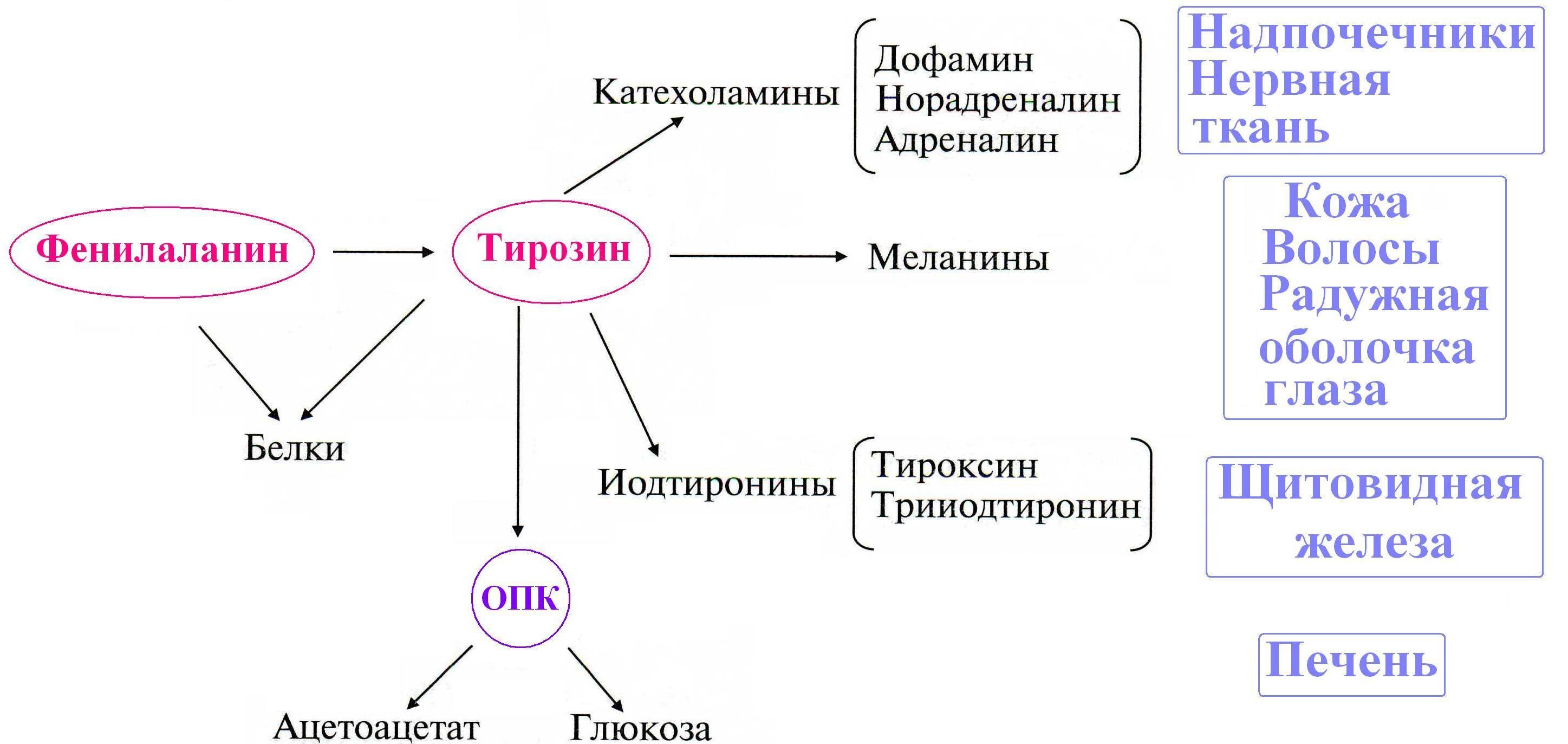
Обмен Фенилаланина и Тирозина
Фенилаланин – незаменимая аминокислота.
Используется в организме только в 2-х процессах:
Субстрат для синтеза белков и превращается в тирозин.
! Превращение Фен в Тир необходимо в 1 очередь для удаления избытка Фен, т.к. его высокие концентрации токсичны для клеток.
В Тир превращается ~ 90% Фен:


Превращение Фен в Тир – это 1-ая реакция основного пути метаболизма Фен.
Все дальнейшие превращения в организме происходят уже с Тир.
При нарушении протекания реакции превращения Фен в Тир возникает заболевание – фенилкетонурия (фенилпировиноградная олигофрения).
