

Химическая технология
.docx
Химическая
технология как наука.
Хим.тех-это
совокупность методов и средств химической
переработки природного сырья, отходов,
предметы потребления и средства
производства, а также наука об экономичных
и наиболее экологичных хим. методах и
средствах.Объект
изучения:
химические производства; Цель
изучения:
создание целесообразных способов
производств, необходимых человеку;
Методы
исследования:
эксперимент, моделирование и системный
анализ.Современные
проблемы хим.тех.:1.увеличение
глубины переработки углеводородного
сырья;2.
повышение качества выпускаемой химич.
продукции;3.повышение
экологической безопасности хим.
производства;4.повышение
ресурсоэффективности ХТ
производств;5.интелектуализация химич.
производств.Особенности
проведения химико-технологич.
процессов:*требования
к чистоте производимых продуктов,*применение
жестких условий при производстве в-в
(высоких или низких давлений, температ.,
агрессивных сред),*энергоемкие
производства(применение энергии
электрич, а не пара, угля и тд),*автоматизация
технологических процессов,*внедрение
новых эффективных методов синтеза.
Общие закономерности химических
процессов.ХТ
процесс-последовательность
химических и физико-химических процессов
целенаправленной переработки исходных
в-в в продукт.1.Механические
и гидромеханические процессы-без
изменения химического и фазового состава
обрабатываемого материала. Используются
дробилки, транспортеры, питатели,
диспергатооры, формователи, компрессоры,
насосы, смесители, фильтры.2.Теплообменные
процессы-нагревание,
охлаждение, изменение фазового состояния.
Химический состав в нем не изменяется.
Протекают в теплообменниках, кипятильниках,
плавильнях, сублиматорах.3.Массообменные
процессы-перенос
ве-ва внутри фазы или между фазами,
вызванный градиентом концентраций и
протекающий без изменения химического
состава (растворение, кристаллизация,
абсорбция, экстракция, десорбция). Для
этого служат: кристаллизатры, сушилки,
дистилляторы, ректификаторы, абсорбенты,
экстракторы, десорбенты.4.Химические
процессы-процессы,
вызывающие коренное изменение химического
состава вещества в химических
реакторах.5.Энергетические
процессы-взаимное
преобразование различных видов энергии
в турбинах, генераторах,
моторах.6.Информационные,
управляющие процессы отвечают за
получение и передачу информации о
состоянии потоков и ве-в на пункт
управления.Классификация
химико-технологических процессов.
1.Отраслевой
тип.Неорганическая
химическая технология:
1)Основной
неорганический синтез: многотоннажные
производства кислот, щелочей, солей,
аммиака, минеральных удобрений 2)Тонкий
неорганический синтез: малотоннажные
производства (катализаторы, неорганические
препараты, реактивы редкие эл-ты,
материалы для электроники, лекарственные
в-ва и др.).3)Ядерно-химические
технологии:
технологии обогащения и получения
радиоактивных веществ и изотопов.4)
Металлургия:
пр-во черных и цветных Ме. 5)Технология
силикатов:
пр-во вяжущих и строительных материалов,
керамических изделий, стекла. Технология
органич/ веществ:
1)Переработка
ископаемого углеродосодержащего сырья:
твердого топлива, нефти и газа.2)Нефтехим.
синтез:
пр-во органич. продуктов на основе УВ,
С, Н2.3)Основной
органический синтез: пр-во
базовых продуктов органич синтеза
4)Биотехнология:
пр-во
кормовых дрожжей, аминокислот, ферментов,
антибиотиков. 5)Тонкий
органический синтез:
пр-во органических препаратов, реактивов,
лекарственных ве-в, душистых ве-в, средств
защиты растений.6)Пр-во
органич. полупродуктов и красителей.7)Технология
высокомолекулярных соединений.
8)Технологии растительного и животного
сырья.Требования
к химич. производству:1.получение
в производстве необходимого продукта.
2.экологич.
безопасность.3.безопасность
и надежность эксплуатации
оборудования.4.максимальное
использование сырья и энергии.
5.максимальная
производительность труда. Компоненты
химич. производства.
Переменные
компоненты:
Сырье-руды,
концентраты-всегда сложная смесь,
которую подвергают действию химич.
реагента, при этом отдельные компоненты
смеси распределяются между фазами.Затем
фазы разделяют и перерабатывают ту из
тех, в которой примеси целевого компонента
*титановые
минералы хлорируют. Тi
превращают в соль TiCl4
и затем отгоняют. *медные
руды подвергают плавлению. Сульфиды Сu
и др ценные Ме переходят в расплав, а в
шлак переходят менее ценные. Вспомогательные
материалы:
*продукт
(основной и вспомогательный)-результат
переработки сырья. *энергия-обеспечивает
функционирование производства.*отходы
производства, не подлежащие дальнейшей
переработки-вещества и материалы,
удаляемые затем в окружающую
среду.Постоянные
компоненты:
*аппаратура
*устройства
контроля и управления*строительные
конструкции (хранилище сырья, продуктов
и др материалов) *система
организации транспортиров. сырья,
продукции вспомогательного материала,
отходов.Разделение
на твердые фазы:
1.Грохотание-ситы
(грохоты) с отверстиями разного диаметра.
2.Гравитационное обогащение-основано
на разности плотности.3.Электромагнит.
или электростатичное обогащение-проходя
на транспортере магнитное или электрическое
поле фракции разделяются (разделение
рудного сырья). 4.Флотация-основана
на разной смачиваемости ве-в, для
ускорения процесса добавляют ПАВ. Во
флотационную машину поступает смесь
воды и мелких частиц обогащаемой рудой
(пульпа) и подается воздух. Гидрофобные
частицы прилипают к пузырькам воздуха
и выносятся вместе с ними на поверхность,
образуя пену, которая удаляется
специальным устройством (обогащают
медные руды или сильвенит).Разделение
жидкости и тв веществ:1.Деконтация.2.
Фильтрация. 3.Центрифугирование.
4.Гидроциклонирование.Разделение
жидкость-жидкость:1.Экстракция
исп. для несмешивающихся жидкостей.
2.Импригирование
(жидким маслом)-твердое тело используют
для извлечения Ме из растворов
(полиметаллические руды).Композиты
«твэксы» (тв экстракты) позволяют
осуществлять более простое
отделение.3.Мембранная
экстракция.4.Высокоскоростное
центрифугирование (разделение изотопов
и газов).5.Процессы
с физическим активированием: ядерные,
ультрозвуковые,
пламенные,микроволновые,высокочастотные.
Сырьевая
база хим пром.Сырье
(переменный компонент пр-ва) это в-ва и
материалы, подвергщиеся ранее воздействию
труда и предназначенные для дальнейшей
переработки. Если при получении продукции
исходные виды тверд. сырья смешиваются,
то такая смесь тв веществ называется
шихтой
(применяется при получении стекла,
чугуна). Если в качестве сырья применяют
смесь тв веществ с жидкостью, то такая
смесь называется пульпа.
Сырье:
1)по
происхождению:
минеральное, растительное, животное.2)по
агрегатному состоянию:
твердое, газообразное, жидкое.3)по
составу:
органич., минеральн, неорганич.
Характеристика
минеральн. сырья.Минеральное-сырье,
доставаемое из недр земли, которое может
быть использовано без предварит.
обработки или с небольшой предварительной
обработкой. Минеральное
сырье: 1.
Рудное (руды, служащие исходным сырьем
для получения черных, цветных или
благородных металлов).2.Нерудное
(горные породы, не служащие специально
для получения Ме).Строительные материалы
(глина, песок, известняк, мрамор,
гравий).Индустриальное сырье (применяется
без хим.обработки (асбест)). 3.Горючее
(сырье, которое может быть использовано,
как топливо: каменный и бурый уголь,
сланец, торф, нефть и газовые гидраты.
Хар-ка
разработок минер. сырья.
Участки земной коры, где имеются скопления
минералов в свободном или связанном
виде-месторождениями.
Добыча тв минерального сырья из недр
земной коры называется горной разработкой.
добыча угля:шахта, добыча руд:рудники,
добыча редких и драг.Ме:прииски, добыча
известняка, песка, мрамора:карьер, добыча
неглубоко залегающего угля:разрез.
Химич.
сырье-это
сырье, которое применяется после химич.
обработки, а также непосредственно (без
обработки): селитра, суперфосфат, апатиты,
самородная S.
Драгоценные и полудрагоцен. камни или
минералы относятся к поделочному
материалу.
Нерудные материалы без обработки, также
могут применяться как поделочный
материал и в качестве украшений. К
горючему
минеральн. сырью относят
сырье, которое применяется в качестве
топлива, но может применяться и как
химическое сырье (нефть, уголь,
газ).Растительное
и животное сырье:1.пищевое:
картофель, зерно, молоко, рыба,
мясо.2.техническое:
древесина, древесная смола, опилки,
солома, лен, шерсть,, кость некоторые
пищев. продукты, применяемые в технических
целях: картофель, растительные масла.
Качество
сырья и методы его обработки.Сортировка-это
разделение сырья на определенные классы;
Брикетирование-укрупнение:
угольную пыль при высоком давлении от
100-200 до 1000 атм подвергают сжатию; получают
угольный брикет, иногда при параллельном
нагревании;Агломерация-процесс
укрупнения частиц, сопровождающийся
термической обработкой, т е. спеканием;
Обогащение
и обезвоживание. Обогащение-это
механич и физико-химич. обработка сырья
с целью получения сырья, обогащенного
ценным компонентом. Обезвоживание-операции
по удалению излишней влаги из материала.
Способы
сортировки сырья:
1Грохочение-процесс
разделения материалов на классы
крупности, осуществляемый на просеивающих
поверхностях;
Гидравлический способ-основан
на различной скорости оседания частиц
в жидкости; Измельчение с помощью
дробилок,
которые бывают молотковые, валковые,
шаровые, вибромельница2.
Способы
обогащения сырья.
Механический-основан
на различных физических свойствах
материалов (гравитационный и воздушный,
грохочение);Физико-химич.:
флотационное обогащение, амальгамирование,
цианизация. Флотация
основана
на различной смачиваемости веществ.
Флотационное
обогащение представл.
собой физико-химич. процесс разделения
компонентов сырья (полезного сырья и
пустой породы), основанный на различной
смачиваемости водой или другими
растворителями. Смачиваемость
минералов характеризуется величиной
краевого угла смачиваемости (Ө), вдоль
границы раздела твердого
вещества-жидкость-воздух.
По
величине Ө минералы подразделяют:
несмачиваемые водой (гидрофобные) Ө>90о;
смачивающиеся водой (гидрофильные)
Ө<90о.Эффективность
флотации определяется рядом
факторов:1)Природа
и состав сырья, в том числе разница в
смачиваемости полезного компонента и
пустой породы; 2)
Дисперсность
сырья 3)Концентрация
пульпы, 4)
Состав
воды 5)
Ассортимент
флотоагентов-в-ва, которые увеличивают
эффективн. флотационного процесса.
Так
как минералы, входящие в состав сырья
обычно не значительно различаются по
смачиваемости, то для увеличения
гидрофобности полезного компонента в
пульпу вводят коллекторы
(собиратели):
ПАВ, адсорбирующиеся на поверхности
частиц и покрывающие ее иономолекулярной
гидрофобной пленкой. Для создания
устойчивой пены в пульпу добавляют
пенообразователи-это
ПАВ, которые образуют на пузырьках
воздуха пены адсорбирующую пленку,
которая препятствует их смачиванию
(терпеновые спирты, сосновая или еловая
живица, сосновое масло, каменноугольная
смола, алифатические спирты, кризолы
или алкиларилсульфанаты). При флотационном
обогащении полиметаллических руд в
пульпу вводят регуляторы.
