Согласно теории Льюиса, основание – любая частица
(атом, молекула или анион), способная отдавать электронную пару для образования ковалентной связи, а кислота – любая частица (атом, молекула, катион), способная принимать пару электронов с образованием ковалентной связи. Основание по теории Льюиса является донором, а кислота – акцептором пары электронов. Из приведённого определения видно, что основания Льюиса тождественны основаниям Бренстеда-Лоури. Однако кислоты Льюиса охватывают более широкий круг органических соединений. Кислотой Льюиса считается любая частица,имеющая вакантную орбиталь. Если в теории Бренстеда-Лоури кислота – это донор протона, то, согласно теории Льюиса, сам протон является кислотой, поскольку имеет вакантную орбиталь. Таким образом, в представлении электронной теории кислота Бренстеда-Лоури является соединением, которое образует кислоту Льюиса. Поэтому, согласно теории Льюиса, к кислотам относят не только соединения, отщепляющие протон (протонные кислоты), но и другие вещества, имеющие вакантную орбиталь и способные принимать пару электронов (апротонные кислоты). Кислотами Льюиса являются такие соединения, как BF3, AlCl3, SbCl3, ZnCl2, HgCl2, Н+, FeCl3, +NO2, Br+.и др. ”. Основания Льюиса – это ионы или молекулы с заполненной электронной конфигурацией, способные отдавать электронную пару с образованием ковалентной связи (доноры электронов):
NH3, HO -, H2O, ROH, F -, Br -, CN -.
Лёгкость протекания кислотно-основной реакции определяется силой кислоты и основания, а также жёсткостью или мягкостью кислоты и основания. Представление о жёстких и мягких кислотах и основаниях (ЖМКО), введённое Р. Пирсоном, по существу является дальнейшим развитием теории Льюиса. Согласно концепции Пирсона, кислоты и основания Льюиса делятся на жёсткие
имягкие.
Кжёстким кислотам относят кислоты Льюиса, в которых атомы-акцепторы имеют малый объём и несут высокий положительный заряд, а следовательно,
103
обладают высокой электроотрицательностью и низкой поляризуемостью (H+, Li+, Na+, K+, Mg2+, Ca2+, Mn2+, Al3+, Fe3+, AlCl3, R-C+ = O). Нижняя свободная молекулярная орбиталь (НСМО) в жёстких кислотах имеет низкую энергию. К мягким кислотам относятся кислоты Льюиса, в которых атомы-акцепторы имеют большой объём и несут низкий положительный заряд, а поэтому обладают низкой электроотрицательностью и высокой поляризуемостью (Cu+, Ag+, Hg2+, Pt2+, I2, Br2 и др.). НСМО в мягких кислотах имеет высокую энергию. К жёстким основаниям относят основания Льюиса, в которых атомы-доноры имеют высокую электроотрицательность и низкую поляризуемость (H2O, OH–, F–, Cl–, CH3COO–, SO42–, CO32–, NO3 – , R-OH, R-O–, R-O-R, NH3, R-NH2, H2N- NH2, NH2– и др.). Верхняя занятая молекулярная орбиталь (ВЗМО) в жёстких основаниях обладает низкой энергией.
К мягким основаниям относят основания Льюиса, в которых атомы-доноры имеют низкую электроотрицательность и высокую поляризуемость (RSH, RS–, R-S-R, HS–, I–, CN–, R-CN, C2H4, C6H6, H–, R– и др.), ВЗМО в мягких основаниях обладает высокой энергией. Исходя из общего положения о том, что более эффективно протекает взаимодействие между орбиталями с близкими энергиями, жёсткие кислоты преимущественно реагируют с жёсткими основаниями, а мягкие кислоты - с мягкими основаниями (принцип ЖМКО).
Следует отметить, что понятия «жёсткие» и «мягкие» кислоты и основания не связаны с понятиями «сильные» и «слабые» кислоты и основания. Так, мягкое основание Н– и жёсткое – С2Н5О– являются сильными, а мягкое основание НS– и жёсткое СН3СОО– являются слабыми основаниями.
В настоящее время кислоты льюиса называют электрофилами, а основаниями Льюиса – н уклеофилами.
Электрофилы: H+, HNO3, H2SO4, HNO2, (т.е. соответственно +NO2, +SO3, +NO),
PhN2+, BF3, AlCl3, ZnCl2, FeCl3, Br2, I*-Cl, H2O2, O3,
Нуклеофилы: H-, H2N-, HO-, RO-, RS-, RCOO-, Hal-, HSO3-, -CN, RC≡C-, -
CH(COOEt)2
104
ЗАКЛЮЧЕНИЕ
От химиков будущего сегодня ожидают инициативности и настойчивости в преодолении косности, способности к риску и активного подхода к решению задач в интересах общества, а также критической оценки своих и чужих результатов работы.
Знания и умения, требуемые от химиков, приобретаются ими уже сейчас.
В обучении технологов и товароведов должны преобладать аспекты, наиболее приемлемые именно для химических дисциплин: методология, систематика и научная постановка проблем. Предпочтение следует отдавать активным методам обучения, применяя лабораторные и экспериментальные занятия.