Из-за невозможности превращения Фен в Тир, катаболизм фенилаланина протекает по альтернативному пути:
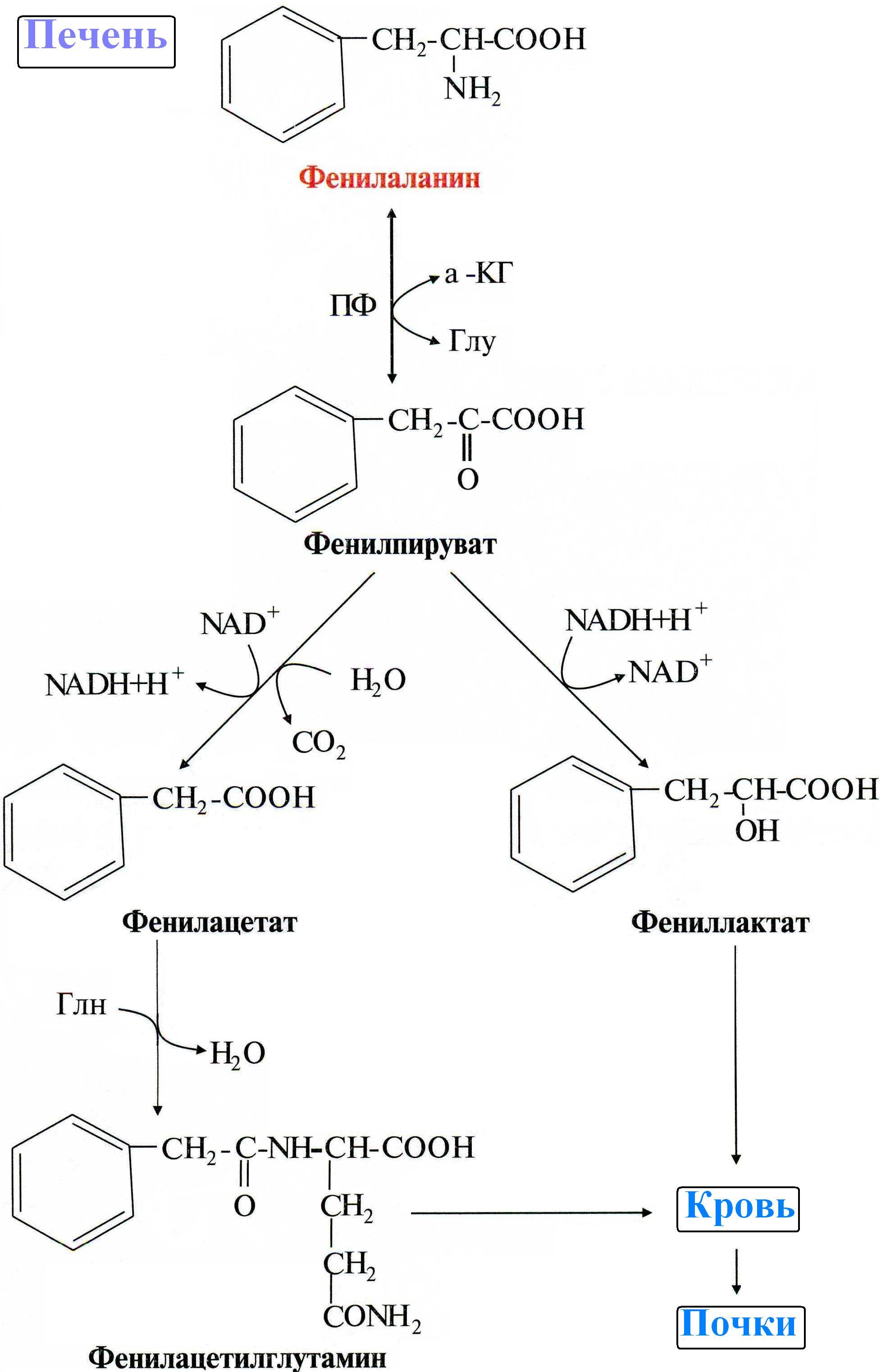
При этом в крови и моче повышается содержание фенилаланина и метаболитов альтернативного пути: фенилпирувата, фениллактата, фенилацетата и др.
Симптомы: Резкое нарушение умственного и физического развития, судорожный синдром, «мышиный» запах.
Также встречается: нарушение пигментации кожи.
Фен и его производные, при их избытке, оказывают токсическое действие на клетки мозга, поскольку:
ограничивают транспорт Тир и Три через гематоэнцефалический барьер и тормозят синтез нейромедиаторов (дофамина, норадреналина, серотонина).
Без лечения больные фенилкетонурией не доживают до 30 лет.
Заболевание наследуется по аутосомно-рецессивному типу.
Выделяют 2 формы фенилкетонурии:
Классическая фенилкетонурия:
Причина: наследственный дефект фермента фенилаланингидроксилазы.
Частота заболевания: 1 случай на ~ 10000 новорожденных.
Вариантная фенилкетонурия:
(коферментзависимая гиперфенилаланинемия)
Причина: мутации в генах, контролирующих метаболизм H4-биоптерина.
Встречается: 1-2 случая на ~ 1 млн. новорожденных.