Регуляторы увеличивают гидрофобное
действие собирателей. В этом случае их
называют активаторами;
Способствуя увеличению гидрофильности
и предотвращая адсорбцию собирателя
на поверхности минерала, они называются
подавителями.Сырьевая
база хим. пр-в.1.Атмосферный
азот и
в очень ограниченной степени натриевая
селитра, запасы которой (Чили, Южная
Африка) быстро истощаются; 2.Водород.
В промышленности производится:-конверсией
метана СН4+2Н2О(пар)→СО2+4Н2;-неполным
окислением метана, который является
комбинацией следующих реакций:
4СН4+2О2→4СО2+8 Н2, 2СН4+О2→2СО2+4Н2 с последующим
взаимодействием СО с водяным паром:
CO+H2O→CO2+H2;-конверсией
твёрдого углеродного топлива:3С+О2+4Н2О→
3СО2+4Н2;- электролиз воды или водных
растворов NaCl. 3.Кислород
или воздух.
Если необходимо иметь чистый кислород,
то его сжижают при высоких давлениях и
пониженной температуре, а затем подвергают
фракционной перегонке; 4.Источником
получения серной кислоты и других
продуктов на её основе является
элементарная
сера, пирит FeS2 и сульфиды цветных
металлов;
5.Источником
получения H3PO4
и фосфат-содержащих удобрений являются
фосфатные
руды: апатиты и фосфориты.
В этих рудах фосфор находится в
нерастворимой форме, главным образом
в виде фторапатита Ca5F(PO4)3 и трикальцийфосфата
Ca3(PO4)2; Фосфориты-породы
осадочного происхождения. Содержание
Р2О5 в фосфоритах колеблется от 20 до 30%.
6.
Первичным сырьём для производства
органических веществ являются природный
газ, нефть, каменный уголь, в меньшей
степени горючие сланцы и торф.
Традиционные способы их первичной
переработки – пиролиз (нагрев без
доступа воздуха). Синтез-газ
(СО
и 3Н2), получаемый из всех перечисленных
видов сырья путём парокислородной
конверсии. 7.
Источником получения металлов в
технически чистом виде являются природные
минералы, содержащие часть пустой
породы.
Минералы руд представляют оксиды и
сульфиды некоторых металлов (Fe3O4, Fe2O3,
Cu2S, CuS, FeCuS2, ZnS и др.), содержащие оксиды
соединений, составляющих пустую породу.
В чёрной металлургии к ним относятся
Al2O3, SiO2, CrO, MgO и т.п.Энергетическая
база хим. пр-в.
Хим. промышленность и смежные с ней
отрасли основанные на химических
превращениях явл. крупнейшими потребителями
энергии. Хим. промышленность и
нефтеперерабатывающие производства
потребляют около 20% от энергопотребления
всей промышленности. Хим.
процессы:
Эндотермические
(Проведение
эндотермических процессов требует
дополнительного подвода тепла из вне
и характеризуется гораздо большим
энергопотреблением); Экзотермические
(Тепло ре-ции может быть использовано
для поддержания необходимого температурного
режима). В химической технологии
используются почти все виды энергии:
электрическая, тепловая, ядерная, хим.,
световая и др. Наиболее широко исп.
тепловая и электрическая
энергия.
Технико-экономические
показатели хим.технолог.Реагент-это
полезный компонент сырья, который
благодаря химическому процессу
превращается в целевой продукт.
Технологический расчет сначала ведут
по реагенту, а затем вычисляют загрузку
сырья. Для
оценки качества ХТП используют
количественные технологические
показатели: Степень
превращения сырья, Селективность
образования и выход продукта,
Производительность реактора или
установки, Интенсивность работы реактора
или катализатора, Расходные коэф. по
реагентам, др ве-вам и энергии. Практическая
целесообразность и рентабельность
всякого процесса производства
характеризуется технико-экономическими
показателями:G =G1+G2+G3 ,G-сырье, G1-целевой
продукт, G2-побочные продукты, G3-отходы.
aA+bB=pP -основная реакция ХТП. Степень
превращения (степень конверсии) реагента
(Х)-это
отношение количества превращенного
реагента к введенному в реакционную
систему количеству этого реагента.
Количества реагента могут быть выражены
в единицах массы, молях, в мольных потоках
и, даже, в единицах объема, взятых при
одинаковых условиях.X(A0)=(G(A0)-G(A))/G(A0);
X(A0)=(N(A0)-N(A))/
N(A0),
G(A)
и G(A0)-массы
введенного и непрореагировавшего
реагента, N(A0)
и N(A)-те
же величины, выраженные в молях.
Расходные
коэф. по сырью.
К основным показателям ХТП относятся
расходные коэффициенты,
характеризующие затраты сырья, воды,
топлива, электроэнергии, пара на единицу
массы целевого продукта. Теоретический
расходный коэф.
 стех.=GA0/Gpстех=(MA*a)/(Mp*p),
G(A0)
и G(р,стехиом.)-массы
реагента и продукта из уравнения реакции.
M(A) и M(P)-молекулярные массы реагента и
продукта, а и р-стехиометрические
коэффициенты. Теоретич. расходный
коэффициент характеризует минимальный
расход сырья на единицу массы продукта.
Практический
расходный коэф.
=GA0,
отражает
реальный расход поступившего в процесс
сырья на единицу массы продукта, т.е.
его рассчитывают как отношение массы
поступившего в процесс сырья G(A0)
к массе получившегося продукта G(Р).
Практический расходный коэффициент по
реагенту можно найти, зная теоретический
расходный коэффициент и выход целевого
продукта по этому реагенту:
стех.=GA0/Gpстех=(MA*a)/(Mp*p),
G(A0)
и G(р,стехиом.)-массы
реагента и продукта из уравнения реакции.
M(A) и M(P)-молекулярные массы реагента и
продукта, а и р-стехиометрические
коэффициенты. Теоретич. расходный
коэффициент характеризует минимальный
расход сырья на единицу массы продукта.
Практический
расходный коэф.
=GA0,
отражает
реальный расход поступившего в процесс
сырья на единицу массы продукта, т.е.
его рассчитывают как отношение массы
поступившего в процесс сырья G(A0)
к массе получившегося продукта G(Р).
Практический расходный коэффициент по
реагенту можно найти, зная теоретический
расходный коэффициент и выход целевого
продукта по этому реагенту: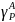 =
стех
рА/
=
стех
рА/ РА,
где
РА
-выход Р по реагенту А в долях единицы.Выход
продукта-отношение
количества практически получаемого
продукта к теоретически возможному
(выход по сырью): β=(G(пр))/(G(m))*100%,
Gт
– определяют из равновесного превращения
исходного вещества или по полному
превращению исходного в продукт
(исключается образование побочных
продуктов). Для
простых реакций выход по сырью
прямопропорционален общей концентрации.
Для
основной реакции химико-технологического
процесса выходы продукта Р на реагенты
А и В выражаются формулами:
РА,
где
РА
-выход Р по реагенту А в долях единицы.Выход
продукта-отношение
количества практически получаемого
продукта к теоретически возможному
(выход по сырью): β=(G(пр))/(G(m))*100%,
Gт
– определяют из равновесного превращения
исходного вещества или по полному
превращению исходного в продукт
(исключается образование побочных
продуктов). Для
простых реакций выход по сырью
прямопропорционален общей концентрации.
Для
основной реакции химико-технологического
процесса выходы продукта Р на реагенты
А и В выражаются формулами:
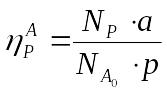
 где N(p),
N(A0), N(B0) количества
молей продукта Р, полученное в ре-ции,
и реагентов А и В, введенные в реакционную
систему, соответственно, а, b и
p-стехиометрические коэффициенты
ре-ции.
Селективность-это
отношение количества полученного
практически продукта, к количеству
этого продукта, которое должно было
быть получено из прореагировавшего
количества реагента в соответствии со
стехиометрией реакции, по которой
образуется этот продукт. Если продукт
образуется по нескольким реакциям, то
селективность, как правило, рассчитать
нельзя. Интегральная
селективность
где N(p),
N(A0), N(B0) количества
молей продукта Р, полученное в ре-ции,
и реагентов А и В, введенные в реакционную
систему, соответственно, а, b и
p-стехиометрические коэффициенты
ре-ции.
Селективность-это
отношение количества полученного
практически продукта, к количеству
этого продукта, которое должно было
быть получено из прореагировавшего
количества реагента в соответствии со
стехиометрией реакции, по которой
образуется этот продукт. Если продукт
образуется по нескольким реакциям, то
селективность, как правило, рассчитать
нельзя. Интегральная
селективность![]()
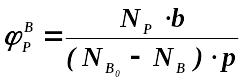 Дифференциальая
селективность-это
отношение скорости расходования реагента
А на образование продукта Р к суммарной
скорости превращения А.
Дифференциальая
селективность-это
отношение скорости расходования реагента
А на образование продукта Р к суммарной
скорости превращения А. ,
,![]() -
скорость расходования А на образование
Р. Количество
готовой продукции-должно
удовлетворять требованиям ГОСТ и
характеризуется содержанием основного
вещества и посторонних примесей и
зависит в первую очередь от характера
побочных реакций и степени очистки
исходных и конечных продуктов.
Производительность
аппарата П-количество
готового продукта, фактически
вырабатываемого данным аппаратом в
единицу времени при данном расчете
процесса производства: П=G/t,
G-масса
(или объем) продукта, полученная за время
t. Мощность-максимальная
производительность аппаратуры: W=Пmax.
Интенсивность
работы процесса или аппарата-производительность
отнесенная к единице полезного объема
или рабочей поверхности аппарата. Обычно
производительность относят к объему
аппарата V или к площади его сечения S:
I=П/V
(кг/м3*ч), I=П/S
(кг/м2*ч).
В каталитич. процессах рассчитывают
интенсивность работы катализатора, для
чего массу полученного за единицу
времени целевого продукта относят к
объему катализатора Vk:
I=П/Vk
(кг/м3к*ч).Производительность
труда-количество
вырабатываемой продукции в единицу
времени на одного рабочего. Себестоимость
продукции-это
сумма затрат в денежном выражении на
единицу продуктов в конкретном
производстве. (заготовка и хранение
сырья и топлива, оплата труда работников,
оплата энергии, амортизация оборудования
и помещений). Себестоимость тем ниже,
чем выше производительность.
Балансы
производства.
1.Материальный баланс. Материальный
баланс ХТП явл. следствием закона
сохранения массы вещества. Это означает,
что масса веществ, поступивших на
технологическую операцию - приход, равна
массе полученных веществ-расходу.
Материальный баланс должен соблюдаться
для ХТП всех типов, синтеза, разделения,
очистки целевых продуктов. Данные о
материальном балансе позволяют дать
оценку целесообразности осуществления
процесса в заданных условиях. По данным
материального баланса рассчитывают
технологические показатели процесса:
выход целевого продукта, степень
превращения сырья, селективность
процесса, расходные коэффициенты по
сырью и т.д. Материальный
баланс является необходимым элементом
при расчете энергетического и
эксергетического балансов.