Использование программированных учебных материалов, в которых предмет представлен отдельными, логически взаимосвязанными частями,
постепенным переходом от простого к сложному, может существенно облегчить и преподавание и обучение. Несмотря на это работа преподавателя с небольшими группами учащихся сохранит своё значение и впредь. Ещё несколько лет назад человек мог иметь одного единственного партнёра для диалога – другого человека. В будущем недостатка в партнёрах у обучающегося не будет: всё большее применение находят технические средства обучения и компьютерные технологии, включающие такие основные программы, как Консультант плюс, Garant, ЭБС «Консультант студента». Благодаря им преподаватель освободится от рутинной учебной работы. У него появится время для того, чтобы сосредоточить своё внимание на научно-исследовательской работе, включая руководство и кураторство студенческими научными работами.
Конечно, сегодня мы не можем наших школьников и студентов обеспечить такими знаниями, которые станут известны лишь через 10 – 20 лет. Но нельзя не считаться с тем фактом, что наши кадры именно сейчас, а не когда-нибудь в будущем, должны получить в руки оружие, с помощью которого они смогут решать возникающие в процессе научно-технического прогресса задачи и проблемы.
Итак, какими же должны быть химики будущего? Они должны сочетать в себе строгую деловитость, фундаментальные знания, творческую фантазию,
105
педантичную точность с торопящей вперёд готовностью к риску. Развитое общество не построят те специалисты, которые говорят:»Это мы сделали так-так и будем делать всегда». Нет, лозунгом трудящихся в науке ли, производстве,
организации или управлении должно стать:» Этого еще никто не делал – попробуем сделать мы!» Ну, а то, что выполнять намеченное они станут не по звездам, а по надёжным указаниям науки – это понятно само собой. Вот тогда путь в третье тысячелетие, на котором нашим верным помощником служит химия, будет успешным для всех нас.
106
БИБЛИОГРАФИЧЕСКИЙ СПИСОК.
1.Будрейко Н. А. Философские вопросы химии / Н. А. Будрейко. – М. : Высшая школа, 1970. – 336 с.
2.Михайличенко Н. И. Общетеоретические основы химии / Н. И. Михайличенко. – Киев : Высшая школа, 1979. – 224 с.
3.Сеничева Л. В. Периодическая система химических элементов Д. И. Менделеева. Прогнозирование свойств элементов и их соединений / Л. В. Сеничева. – Хабаровск : Изд. ТОГУ, 2005. – 24 с.
4.Бутуханов В. Л. Физическая и коллоидная химия : учеб. пособие / В. Л. Бутуханов. – Хабаровск : РИЦ ХГАЭП, 2009. – 96 с.
5.Бутуханов В. Л. Химия / В. Л. Бутуханов, Л. П. Павлюченкова,
И. А. Жарская. – Хабаровск : РИЦ ХГАЭП, 2008, – 68 с.
6.Органическая химия : в 2 кн. Кн. 2: Специальный курс / В. Л. Белобородов , С. Э. Зурабян, А. П. Лузин, Н. А. Тюкавкина; под ред. Н. А. Тюкавкиной. – М. :
Дрофа,2008. – 592 с.
7.Органическая химия : в 2 кн. Кн.1: Основной курс / В. Л. Белобородов, С. Э. Зурабян, А. П. Лузин, Н. А. Тюкавкина; под ред. Н. А. Тюкавкиной. – М. :
Дрофа,2002. – 640 с.
8.Тюкавкина Н. А. Биоорганическая химия / Н. А. Тюкавкина, Ю. И. Бауков. –
М. : Дрофа, 2008. – 542 с.
9.Руководство к лабораторным занятиям по органической химии / Н. Н. Артемьева, В. Л. Белобородов, С. Э. Зурабян и др.; под ред. Н. А. Тюкавкиной. – 2-е изд.,перераб. и доп. – М. : Дрофа, 2002. – 384 с.
10.Зеленин К. Н. Химия общая и биоорганическая / К. Н. Зеленин, В. В. Алексеев. – СПб. : ЭЛБИ-СПб., 2003. – 712 с.
11.Хаускрофт К. Современный курс общей химии : в 2 т. Т. 1 : пер. с англ. / –
М. : Мир, 2002. – 540 с.
Хаускрофт К. Современный курс общей химии : в 2 т. Т. 2 : пер. с англ. / – М. :
Мир, 2002. – 528 с.
12.Оганесян Э. Т. Органическая химия / Э. Т. Оганесян. – М. : – Академия, 2011. – 432 с.
107
Владимир Лаврентьевич Бутуханов, Нина Николаевна Минаева
Учебное издание
ХИМИЯ
Учебное пособие для студентов направлений 38.03.07 «Товароведение» и
19.03.04 «Технология продукции и организация общественного питания»
Редактор Г.С. Одинцова
Подписано в печать |
|
|
. Формат 60 х 84/16. |
|
|
|||
Бумага писчая. Цифровая печать. Усл.печ.л. |
|
. Уч-изд.л. |
|
. |
||||
Тираж 50 экз. Заказ ____. |
|
|
|
|||||
|
|
|
|
|
|
|
|
|
680042, г. Хабаровск, ул. Тихоокеанская, 134, ХГАЭП, РИЦ
108