H4-биоптерин необходим для гидроксилирования не только Фен, но и Тир и Три, поэтому, при этой форме заболевания нарушен метаболизм всех 3 аминокислот, а также синтез многих нейромедиаторов.
При этой форме заболевания возникают тяжелые неврологические нарушения и ранняя смерть.
Лечение фенилкетонурии: диета, с почти полным исключением из пищи фенилаланина.
! Начинать: сразу после рождения ребенка.
Для диагностики фенилкетонурии определяют концентрацию фенилаланина и патологических метаболитов в крови и моче больного.
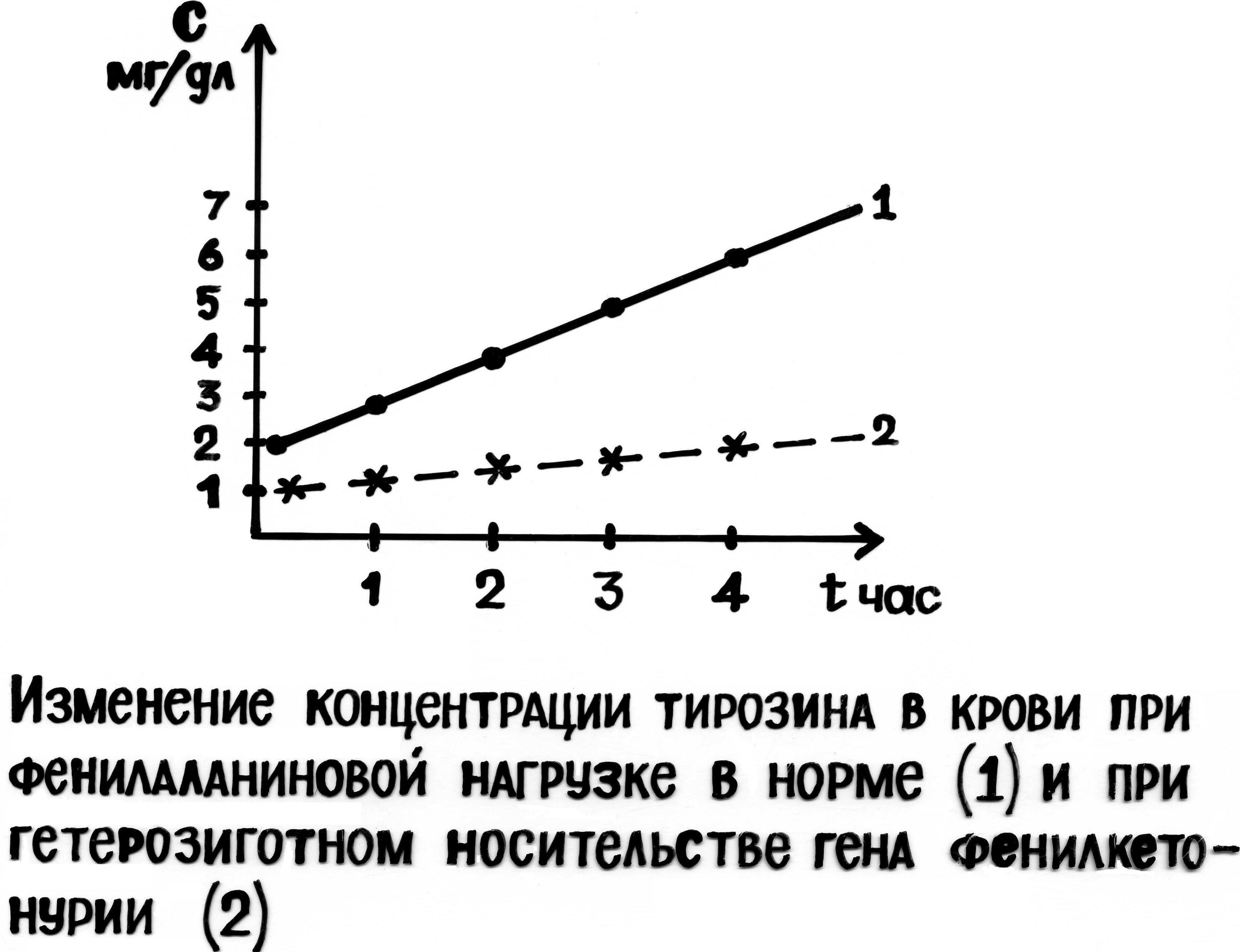
В генетической консультации, для выявления гетерозиготного носителя заболевания, используют тест толерантности к фенилаланину:
Разработаны специальные схемы скрининга для выявления новорождённых детей с ФКУ.
В настоящее время для выявления мутантного гена фенилаланингидроксилазы у гетерозиготных носителей ФКУ, используют также ПЦР-диагностику.
Особенности обмена тирозина в разных тканях
Кроме использования в синтезе белков, Тир в разных тканях используется для синтеза многих биологически-активных соединений.
Катаболизм Тир до конечных продуктов происходит в печени.