три основные формы составления
материального баланса: Мистема уравнений,
Таблица, Поточная диаграмма. Уравнение
материального баланса в общем виде:
G(AO)+G(BO)+ ...=G(R)+...+G(D)+...+
G(A)
+G(B)+∆G;
G(AO),
G(BO)-массы
исходных реагентов (сырья). G(R)-масса
целевого продукта, G(D)-масса побочного
продукта, G(А), G(В)-массы не прореагировавших
веществ, ∆G-невязка баланса. Невязка
баланса является следствием потерь
сырья и продуктов при проведении
процесса, неточности эксперимента и
расчета, а также принятых допущений.
Поточная
диаграмма материального баланса.Поточная
диаграмма это блок-схема. Материальн.
потоки изображают в виде полос, ширина
которых пропорциональна массе в выбранном
масштабе. Основным преимуществом данного
способа изображения является его
наглядность. Поточная
диаграмма колонны синтеза NH3:31-свежий
газ; 2-циркулирующий газ; 3-жидкий NH3
и растворенные в нем газы; 4-отдувочные
газы.2.Энергетический
(тепловой баланс). Составляется
на основе закона сохранения энергии.
Наиболее распространенные статьи:
1.Тепловой эффект химической реакции:
экзотермических веществ-приход;
эндотермических веществ-расход; 2.
Физическое тепло (теплосодержание)
исходных веществ-приход или конечных-расход
продуктов: Q=
c*m*∆t,
с-теплоемкость вещества. 3.Теплота,
вносимая из вне если нагреваем-приход;
охлаждаем-расход;4.Потери в окружающую
среду: Qприхода=
Qрасхода.3.Экономический
баланс.
Выражен в денежном эквиваленте,cоставляется
на основе материального и энергетических
балансов; бух. Отчетах о стоимости сырья,
заработной платы. Рассчитывают
себестоимость и сравнивают рентабельность
производств. Технологические
параметры хим.технолог. процессов.1.
Время
пребывания исходных в-в в реакционной
зоне.
В аппаратах периодического
действия
время пребывания реагентов - это интервал
времени между загрузкой и выгрузкой
аппарата. В аппаратах непрерывного
действия время пребывания исходных
веществ в реакционной зоне определяют
следующим образом: выход целевого
продукта и интенсивность работы аппарата
влияют не только температура.
-
скорость расходования А на образование
Р. Количество
готовой продукции-должно
удовлетворять требованиям ГОСТ и
характеризуется содержанием основного
вещества и посторонних примесей и
зависит в первую очередь от характера
побочных реакций и степени очистки
исходных и конечных продуктов.
Производительность
аппарата П-количество
готового продукта, фактически
вырабатываемого данным аппаратом в
единицу времени при данном расчете
процесса производства: П=G/t,
G-масса
(или объем) продукта, полученная за время
t. Мощность-максимальная
производительность аппаратуры: W=Пmax.
Интенсивность
работы процесса или аппарата-производительность
отнесенная к единице полезного объема
или рабочей поверхности аппарата. Обычно
производительность относят к объему
аппарата V или к площади его сечения S:
I=П/V
(кг/м3*ч), I=П/S
(кг/м2*ч).
В каталитич. процессах рассчитывают
интенсивность работы катализатора, для
чего массу полученного за единицу
времени целевого продукта относят к
объему катализатора Vk:
I=П/Vk
(кг/м3к*ч).Производительность
труда-количество
вырабатываемой продукции в единицу
времени на одного рабочего. Себестоимость
продукции-это
сумма затрат в денежном выражении на
единицу продуктов в конкретном
производстве. (заготовка и хранение
сырья и топлива, оплата труда работников,
оплата энергии, амортизация оборудования
и помещений). Себестоимость тем ниже,
чем выше производительность.
Балансы
производства.
1.Материальный баланс. Материальный
баланс ХТП явл. следствием закона
сохранения массы вещества. Это означает,
что масса веществ, поступивших на
технологическую операцию - приход, равна
массе полученных веществ-расходу.
Материальный баланс должен соблюдаться
для ХТП всех типов, синтеза, разделения,
очистки целевых продуктов. Данные о
материальном балансе позволяют дать
оценку целесообразности осуществления
процесса в заданных условиях. По данным
материального баланса рассчитывают
технологические показатели процесса:
выход целевого продукта, степень
превращения сырья, селективность
процесса, расходные коэффициенты по
сырью и т.д. Материальный
баланс является необходимым элементом
при расчете энергетического и
эксергетического балансов.
три основные формы составления
материального баланса: Мистема уравнений,
Таблица, Поточная диаграмма. Уравнение
материального баланса в общем виде:
G(AO)+G(BO)+ ...=G(R)+...+G(D)+...+
G(A)
+G(B)+∆G;
G(AO),
G(BO)-массы
исходных реагентов (сырья). G(R)-масса
целевого продукта, G(D)-масса побочного
продукта, G(А), G(В)-массы не прореагировавших
веществ, ∆G-невязка баланса. Невязка
баланса является следствием потерь
сырья и продуктов при проведении
процесса, неточности эксперимента и
расчета, а также принятых допущений.
Поточная
диаграмма материального баланса.Поточная
диаграмма это блок-схема. Материальн.
потоки изображают в виде полос, ширина
которых пропорциональна массе в выбранном
масштабе. Основным преимуществом данного
способа изображения является его
наглядность. Поточная
диаграмма колонны синтеза NH3:31-свежий
газ; 2-циркулирующий газ; 3-жидкий NH3
и растворенные в нем газы; 4-отдувочные
газы.2.Энергетический
(тепловой баланс). Составляется
на основе закона сохранения энергии.
Наиболее распространенные статьи:
1.Тепловой эффект химической реакции:
экзотермических веществ-приход;
эндотермических веществ-расход; 2.
Физическое тепло (теплосодержание)
исходных веществ-приход или конечных-расход
продуктов: Q=
c*m*∆t,
с-теплоемкость вещества. 3.Теплота,
вносимая из вне если нагреваем-приход;
охлаждаем-расход;4.Потери в окружающую
среду: Qприхода=
Qрасхода.3.Экономический
баланс.
Выражен в денежном эквиваленте,cоставляется
на основе материального и энергетических
балансов; бух. Отчетах о стоимости сырья,
заработной платы. Рассчитывают
себестоимость и сравнивают рентабельность
производств. Технологические
параметры хим.технолог. процессов.1.
Время
пребывания исходных в-в в реакционной
зоне.
В аппаратах периодического
действия
время пребывания реагентов - это интервал
времени между загрузкой и выгрузкой
аппарата. В аппаратах непрерывного
действия время пребывания исходных
веществ в реакционной зоне определяют
следующим образом: выход целевого
продукта и интенсивность работы аппарата
влияют не только температура.
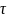 =V/Vt,p,
V-реакционный
объем, м3; V(t,p)-расход
исходных в-в, поступающих в реакционный
аппарат при температуре и давлении в
аппарате, м3/с. Для каталитических
процессов
рассчитывают время соприкосновения
исходных веществ с катализатором. Его
называют временем контактирования.
Время контактирования находят из
отношения свободного объема катализатора
V(св) к расходу исходных веществ V(t,p),
проходящих через катализатор при
условиях процесса:
=Vсв/Vt,p,
Свободный
объем катализатора
- это объем пустот между зернами и в
сетках катализатора. Свободный объем
V(св) рассчитывают как произведение
объема катализатора V(k) на долю свободного
объема ℰ:
V(св)=V(k)ℰ.
Если катализатор выполняется в виде
сетки, его свободный объем рассчитывают
по формуле: Vсв=3
=V/Vt,p,
V-реакционный
объем, м3; V(t,p)-расход
исходных в-в, поступающих в реакционный
аппарат при температуре и давлении в
аппарате, м3/с. Для каталитических
процессов
рассчитывают время соприкосновения
исходных веществ с катализатором. Его
называют временем контактирования.
Время контактирования находят из
отношения свободного объема катализатора
V(св) к расходу исходных веществ V(t,p),
проходящих через катализатор при
условиях процесса:
=Vсв/Vt,p,
Свободный
объем катализатора
- это объем пустот между зернами и в
сетках катализатора. Свободный объем
V(св) рассчитывают как произведение
объема катализатора V(k) на долю свободного
объема ℰ:
V(св)=V(k)ℰ.
Если катализатор выполняется в виде
сетки, его свободный объем рассчитывают
по формуле: Vсв=3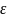 mSd,
ℰ-доля
свободного объема катализатора, m - число
сеток, S - рабочая площадь сетки, м2,
d-диаметр нити сетки, м, 3 - числовой
коэффициент, учитывающий увеличение
толщины сетки при переплетении нитей:
ℰ=1-1,57*d*
mSd,
ℰ-доля
свободного объема катализатора, m - число
сеток, S - рабочая площадь сетки, м2,
d-диаметр нити сетки, м, 3 - числовой
коэффициент, учитывающий увеличение
толщины сетки при переплетении нитей:
ℰ=1-1,57*d* ,
d-диаметр нити сетки, см; n-число плетений
сетки на 1 см2. Долю свободного объема
катализатора называют также порозностью.
При расчете времени контактирования
во взвешенном слое катализатора свободный
объем находят как разность между объемом,
занимаемым катализатором во взвешенном
состоянии, V(взв), и объемом тв частиц
катализатора Vk(1-ε):Vсв-Vвзв-Vk(1-ε).
2.
Объемная скорость
W-это
объем исходных веществ, поступающих в
реакционный аппарат в единицу времени
в фазовом состоянии, соответствующем
условиям проведения процесса (для
газофазных процессов – в газообразном
состоянии), отнесенный к единице
реакционного объема. Wt,p=Vt,p/V
(м3/м3∙ч=ч(-1)) или Wну=Vну/V,
V-объем
реактора. Объемная скорость и время
пребывания исходных веществ в реакционной
зоне связаны соотношением: Wt,p=1/
Аналогично рассчитывают условную
объемную скорость:Wну=Vну/Vk.
В каталитических процессах вместо
реакционного объема в расчете объемной
скорости используют объем катализатора
Vk: Vt,p=Vt,p/Vk
или с учетом Wt,p=(Vt,p*ε)/
(
*Vt,p)=ε/
,
ℰ-доля
свободного объема катализатора;
-время
контактирования.
Химические
реакторы-основные элементы ХТС.Признаки
классификации химических реакторов и
режимов их работы:
Режим
движения реакционной среды; Условия
теплообмена в реакторе; Фазовый состав
реакционной смеси; Способ организации
процесса; Характер изменения параметров
процесса во времени; Конструктивные
характеристики.1.Классификация
химических реакторов по гидродинамической
обстановке.Реакторы
смешения-это
емкостные аппараты с перемешиванием
механической мешалкой или циркуляционным
насосом.
Реакторы вытеснения-трубчатые
аппараты, достаточно большой длины по
сравнению с диаметром. В таких аппаратах
течение реакционного потока имеет
поршнеобразный характер. В теории
реакторов обычно рассматривают идеальные
варианты этих аппаратов – реактор
идеального или полного смешения и
реактор идеального или полного вытеснения.
По длине реактора в соответствии с
закономерностями протекания реакции
устанавливается определённое распределение
концентраций участников реакции, t и
других параметров.2.
Классификация химических реакторов по
условиям теплообмена.Адиабатические
реакторы-при
отсутствии теплообмена между реактором
и окружающей средой химический реактор
является адиабатическим. Вся теплота,
выделяющаяся или поглощающаяся в
результате химических реакций, расходуется
на внутренний теплообмен, т.е. на нагрев
или охлаждение реакционной
смеси.Изотермические
реакторы-если
теплообмен с окружающей средой протекает
гораздо быстрее, чем тепловыделение
или теплопоглощение, то во всех точках
реакционной зоны обеспечивается
постоянство температуры и такой реактор
называется изотермическим.Политермические
реакторы-реакторы,
в которых скорости тепловыделения или
теплопоглощения соизмеримы со скоростями
теплообмена с окружающей средой,
температурный режим представляет собой
результат баланса между этими процессами
и в общем случае это обусловливает
неравномерность распределения температуры
в реакционной зоне. Автотермические
реакторы-в
них поддержание необходимой температуры
процесса осуществляется исключительно
за счёт теплоты химического процесса
без использования внешних источников
энергии. В практике химической технологии
стремятся к тому, чтобы химические
реакторы, особенно в крупнотоннажных
производствах, были автотермическими.3.
Классификация
химических реакторов по фазовому составу
реакционной массы.а)Реакторы
для проведения гомогенных процессов
подразделяют на аппараты для газофазных
и жидкофазных реакций.б) Аппараты для
проведения реакций с двухфазными
системами подразделяют на газо-жидкостные,
реакции для систем газ – твёрдое тело,
жидкость твёрдое тело.в) Особо выделяют
реакторы для гетерогенно-каталитических
процессов.4.
Классификация
химических реакторов по способу
организации процесса.
Периодические-В
реакторе периодического действия все
реагенты вводят в реактор до начала
реакции, смесь выдерживают в реакторе
необходимое время, после чего производится
выгрузка продуктов. Продолжительность
операции от момента загрузки до момента
выгрузки соответствует времени реакции.
Недостатки периодических реакторов-цикличность
работы, низкая производительность,
большие затраты ручного труда. Такие
реакторы выгодны при организации
малотоннажных производств.
Непрерывныe-в
нем производится непрерывная подача
реагентов в реакционную зону и непрерывный
отвод продуктов. Эти реакторы обеспечивают
высокую производительность и их
применение особенно выгодно при
организации крупнотоннажных производств.
Полунепрерывные-характеризуется
тем, что один из реагентов поступает в
него непрерывно, а другой – периодически.
Возможны варианты, когда реагенты
поступают в реактор периодически, а
продукты реакции выводятся непрерывно,
или наоборот.5.Классификация
хим. реакторов по характеру изменения
параметров процесса во времени. Реакторы,
работающие в стационарном режиме. Режим
работы реактора называют стационарным,
если протекание химической реакции в
произвольно выбранной точке реактора
характеризуется постоянством концентраций
реагентов и продуктов, скорости и других
показателей во времени. В стационарном
режиме показатели потока на выходе из
реактора не зависят от времени. Это
постоянство показателей определяется
двумя факторами: стационарностью режима
и постоянством состава параметров
потока на входе в реактор.
Стационарный
режим обычно выдерживается в непрерывно
действующих проточных реакторах.
Стационарные процессы легче
автоматизировать. Реакторы,
работающие в нестационарном режиме.
Если
в произвольно выбранной точке происходит
изменение параметров химического
процесса во времени, режим работы
реактора называется нестационарным.
Нестационарными являются все периодические
процессы. 6.
Классификация химических реакторов по
конструктивным характеристикам.Реакторы
для гомогенных процессов, гетерогенных,
гетерофазных.
Промышленные химические реакторы.
Реакторы для гомогенных процессов.Емкостные
реакторы.4Емкостные
аппараты-периодические и проточные-снабжены
мешалками, конструкции которых
разнообразны(а,б,в). Особый тип мешалок-для
вязких жидкостей (г,д.). Реакторы а,б,г)
используются в периодических процессах,
реакторы в),д)-в непрерывных. В то же
время реактор а) может быть приспособлен
для непрерывного режима, однако в этом
случае необходимо изменить порядок
подачи реагентов и отвода продуктов
реакции: снизу-реагенты, сверху-продукты.
В последнем случае необходимо организовать
отвод через боковой штуцер.Трубчатые
реакторы.5Типичным
примером трубчатого реактора является
реактор типа “труба в трубе” (з.). В
таком реакторе теплоноситель циркулирует
через рубашку реактора. В случае
необходимости подвода или отвода тепла
предусматриваются теплообменные
устройства в реакторах. В качестве
таковых используются рубашки или
теплообменники внутри реактора в виде
змеевиков или секций трубок.В
высокотемпературных процессах (например)
в термическом крекинге УВ проще помещать
секцию трубок в камеру сгорания, в
которой аккумулируется тепло, необходимое
для обеспечения необходимой температуры
(и,к). Такие реакторы называются трубчатыми
печами.Реакторы
для гетерогенных процессов с твердой
фазой.
6Простейшим
вариант-цилиндрический реактор,
заполненный твердой фазой, через которую
циркулирует газообразный реагент (а).
В таком реакторе проводят процессы
адсорбционной очистки газов и жидкостей,
Недостаток-цикличность. Для организации
непрерывного процесса обновления
твердой фазы предлагается процесс,
осуществляемый в многоколоночном
реакторе, снабженном скребком на каждой
полке (б).Так устроен реактор обжига
серного колчедана.
Удобен и распространен
процесс непрерывного движения твердого
материала на вращающейся наклонной
трубе (в). Такого же типа реактор аммиачный
нейтрализатор в
производстве двойного суперфосфата.
7Химич.
процессы "газ-твердое тело" протекают
значительно интенсивнее при ↑ степени
дробления твердого реагента. В реакторах
с неподвижным слоем твердого реагента
это сделать практически невозможно, тк
с увеличением степени дисперсности
резко возрастает гидравлическое
сопротивление слоя, возрастает вероятность
слипания и комкования мелких частиц-в
псевдоожиженном (г) или фонтанирующем
(д) слоях, с распылительным инжектированием
твердого материала через специальную
форсунку (е) и в режиме пневмотранспорта
(ж).Недостатки этих способов-эрозия
стенок аппарата, дробление частиц, унос
пыли и загрязнение его газового
потока.Если используются системы
ж-твердое тело, то организовать процесс
суспензирования гораздо легче из-за
близости плотностей компонентов.Реакторы
для гетерогенно-каталитических процессов.
8Наиболее
распространенными явл. реакторы с
неподвижным слоем kat.
Для адиабатического режима kat в виде
частиц различной формы засыпают в
аппарат (а). Слой располагается на жесткой
опорной решетке, выдерживающей массу
kat и перепад давления в слое. Чтобы мелкие
зерна kat не проваливались, на нее обычно
насыпают тонкий слой крупнокускового
материала, а сверху-kat.
Kat
насыпают "в навал", после чего
требуется выравнивание слоя для
обеспечения равномерного прохождения
через него реакционной смеси.Использование
варианта адиабатического реактора
приводит к большим перепадам t
по высоте каталитического слоя и
отклонения средней t от оптим. значения.
Для того, чтобы приблизить t режим к
оптимальному-многотоннажные реакторы,
в которых выравнивание температурн.
режима осуществляют съемом или подводом
тепла с помощью теплообменных устройств
между секциями или путем подачи между
секциями захоложенного или перегретого
реагента (компонента) (б,в).
9Часто
необходим отвод тепла непосредственно
из реакционной зоны. С этой целью
используют реакторы типа кожухотрубных
теплообменников-универсальный
тип каталитич. реактора (г). Обычно
в трубки загружают kat, а в межтрубное
пространство поступает теплоноситель.
Если необходимо снимать тепло, то в
межтрубное пространство снизу поступает
конденсат, а сверху отводится водяной
пар. В другом варианте в межтрубном
пространстве циркулирует
смесь-NaNO3-NaNO2-KNO3. Аккумулированное им
тепло далее используется для генерирования
водяного пара. Для обеспечения теплотой
эндотермических процессов используют
горячие дымовые газы. В последнем случае
реактор представляет собой трубчатую
печь (д). Отводить тепло реакции можно
не только теплоносителем, но и свежей
реакционной смесью.
Другая
группа реакторов-со взвешенным (кипящим)
или восходящим слоем катализатора.10
При подаче реакционной смеси снизу слоя
с достаточной скоростью твердые частицы
будут витать в воздухе, не уносясь с ним
(е); в этом случае применяют частицы не
крупнее 1 мм. Это обеспечивает полное
использование их внутренней поверхности.
Циркулирующие частицы выравнивают t в
слое -процесс в нем протекает практически
изотермически. Система "реактор-регенератор"
обеспечивает непрерывность процесса
в целом. Такой тип реакционного узла
можно организовать в процессе
каталитического крекинга НП, в котором
kat
быстро закоксовывается, теряя свою
активность. При организации псевдоожиженного
слоя часть газа проходит слой в виде
пузырей. Коэффициент массопередачи
между пузырями и остальной частью слоя
невысокий-фактически образование
пузырей газа-это образование байпасных
потоков.
Для очистки газа от пыли после реактора
устанавливают циклоны.
Если
скорость газового потока будет такой,
что твердые частицы будут увлекаться
им, то реализуется режим пневмотранспорта
(з) и реакция в восходящем потоке kat.
Такая организация процесса эффективна
для быстрых реакций-т.к. время прохождения
реакционной смеси в длинном узком
реакторе небольшое. Реакторы
для геторофазных процессов.11В
трубчатом реакторе (а) жидкость стекает
по стенкам трубок и контактирует со
встречным потоком или попутным потоком
газа. Это наиболее организованный
процесс, т.к. здесь строго поддерживается
поверхность контакта фаз, равная
поверхности трубок. Тепловой режим
поддерживается регулированием t трубок.
Такие реакторы используют в малотоннажных
производствах тонкого органич. и
неорганич. синтеза. Эти
реакторы характеризуются устойчивостью
гидродинамического и температурного
режимов, однако у них низкая
производительность из-за низкой
поверхности контакта фаз.В
зависимости от скорости потоков возможны
различные режимы течения: полное
расслоение фаз, раздельное течение с
сильно возмущенной развитой поверхностью
контакта фаз, хорошо перемешанный
газожидкостной поток.
Указанные режимы наблюдаются
последовательно с возрастанием скорости
потоков, особенно газового. Такие
реакторы компактны даже при необходимости
большого времени контакта-их делают в
виде змеевика. У них высокий коэффициент
массопередачи. Такого типа реактор
используется в производстве полиэтилена
и окислении этилена в ацетальдегид.
Более
распространены насадочные реакторы
(в).
12В
такой аппарат засыпается инертная
насадка-сравнительно небольшие элементы,
по поверхности которых стекает жидкость,
а в пространстве между ними, противотоком
движется газ. Взаимодействие фаз
происходит интенсивно. Производительность
таких реакторов ограничивается
недостаточно интенсивным режимом
движения, т.к. жидкость движется не
принудительно, а за счет силы тяжести.
Простейшее взаимодействие жидкости и
газа-барботаж последнего через жидкость
(г, д).(г) для периодического процесса,
(д)-для непрерывного.Многосекционные
реакторы:13Это
могут быть колонны с колпачковыми
тарелками, снабженными переливными
устройствами (е) или ситчатыми провальными
тарелками (ж). В реакторе (з) жидкая фаза
диспергируется в потоке газа. Для
диспергирования жидкости используются
специальные разбрызгиватели (форсунки).
Мелкие капли более устойчивы в размерах,
но скорость их падения определяется
силами гравитации и скоростью встречного
газа. Поэтому степень диспергирования
должна быть оптимальной.Энергия
в химическом производстве. Тепловой
эффект реакции в технологических
расчетах. Направленность реакции в
технологических расчетах.
Изменение
хим. состава реагирующей смеси приводит
к изменению ее теплосодержания ∆
НТ которое
можно рассчитать через энтальпии
образования компонентов (∆
НТ )обр:∆
Н(Т)
=
Σv(i)(∆
Н(T))обр.i
Если энтальпия образования продуктов
меньше, чем энтальпия образования
исходных веществ, (∆
Н(Т)
< 0),
то
выделяется теплота Q(p)=-∆Н(T),
называемая теплотой реакции. Если при
химическом превращении теплосодержание
смеси увеличивается (∆Н(T)
> 0),
то
происходит поглощение теплоты.В
зависимости от знака ∆Н
(или
Q(p))
реакции
бывают экзотермические (∆Н<0,Q(p)>0)
и
эндотермические (∆Н>0,
Qр<0).
Тепловой
эффект реакции входит в запись
термохимического уравнения,
представляющего собой стехиометрическое
уравнение с указанием его теплового
результата: V(A)А
+v(B)В+…=
v(R)R+v(S)S+…+Q(p).
Значение
Q(p)
в уравнении зависит от записи химического
уравнения. Например,
тепловой эффект реакции, записанной
следующим образом: N2+3Н2=2NН3
в два раза больше, нежели для той же
реакции, записанной по-другому:
0,5N2+1,5Н2=NН3,
поэтому в справочной литературе Q(p)
приводят прямо в уравнениях, как это
сделано в, или указывают изменение
энтальпии, соответствующее превращению
1
моля вещества.Тепловой
эффект реакции в технологических
расчетах.
Знание
теплового эффекта реакции необходимо
для определения тепловых явлений в
технологических процессах. Количество
выделившейся (или поглощенной) теплоты
qр
зависит от количества превращенного
вещества
∆N:
q(p)
=Q(p)
∆N(A)/v(a).
В
зависимости от знака Q(p)
(экзо-
или эндотермическая реакция),
теплота в ходе протекания процесса
будет выделяться или поглощаться.Возможность
химического превращения.
Химический
процесс принципиально осуществим, если
реакция протекает с уменьшением
химического потенциала, называемого
также изобарным потенциалом, или энергией
Гиббса, т.е. возможность протекания
реакции определяется из след. условий:1.при
∆G(T,P)>0
протекание
реакции не возможно; 2.при
∆G(T,P)<0
протекание
реакции возможно; 3.
при
∆G(Т,Р)=0
реакционная
система находится в термодинамическом
равновесии, где ∆G(Т,Р)
-
изменение энергии Гиббса при превращении
исходных веществ в продукты при
температуре
Т
и давлении Р.
Изменение энергии Гиббса реакции можно
рассчитать по уравнению:
,
d-диаметр нити сетки, см; n-число плетений
сетки на 1 см2. Долю свободного объема
катализатора называют также порозностью.
При расчете времени контактирования
во взвешенном слое катализатора свободный
объем находят как разность между объемом,
занимаемым катализатором во взвешенном
состоянии, V(взв), и объемом тв частиц
катализатора Vk(1-ε):Vсв-Vвзв-Vk(1-ε).
2.
Объемная скорость
W-это
объем исходных веществ, поступающих в
реакционный аппарат в единицу времени
в фазовом состоянии, соответствующем
условиям проведения процесса (для
газофазных процессов – в газообразном
состоянии), отнесенный к единице
реакционного объема. Wt,p=Vt,p/V
(м3/м3∙ч=ч(-1)) или Wну=Vну/V,
V-объем
реактора. Объемная скорость и время
пребывания исходных веществ в реакционной
зоне связаны соотношением: Wt,p=1/
Аналогично рассчитывают условную
объемную скорость:Wну=Vну/Vk.
В каталитических процессах вместо
реакционного объема в расчете объемной
скорости используют объем катализатора
Vk: Vt,p=Vt,p/Vk
или с учетом Wt,p=(Vt,p*ε)/
(
*Vt,p)=ε/
,
ℰ-доля
свободного объема катализатора;
-время
контактирования.
Химические
реакторы-основные элементы ХТС.Признаки
классификации химических реакторов и
режимов их работы:
Режим
движения реакционной среды; Условия
теплообмена в реакторе; Фазовый состав
реакционной смеси; Способ организации
процесса; Характер изменения параметров
процесса во времени; Конструктивные
характеристики.1.Классификация
химических реакторов по гидродинамической
обстановке.Реакторы
смешения-это
емкостные аппараты с перемешиванием
механической мешалкой или циркуляционным
насосом.
Реакторы вытеснения-трубчатые
аппараты, достаточно большой длины по
сравнению с диаметром. В таких аппаратах
течение реакционного потока имеет
поршнеобразный характер. В теории
реакторов обычно рассматривают идеальные
варианты этих аппаратов – реактор
идеального или полного смешения и
реактор идеального или полного вытеснения.
По длине реактора в соответствии с
закономерностями протекания реакции
устанавливается определённое распределение
концентраций участников реакции, t и
других параметров.2.
Классификация химических реакторов по
условиям теплообмена.Адиабатические
реакторы-при
отсутствии теплообмена между реактором
и окружающей средой химический реактор
является адиабатическим. Вся теплота,
выделяющаяся или поглощающаяся в
результате химических реакций, расходуется
на внутренний теплообмен, т.е. на нагрев
или охлаждение реакционной
смеси.Изотермические
реакторы-если
теплообмен с окружающей средой протекает
гораздо быстрее, чем тепловыделение
или теплопоглощение, то во всех точках
реакционной зоны обеспечивается
постоянство температуры и такой реактор
называется изотермическим.Политермические
реакторы-реакторы,
в которых скорости тепловыделения или
теплопоглощения соизмеримы со скоростями
теплообмена с окружающей средой,
температурный режим представляет собой
результат баланса между этими процессами
и в общем случае это обусловливает
неравномерность распределения температуры
в реакционной зоне. Автотермические
реакторы-в
них поддержание необходимой температуры
процесса осуществляется исключительно
за счёт теплоты химического процесса
без использования внешних источников
энергии. В практике химической технологии
стремятся к тому, чтобы химические
реакторы, особенно в крупнотоннажных
производствах, были автотермическими.3.
Классификация
химических реакторов по фазовому составу
реакционной массы.а)Реакторы
для проведения гомогенных процессов
подразделяют на аппараты для газофазных
и жидкофазных реакций.б) Аппараты для
проведения реакций с двухфазными
системами подразделяют на газо-жидкостные,
реакции для систем газ – твёрдое тело,
жидкость твёрдое тело.в) Особо выделяют
реакторы для гетерогенно-каталитических
процессов.4.
Классификация
химических реакторов по способу
организации процесса.
Периодические-В
реакторе периодического действия все
реагенты вводят в реактор до начала
реакции, смесь выдерживают в реакторе
необходимое время, после чего производится
выгрузка продуктов. Продолжительность
операции от момента загрузки до момента
выгрузки соответствует времени реакции.
Недостатки периодических реакторов-цикличность
работы, низкая производительность,
большие затраты ручного труда. Такие
реакторы выгодны при организации
малотоннажных производств.
Непрерывныe-в
нем производится непрерывная подача
реагентов в реакционную зону и непрерывный
отвод продуктов. Эти реакторы обеспечивают
высокую производительность и их
применение особенно выгодно при
организации крупнотоннажных производств.
Полунепрерывные-характеризуется
тем, что один из реагентов поступает в
него непрерывно, а другой – периодически.
Возможны варианты, когда реагенты
поступают в реактор периодически, а
продукты реакции выводятся непрерывно,
или наоборот.5.Классификация
хим. реакторов по характеру изменения
параметров процесса во времени. Реакторы,
работающие в стационарном режиме. Режим
работы реактора называют стационарным,
если протекание химической реакции в
произвольно выбранной точке реактора
характеризуется постоянством концентраций
реагентов и продуктов, скорости и других
показателей во времени. В стационарном
режиме показатели потока на выходе из
реактора не зависят от времени. Это
постоянство показателей определяется
двумя факторами: стационарностью режима
и постоянством состава параметров
потока на входе в реактор.
Стационарный
режим обычно выдерживается в непрерывно
действующих проточных реакторах.
Стационарные процессы легче
автоматизировать. Реакторы,
работающие в нестационарном режиме.
Если
в произвольно выбранной точке происходит
изменение параметров химического
процесса во времени, режим работы
реактора называется нестационарным.
Нестационарными являются все периодические
процессы. 6.
Классификация химических реакторов по
конструктивным характеристикам.Реакторы
для гомогенных процессов, гетерогенных,
гетерофазных.
Промышленные химические реакторы.
Реакторы для гомогенных процессов.Емкостные
реакторы.4Емкостные
аппараты-периодические и проточные-снабжены
мешалками, конструкции которых
разнообразны(а,б,в). Особый тип мешалок-для
вязких жидкостей (г,д.). Реакторы а,б,г)
используются в периодических процессах,
реакторы в),д)-в непрерывных. В то же
время реактор а) может быть приспособлен
для непрерывного режима, однако в этом
случае необходимо изменить порядок
подачи реагентов и отвода продуктов
реакции: снизу-реагенты, сверху-продукты.
В последнем случае необходимо организовать
отвод через боковой штуцер.Трубчатые
реакторы.5Типичным
примером трубчатого реактора является
реактор типа “труба в трубе” (з.). В
таком реакторе теплоноситель циркулирует
через рубашку реактора. В случае
необходимости подвода или отвода тепла
предусматриваются теплообменные
устройства в реакторах. В качестве
таковых используются рубашки или
теплообменники внутри реактора в виде
змеевиков или секций трубок.В
высокотемпературных процессах (например)
в термическом крекинге УВ проще помещать
секцию трубок в камеру сгорания, в
которой аккумулируется тепло, необходимое
для обеспечения необходимой температуры
(и,к). Такие реакторы называются трубчатыми
печами.Реакторы
для гетерогенных процессов с твердой
фазой.
6Простейшим
вариант-цилиндрический реактор,
заполненный твердой фазой, через которую
циркулирует газообразный реагент (а).
В таком реакторе проводят процессы
адсорбционной очистки газов и жидкостей,
Недостаток-цикличность. Для организации
непрерывного процесса обновления
твердой фазы предлагается процесс,
осуществляемый в многоколоночном
реакторе, снабженном скребком на каждой
полке (б).Так устроен реактор обжига
серного колчедана.
Удобен и распространен
процесс непрерывного движения твердого
материала на вращающейся наклонной
трубе (в). Такого же типа реактор аммиачный
нейтрализатор в
производстве двойного суперфосфата.
7Химич.
процессы "газ-твердое тело" протекают
значительно интенсивнее при ↑ степени
дробления твердого реагента. В реакторах
с неподвижным слоем твердого реагента
это сделать практически невозможно, тк
с увеличением степени дисперсности
резко возрастает гидравлическое
сопротивление слоя, возрастает вероятность
слипания и комкования мелких частиц-в
псевдоожиженном (г) или фонтанирующем
(д) слоях, с распылительным инжектированием
твердого материала через специальную
форсунку (е) и в режиме пневмотранспорта
(ж).Недостатки этих способов-эрозия
стенок аппарата, дробление частиц, унос
пыли и загрязнение его газового
потока.Если используются системы
ж-твердое тело, то организовать процесс
суспензирования гораздо легче из-за
близости плотностей компонентов.Реакторы
для гетерогенно-каталитических процессов.
8Наиболее
распространенными явл. реакторы с
неподвижным слоем kat.
Для адиабатического режима kat в виде
частиц различной формы засыпают в
аппарат (а). Слой располагается на жесткой
опорной решетке, выдерживающей массу
kat и перепад давления в слое. Чтобы мелкие
зерна kat не проваливались, на нее обычно
насыпают тонкий слой крупнокускового
материала, а сверху-kat.
Kat
насыпают "в навал", после чего
требуется выравнивание слоя для
обеспечения равномерного прохождения
через него реакционной смеси.Использование
варианта адиабатического реактора
приводит к большим перепадам t
по высоте каталитического слоя и
отклонения средней t от оптим. значения.
Для того, чтобы приблизить t режим к
оптимальному-многотоннажные реакторы,
в которых выравнивание температурн.
режима осуществляют съемом или подводом
тепла с помощью теплообменных устройств
между секциями или путем подачи между
секциями захоложенного или перегретого
реагента (компонента) (б,в).
9Часто
необходим отвод тепла непосредственно
из реакционной зоны. С этой целью
используют реакторы типа кожухотрубных
теплообменников-универсальный
тип каталитич. реактора (г). Обычно
в трубки загружают kat, а в межтрубное
пространство поступает теплоноситель.
Если необходимо снимать тепло, то в
межтрубное пространство снизу поступает
конденсат, а сверху отводится водяной
пар. В другом варианте в межтрубном
пространстве циркулирует
смесь-NaNO3-NaNO2-KNO3. Аккумулированное им
тепло далее используется для генерирования
водяного пара. Для обеспечения теплотой
эндотермических процессов используют
горячие дымовые газы. В последнем случае
реактор представляет собой трубчатую
печь (д). Отводить тепло реакции можно
не только теплоносителем, но и свежей
реакционной смесью.
Другая
группа реакторов-со взвешенным (кипящим)
или восходящим слоем катализатора.10
При подаче реакционной смеси снизу слоя
с достаточной скоростью твердые частицы
будут витать в воздухе, не уносясь с ним
(е); в этом случае применяют частицы не
крупнее 1 мм. Это обеспечивает полное
использование их внутренней поверхности.
Циркулирующие частицы выравнивают t в
слое -процесс в нем протекает практически
изотермически. Система "реактор-регенератор"
обеспечивает непрерывность процесса
в целом. Такой тип реакционного узла
можно организовать в процессе
каталитического крекинга НП, в котором
kat
быстро закоксовывается, теряя свою
активность. При организации псевдоожиженного
слоя часть газа проходит слой в виде
пузырей. Коэффициент массопередачи
между пузырями и остальной частью слоя
невысокий-фактически образование
пузырей газа-это образование байпасных
потоков.
Для очистки газа от пыли после реактора
устанавливают циклоны.
Если
скорость газового потока будет такой,
что твердые частицы будут увлекаться
им, то реализуется режим пневмотранспорта
(з) и реакция в восходящем потоке kat.
Такая организация процесса эффективна
для быстрых реакций-т.к. время прохождения
реакционной смеси в длинном узком
реакторе небольшое. Реакторы
для геторофазных процессов.11В
трубчатом реакторе (а) жидкость стекает
по стенкам трубок и контактирует со
встречным потоком или попутным потоком
газа. Это наиболее организованный
процесс, т.к. здесь строго поддерживается
поверхность контакта фаз, равная
поверхности трубок. Тепловой режим
поддерживается регулированием t трубок.
Такие реакторы используют в малотоннажных
производствах тонкого органич. и
неорганич. синтеза. Эти
реакторы характеризуются устойчивостью
гидродинамического и температурного
режимов, однако у них низкая
производительность из-за низкой
поверхности контакта фаз.В
зависимости от скорости потоков возможны
различные режимы течения: полное
расслоение фаз, раздельное течение с
сильно возмущенной развитой поверхностью
контакта фаз, хорошо перемешанный
газожидкостной поток.
Указанные режимы наблюдаются
последовательно с возрастанием скорости
потоков, особенно газового. Такие
реакторы компактны даже при необходимости
большого времени контакта-их делают в
виде змеевика. У них высокий коэффициент
массопередачи. Такого типа реактор
используется в производстве полиэтилена
и окислении этилена в ацетальдегид.
Более
распространены насадочные реакторы
(в).
12В
такой аппарат засыпается инертная
насадка-сравнительно небольшие элементы,
по поверхности которых стекает жидкость,
а в пространстве между ними, противотоком
движется газ. Взаимодействие фаз
происходит интенсивно. Производительность
таких реакторов ограничивается
недостаточно интенсивным режимом
движения, т.к. жидкость движется не
принудительно, а за счет силы тяжести.
Простейшее взаимодействие жидкости и
газа-барботаж последнего через жидкость
(г, д).(г) для периодического процесса,
(д)-для непрерывного.Многосекционные
реакторы:13Это
могут быть колонны с колпачковыми
тарелками, снабженными переливными
устройствами (е) или ситчатыми провальными
тарелками (ж). В реакторе (з) жидкая фаза
диспергируется в потоке газа. Для
диспергирования жидкости используются
специальные разбрызгиватели (форсунки).
Мелкие капли более устойчивы в размерах,
но скорость их падения определяется
силами гравитации и скоростью встречного
газа. Поэтому степень диспергирования
должна быть оптимальной.Энергия
в химическом производстве. Тепловой
эффект реакции в технологических
расчетах. Направленность реакции в
технологических расчетах.
Изменение
хим. состава реагирующей смеси приводит
к изменению ее теплосодержания ∆
НТ которое
можно рассчитать через энтальпии
образования компонентов (∆
НТ )обр:∆
Н(Т)
=
Σv(i)(∆
Н(T))обр.i
Если энтальпия образования продуктов
меньше, чем энтальпия образования
исходных веществ, (∆
Н(Т)
< 0),
то
выделяется теплота Q(p)=-∆Н(T),
называемая теплотой реакции. Если при
химическом превращении теплосодержание
смеси увеличивается (∆Н(T)
> 0),
то
происходит поглощение теплоты.В
зависимости от знака ∆Н
(или
Q(p))
реакции
бывают экзотермические (∆Н<0,Q(p)>0)
и
эндотермические (∆Н>0,
Qр<0).
Тепловой
эффект реакции входит в запись
термохимического уравнения,
представляющего собой стехиометрическое
уравнение с указанием его теплового
результата: V(A)А
+v(B)В+…=
v(R)R+v(S)S+…+Q(p).
Значение
Q(p)
в уравнении зависит от записи химического
уравнения. Например,
тепловой эффект реакции, записанной
следующим образом: N2+3Н2=2NН3
в два раза больше, нежели для той же
реакции, записанной по-другому:
0,5N2+1,5Н2=NН3,
поэтому в справочной литературе Q(p)
приводят прямо в уравнениях, как это
сделано в, или указывают изменение
энтальпии, соответствующее превращению
1
моля вещества.Тепловой
эффект реакции в технологических
расчетах.
Знание
теплового эффекта реакции необходимо
для определения тепловых явлений в
технологических процессах. Количество
выделившейся (или поглощенной) теплоты
qр
зависит от количества превращенного
вещества
∆N:
q(p)
=Q(p)
∆N(A)/v(a).
В
зависимости от знака Q(p)
(экзо-
или эндотермическая реакция),
теплота в ходе протекания процесса
будет выделяться или поглощаться.Возможность
химического превращения.
Химический
процесс принципиально осуществим, если
реакция протекает с уменьшением
химического потенциала, называемого
также изобарным потенциалом, или энергией
Гиббса, т.е. возможность протекания
реакции определяется из след. условий:1.при
∆G(T,P)>0
протекание
реакции не возможно; 2.при
∆G(T,P)<0
протекание
реакции возможно; 3.
при
∆G(Т,Р)=0
реакционная
система находится в термодинамическом
равновесии, где ∆G(Т,Р)
-
изменение энергии Гиббса при превращении
исходных веществ в продукты при
температуре
Т
и давлении Р.
Изменение энергии Гиббса реакции можно
рассчитать по уравнению:
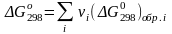 Тепловой
эффект реакции в технологических
расчетах.
Значения стандартной энергии Гиббса
образования веществ при стандартных
температуре 298 К и давлении Р=1 атм
приведены в справочной литературе по
термодинамике и означают изменение
энергии Гиббса при превращении такого
количества вещества, находящегося в
стандартном состоянии, которое записано
в стехиометрическом уравнении.
Для расчета в условиях, отличающихся
от стандартных, используют зависимость
энергии Гиббса от температуры:
Тепловой
эффект реакции в технологических
расчетах.
Значения стандартной энергии Гиббса
образования веществ при стандартных
температуре 298 К и давлении Р=1 атм
приведены в справочной литературе по
термодинамике и означают изменение
энергии Гиббса при превращении такого
количества вещества, находящегося в
стандартном состоянии, которое записано
в стехиометрическом уравнении.
Для расчета в условиях, отличающихся
от стандартных, используют зависимость
энергии Гиббса от температуры:
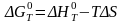 где (после = без Т),
S
изменение
энтальпии и энтропии при стандартном
давлении, которые можно рассчитать
по формулам, аналогичным. Зависимость
энергии Гиббса от состава реакционной
смеси отражает уравнение Вант-Гоффа:
где (после = без Т),
S
изменение
энтальпии и энтропии при стандартном
давлении, которые можно рассчитать
по формулам, аналогичным. Зависимость
энергии Гиббса от состава реакционной
смеси отражает уравнение Вант-Гоффа:
 где
R-универсальная
газовая постоянная, равная 8,314 Дж/моль∙К;
П-знак
произведения; С(i)-
концентрации компонентов; v(i)
- стехиометрические коэффициенты
уравнения реакции в алгебраической
форме. Направленность
реакции в технологических расчетах.
Использование
условий позволяет определить возможность
получения желаемого продукта-с этого
начинают разработку нового способа
производства. Другой вариант
применения-поиск возможностей,
предотвращающих протекание
нежелательных реакций. Продемонстрируем
это на следующем примере. При конверсии
метана водяным паром возможно образование
сажи. Одна
из вероятных реакций: СО
+ Н2
= С + Н2О.
Конверсия
метана в промышленном реакторе протекает
вблизи равновесия. Соответствующее
содержание СО, Н2
и Н2О,
ответственных за образование сажи
(углерода), для исходной смеси с
стехиометрическим соотношением пар:
метан
= 2:1.
(Зависимость
энергии Гиббса ∆G от температуры Т для
реакции образования углерода в реакторе
конверсии метана при начальном соотношении
пар:газ = 1:1(1); 2:1 (2); 4:1 (3))14
Массообменные
процессы. Основные принципы массообменных
процессов. Моделирование процессов
теплообмена.Для
математического описания того или иного
массообменного процесса предложены
несколько теоретических моделей.
Общепринятыми
считаются две теории: 1.
Двухпленочная модель Льюиса и Уитмена;
2.
Теория граничного диффузионного слоя
Ландау и Девиса.
В соответствии с ними процесс переноса
в-ва из одной фазы в др можно представить
происходящим в три последовательные
стадии. Пусть вещество переходит из
фазы Ф(у)
в
фазу Ф(х)
(например,
аммиак из воздуха в воду). Движение фаз
турбулентное. Между фазами существует
поверхность раздела фаз (ПРФ),
которая обусловлена тормозящим действием
сил трения, возникающих при движении
фаз относительно друг друга и действием
сил поверхностного натяжения, имеющих
место на поверхности жидкой фазы.
Турбулентный
поток каждой фазы имеет следующую
структуру: ядро
потока (основная масса фазы) и пограничный
диффузионный слой у границы фазы
(пограничная пленка). В ядре потока
вследствие интенсивного перемешивания
концентрация вещества постоянная. Здесь
преобладает конвективная (турбулентная)
диффузия. В пограничном слое движение
потока замедляется, так как турбулентный
поток заменяется ламинарным. 15
Конвективная диффузия уступает место
молекулярной диффузии. Вследствие этого
в пограничном слое происходит резкое
изменение концентрации от С(0у)
до С(у)*
и
от С(х)*
до
С(0х).
Перенос
вещества замедляется. Вот почему у
границы раздела наблюдается почти
линейное изменение концентрации
вещества.
Согласно
названных выше моделей процесс
массопередачи представляет сумму
процессов массоотдачи из основной массы
фазы
Ф(у)
к
поверхности раздела фаз и процесса
массоотдачи от поверхности раздела к
основной массе фазы Ф(х).В
итоге в основе пленочной и других теорий
массообмена предложены следующие
допущения:
*Общее
диффузионное сопротивление переносу
из фазы в фазу складывается из диффузионного
сопротивления двух фаз и сопротивления
поверхности раздела фаз.
Причем
сопротивление на поверхности раздела
фаз принимается равным нулю. Таким
образом, общее сопротивление переносу
массы можно рассматривать как сумму
фазовых сопротивлений.
*На
поверхности раздела фазы находятся в
равновесии.
Предложены
и другие модели механизма массопереноса.
Но вследствие большой сложности
турбулентных двухфазных потоков
практически невозможно определение с
концентраций (у(гр),
х(гр),
у*,
x*)
в фазах, поверхности контакта и т.д.
Молекулярная
диффузия
обусловлена
беспорядочным перемещением молекул
вещества в неподвижной среде. Конвективная
диффузия
имеет
место в движущейся среде и осуществляется
движущимися частицами носителя или
распределяемого вещества. При турбулентном
движении перенос происходит главным
образом путем турбулентной
диффузии,
а роль молекулярной диффузии незначительна,
за исключением области вблизи поверхности
раздела фаз.
Молекулярная
диффузия. Первый закон Фика.
Молекулярная
диффузия описывается первым законом
Фика:
где
R-универсальная
газовая постоянная, равная 8,314 Дж/моль∙К;
П-знак
произведения; С(i)-
концентрации компонентов; v(i)
- стехиометрические коэффициенты
уравнения реакции в алгебраической
форме. Направленность
реакции в технологических расчетах.
Использование
условий позволяет определить возможность
получения желаемого продукта-с этого
начинают разработку нового способа
производства. Другой вариант
применения-поиск возможностей,
предотвращающих протекание
нежелательных реакций. Продемонстрируем
это на следующем примере. При конверсии
метана водяным паром возможно образование
сажи. Одна
из вероятных реакций: СО
+ Н2
= С + Н2О.
Конверсия
метана в промышленном реакторе протекает
вблизи равновесия. Соответствующее
содержание СО, Н2
и Н2О,
ответственных за образование сажи
(углерода), для исходной смеси с
стехиометрическим соотношением пар:
метан
= 2:1.
(Зависимость
энергии Гиббса ∆G от температуры Т для
реакции образования углерода в реакторе
конверсии метана при начальном соотношении
пар:газ = 1:1(1); 2:1 (2); 4:1 (3))14
Массообменные
процессы. Основные принципы массообменных
процессов. Моделирование процессов
теплообмена.Для
математического описания того или иного
массообменного процесса предложены
несколько теоретических моделей.
Общепринятыми
считаются две теории: 1.
Двухпленочная модель Льюиса и Уитмена;
2.
Теория граничного диффузионного слоя
Ландау и Девиса.
В соответствии с ними процесс переноса
в-ва из одной фазы в др можно представить
происходящим в три последовательные
стадии. Пусть вещество переходит из
фазы Ф(у)
в
фазу Ф(х)
(например,
аммиак из воздуха в воду). Движение фаз
турбулентное. Между фазами существует
поверхность раздела фаз (ПРФ),
которая обусловлена тормозящим действием
сил трения, возникающих при движении
фаз относительно друг друга и действием
сил поверхностного натяжения, имеющих
место на поверхности жидкой фазы.
Турбулентный
поток каждой фазы имеет следующую
структуру: ядро
потока (основная масса фазы) и пограничный
диффузионный слой у границы фазы
(пограничная пленка). В ядре потока
вследствие интенсивного перемешивания
концентрация вещества постоянная. Здесь
преобладает конвективная (турбулентная)
диффузия. В пограничном слое движение
потока замедляется, так как турбулентный
поток заменяется ламинарным. 15
Конвективная диффузия уступает место
молекулярной диффузии. Вследствие этого
в пограничном слое происходит резкое
изменение концентрации от С(0у)
до С(у)*
и
от С(х)*
до
С(0х).
Перенос
вещества замедляется. Вот почему у
границы раздела наблюдается почти
линейное изменение концентрации
вещества.
Согласно
названных выше моделей процесс
массопередачи представляет сумму
процессов массоотдачи из основной массы
фазы
Ф(у)
к
поверхности раздела фаз и процесса
массоотдачи от поверхности раздела к
основной массе фазы Ф(х).В
итоге в основе пленочной и других теорий
массообмена предложены следующие
допущения:
*Общее
диффузионное сопротивление переносу
из фазы в фазу складывается из диффузионного
сопротивления двух фаз и сопротивления
поверхности раздела фаз.
Причем
сопротивление на поверхности раздела
фаз принимается равным нулю. Таким
образом, общее сопротивление переносу
массы можно рассматривать как сумму
фазовых сопротивлений.
*На
поверхности раздела фазы находятся в
равновесии.
Предложены
и другие модели механизма массопереноса.
Но вследствие большой сложности
турбулентных двухфазных потоков
практически невозможно определение с
концентраций (у(гр),
х(гр),
у*,
x*)
в фазах, поверхности контакта и т.д.
Молекулярная
диффузия
обусловлена
беспорядочным перемещением молекул
вещества в неподвижной среде. Конвективная
диффузия
имеет
место в движущейся среде и осуществляется
движущимися частицами носителя или
распределяемого вещества. При турбулентном
движении перенос происходит главным
образом путем турбулентной
диффузии,
а роль молекулярной диффузии незначительна,
за исключением области вблизи поверхности
раздела фаз.
Молекулярная
диффузия. Первый закон Фика.
Молекулярная
диффузия описывается первым законом
Фика: .
Количество вещества dM,
продиффундировавшего
за время d
через элементарную поверхность dF
(нормальную
к направлению диффузии) пропорционально
градиенту концентрации dc/dn
этого
вещества. Или через всю поверхность
F
диффундирует
количество вещества:
.
Количество вещества dM,
продиффундировавшего
за время d
через элементарную поверхность dF
(нормальную
к направлению диффузии) пропорционально
градиенту концентрации dc/dn
этого
вещества. Или через всю поверхность
F
диффундирует
количество вещества:
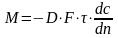 ,
,
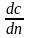 -
градиент концентрации характеризует
изменение концентрации диффундирующего
вещества на единицу длины нормали между
двумя поверхностями постоянных
концентраций; D-коэффициент
пропорциональности, который называют
коэффициентом
молекулярной диффузии. Знак
(-) перед D
указывает на то, что молекулярная
диффузия всегда протекает в направлении
уменьшения концентрации распределяемого
компонента. Коэффициент
D
есть
функция свойств распределяемого
вещества, свойств среды, через которую
оно диффундирует, температуры, t,
и давления, р.
Например, коэффициент D
для
газов возрастает c увеличением температуры
и понижением давления.
Размерность
[D]:
-
градиент концентрации характеризует
изменение концентрации диффундирующего
вещества на единицу длины нормали между
двумя поверхностями постоянных
концентраций; D-коэффициент
пропорциональности, который называют
коэффициентом
молекулярной диффузии. Знак
(-) перед D
указывает на то, что молекулярная
диффузия всегда протекает в направлении
уменьшения концентрации распределяемого
компонента. Коэффициент
D
есть
функция свойств распределяемого
вещества, свойств среды, через которую
оно диффундирует, температуры, t,
и давления, р.
Например, коэффициент D
для
газов возрастает c увеличением температуры
и понижением давления.
Размерность
[D]: Физический
смысл D
можно
выяснить из предыдущего выражения.
Коэффициент
диффузии
показывает,
какое количество вещества диффундирует
в единицу времени через единицу
поверхности при градиенте концентрации,
равном единице.Коэффициент
диффузии D-физическая
константа, которая характеризует
способность данного вещества проникать
в неподвижную среду. Величина D
не зависит от гидродинамических условий,
в которых протекает процесс.
Турбулентная
диффузия.
Количество
вещества dMТ,
которое
переносится в пределах фазы турбулентной
диффузией, может быть рассчитано по
уравнению:
Физический
смысл D
можно
выяснить из предыдущего выражения.
Коэффициент
диффузии
показывает,
какое количество вещества диффундирует
в единицу времени через единицу
поверхности при градиенте концентрации,
равном единице.Коэффициент
диффузии D-физическая
константа, которая характеризует
способность данного вещества проникать
в неподвижную среду. Величина D
не зависит от гидродинамических условий,
в которых протекает процесс.
Турбулентная
диффузия.
Количество
вещества dMТ,
которое
переносится в пределах фазы турбулентной
диффузией, может быть рассчитано по
уравнению:
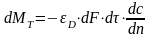 где
D
- коэффициент турбулентной диффузии.
Или для всей поверхности (F
= dF)
где
D
- коэффициент турбулентной диффузии.
Или для всей поверхности (F
= dF)
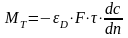 Коэффициент
турбулентной диффузии ,D,
показывает,
какое количество вещества передается
посредством турбулентной диффузии в
единицу времени через единицу поверхности
при градиенте концентраций, равном
единице.Коэффициент
D
не
является физической константой, он
зависит от гидродинамических условий
(скорости потока и масштаба турбулентности).
Уравнение
массоотдачи.
В
связи с большой сложностью процессов
массопереноса при расчетах принимают,
что количество вещества, перенесенного
от границы раздела фаз в другую фазу,
пропорционально движущей силе, которая
равна разности концентраций в ядре и
на границе фазы, поверхности контакта
фаз и времени. Тогда если массоперенос
происходит из фазы Фу в фазу Фх, то
количество вещества М, переносимого в
единицу времени в каждой из фаз (к границе
раздела фаз и в обратном направлении),
может быть посчитано по основному
уравнению массоотдачи:
для
фазы Фу:
Коэффициент
турбулентной диффузии ,D,
показывает,
какое количество вещества передается
посредством турбулентной диффузии в
единицу времени через единицу поверхности
при градиенте концентраций, равном
единице.Коэффициент
D
не
является физической константой, он
зависит от гидродинамических условий
(скорости потока и масштаба турбулентности).
Уравнение
массоотдачи.
В
связи с большой сложностью процессов
массопереноса при расчетах принимают,
что количество вещества, перенесенного
от границы раздела фаз в другую фазу,
пропорционально движущей силе, которая
равна разности концентраций в ядре и
на границе фазы, поверхности контакта
фаз и времени. Тогда если массоперенос
происходит из фазы Фу в фазу Фх, то
количество вещества М, переносимого в
единицу времени в каждой из фаз (к границе
раздела фаз и в обратном направлении),
может быть посчитано по основному
уравнению массоотдачи:
для
фазы Фу:
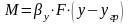 для фазы Фх
:
для фазы Фх
:
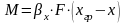 где
у
и х
– средние концентрации в ядре каждой
фазы; у(гр)
и х(гр)
– концентрации у границы соответствующей
фазы; у
и х
– коэффициенты пропорциональности,
коэффициентами
массоотдачи;
F-поверхность
контакта фаз, м(2).
где
у
и х
– средние концентрации в ядре каждой
фазы; у(гр)
и х(гр)
– концентрации у границы соответствующей
фазы; у
и х
– коэффициенты пропорциональности,
коэффициентами
массоотдачи;
F-поверхность
контакта фаз, м(2).
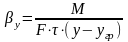 ,
,
 Если
принять F=1м(2);
=
1с; у-у(гр).
= 1 (или х(гр).
– х=1),
то физическую сущность коэффициентов
массоотдачи выразим следующим образом:
они показывают, какое количество вещества
переходит от поверхности раздела фаз
в ядро фазы (или в обратном направлении)
через единицу поверхности в единицу
времени при движущей силе, равной 1.
Коэффициенты массоотдачи не являются
физической константой. Это кинетическая
характеристика. Они
зависят от следующих факторов:1.физико-хим.
свойств фазы (,
и других):2.гидродинамических
условий в ней (ламинарный или турбулентный
режимы движения жидкости);3.геометрич.
факторов, в том числе конструкция и
размеры аппарата. Коэффициент массоотдачи
учитывает перенос вещества как
конвективной, так и молекулярной
диффузией.Размерность коэффициента
массоотдачи находим:
Если
принять F=1м(2);
=
1с; у-у(гр).
= 1 (или х(гр).
– х=1),
то физическую сущность коэффициентов
массоотдачи выразим следующим образом:
они показывают, какое количество вещества
переходит от поверхности раздела фаз
в ядро фазы (или в обратном направлении)
через единицу поверхности в единицу
времени при движущей силе, равной 1.
Коэффициенты массоотдачи не являются
физической константой. Это кинетическая
характеристика. Они
зависят от следующих факторов:1.физико-хим.
свойств фазы (,
и других):2.гидродинамических
условий в ней (ламинарный или турбулентный
режимы движения жидкости);3.геометрич.
факторов, в том числе конструкция и
размеры аппарата. Коэффициент массоотдачи
учитывает перенос вещества как
конвективной, так и молекулярной
диффузией.Размерность коэффициента
массоотдачи находим: Коэффициент
массоотдачи определяется при помощи
теории подобия. Сложность расчета по
уравнениям массоотдачи состоит в том,
что практически невозможно измерить
концентрации фаз непосредственно у
границы их раздела. По этой причине
количество вещества М, переносимого из
фазы в фазу, рассчитывают по основному
уравнению массопередачи, которое
записывают в форме:
Коэффициент
массоотдачи определяется при помощи
теории подобия. Сложность расчета по
уравнениям массоотдачи состоит в том,
что практически невозможно измерить
концентрации фаз непосредственно у
границы их раздела. По этой причине
количество вещества М, переносимого из
фазы в фазу, рассчитывают по основному
уравнению массопередачи, которое
записывают в форме: ,
, здесь К(х)
и К(у)
– коэффициенты массопередачи, выраженные,
соответственно, через концентрации фаз
Ф(х) и Ф(у), у*
и х*
- равновесные концентрации в данной
фазе, соответствующие концентрациям
распределяемого вещества в ядре другой
фазы. Физический
смысл коэффициента массопередачи
(К(х)
и К(у)).
Он показывает, какое количество вещества
переходит из фазы в фазу за единицу
времени через единицу поверхности
контакта фаз при движущей силе
массопередачи, равной 1. Коэффициенты
и К
различаются по физической сущности, но
выражаются одинаковыми единицами
измерения.
здесь К(х)
и К(у)
– коэффициенты массопередачи, выраженные,
соответственно, через концентрации фаз
Ф(х) и Ф(у), у*
и х*
- равновесные концентрации в данной
фазе, соответствующие концентрациям
распределяемого вещества в ядре другой
фазы. Физический
смысл коэффициента массопередачи
(К(х)
и К(у)).
Он показывает, какое количество вещества
переходит из фазы в фазу за единицу
времени через единицу поверхности
контакта фаз при движущей силе
массопередачи, равной 1. Коэффициенты
и К
различаются по физической сущности, но
выражаются одинаковыми единицами
измерения.
 Так
как концентрации фаз изменяются при их
движении вдоль поверхности раздела,
изменяется и движущая сила массопередачи.
Поэтому в уравнение массопередачи
вводят среднюю движущую силу (х(ср)
или
у(ср))
и
уравнения принимают вид: М=Ку*F*∆yср*
,
M=Kx*F*∆xср*
.
Из
уравнений массопередачи можно рассчитать
скорость процесса
Так
как концентрации фаз изменяются при их
движении вдоль поверхности раздела,
изменяется и движущая сила массопередачи.
Поэтому в уравнение массопередачи
вводят среднюю движущую силу (х(ср)
или
у(ср))
и
уравнения принимают вид: М=Ку*F*∆yср*
,
M=Kx*F*∆xср*
.
Из
уравнений массопередачи можно рассчитать
скорость процесса
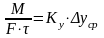 ,
,
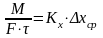 Из основного уравнения массопередачи
обычно рассчитывают размеры аппарата,
если можно определить К(х)
и К(у).
Связь
коэффициента массопередачи и коэффициентов
массоотдачи (или уравнение аддитивности
фазовых сопротивлений).
Перенос
вещества из фазы Ф(у) в фазу Ф(х).
Движущая
сила массопередачи выражена в единицах
концентрации фазы Ф(у).
Количество
вещества
М,
переносимое из фазы в фазу, рассчитываем
из уравнения массопередачи. Допустим,
что равновесная зависимость между
концентрациями в фазах линейна у*=mx,
где m–tg
угла наклона линии равновесия.
Примем,
что концентрации распределяемого
вещества в фазах у границы раздела
(х(гр).,
у(гр).)
равновесны
друг другу.Тогда из уравнения линии
равновесия следует, что: х(гр).=у(гр.)/m
и х
= у*/m,
где х(гр.)
и
у(гр)-
концентрации каждой фазы, у*-
концентрация фазы Ф(у), равновесная с
концентрацией
х
фазы
Ф(х).
Подставляя
значения х(гр.)
и
х
в уравнение массоотдачи , получим
Из основного уравнения массопередачи
обычно рассчитывают размеры аппарата,
если можно определить К(х)
и К(у).
Связь
коэффициента массопередачи и коэффициентов
массоотдачи (или уравнение аддитивности
фазовых сопротивлений).
Перенос
вещества из фазы Ф(у) в фазу Ф(х).
Движущая
сила массопередачи выражена в единицах
концентрации фазы Ф(у).
Количество
вещества
М,
переносимое из фазы в фазу, рассчитываем
из уравнения массопередачи. Допустим,
что равновесная зависимость между
концентрациями в фазах линейна у*=mx,
где m–tg
угла наклона линии равновесия.
Примем,
что концентрации распределяемого
вещества в фазах у границы раздела
(х(гр).,
у(гр).)
равновесны
друг другу.Тогда из уравнения линии
равновесия следует, что: х(гр).=у(гр.)/m
и х
= у*/m,
где х(гр.)
и
у(гр)-
концентрации каждой фазы, у*-
концентрация фазы Ф(у), равновесная с
концентрацией
х
фазы
Ф(х).
Подставляя
значения х(гр.)
и
х
в уравнение массоотдачи , получим
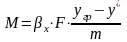 Определим
движущую силу процесса:
Определим
движущую силу процесса:
 Из
уравнения движущая сила процесса в фазе
Ф(у)
Из
уравнения движущая сила процесса в фазе
Ф(у)
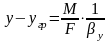 .
Суммируем почленно получим :
.
Суммируем почленно получим :
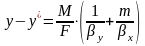 Уравнение
массопередачи для рассматриваемого
случая имеет следующий вид:
или
Уравнение
массопередачи для рассматриваемого
случая имеет следующий вид:
или
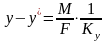 .
Приравнивая правые части уравнений,
находим зависимость между коэффициентами
массопередачи К(у)
и массоотдачи (х)
и (у):
.
Приравнивая правые части уравнений,
находим зависимость между коэффициентами
массопередачи К(у)
и массоотдачи (х)
и (у): .
При выражении коэффициента массопередачи
в конценрациях фазы Ф(х)
будем
иметь
.
При выражении коэффициента массопередачи
в конценрациях фазы Ф(х)
будем
иметь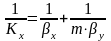 Левые
части уравнений представляют собой
общее сопротивление массопередаче, а
правые части-сумму сопротивлений
массоотдаче в фазах. При m
= сonst
разделим уравнение на m,
получим:
Левые
части уравнений представляют собой
общее сопротивление массопередаче, а
правые части-сумму сопротивлений
массоотдаче в фазах. При m
= сonst
разделим уравнение на m,
получим: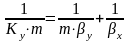 или
или
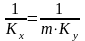 т.е.
т.е.
 В
уравнении член 1/(у)
выражает
диффузионное сопротивление переходу
вещества в фазе Ф(у),
член
m/y
– сопротивление в фазе Ф(х).
Если
коэффициент (у)
велик,
то член 1/(у)
мал
и, как видно из уравнения,
В
уравнении член 1/(у)
выражает
диффузионное сопротивление переходу
вещества в фазе Ф(у),
член
m/y
– сопротивление в фазе Ф(х).
Если
коэффициент (у)
велик,
то член 1/(у)
мал
и, как видно из уравнения,
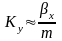 В этом случае сопротивлением в фазе
Ф(у) можно пренебречь.
При
большом коэффициенте (х)
член
m/(х)
мал
и как видно из уравнения, . В этом случае
сопротивлением Ф(х) можно пренебречь.
При выводе уравнения было принято
условие линейной зависимости между
равновесными концентрациями у*
и
х.
В
случае отсутствия такой зависимости
линия равновесия не будет прямой и
константу равновесия надо брать как
тангенс угла наклона касательной к
линии равновесия в данной точке. При
этом величины m
и
К(у)
будут
изменяться по длине аппарата. При
расчетах берут среднее значение
коэффициента распределения m.
Пр-во
H2SO4. По
объему производ. и потреб. занимает 1-ое
место. Из-за: ее дешевизны по сравн. со
всеми др. ки-тами, и ее св-в. H2SO4
не дымит, в конц. виде не разрушает черные
Ме, явл. одной из сильных кислот, в широком
диапазоне температур находится в жидком
состоянии. Безводная H2SO4
(моногидрат)-тяжелая маслянистая
жидкость, которая смешивается с водой
с выделением большого кол-ва
теплоты.Тривиальное название «купоросное
масло». Крупн. потреб.: пр-ва минер.
употреб.: суперфосфата, сульфата аммония.
Многие ки-ты и соли производ. в знач.
части при помощи серной ки-ты. Применение:
*в производстве цветных и редких Ме, а
также Ме-обраб. промышлен. *получение
ряда красителей (для тканей), лаков и
красок (для зданий и машин) *производ.
лекарств. ве-в *производ. пластмасс. При
помощи H2SO4
производят этиловый и др. спирты,
некоторые эфиры, ПАВ, ряд ядохимикатов
для борьбы с вредителями сельского
хозяйства и сорными травами. Разбавленные
ра-ры H2SO4
и её солей применяют в текстильной, а
также др. отраслях легкой промышленности.
В пищевой промышленности применяется
для получения крахмала, патоки и ряда
др. продуктов. В электротехнике она исп.
В качестве электролита в аккумуляторах.
Серную ки-ту исп. Для осушки газов и при
концентрации ки-т. Так же её применяют
как компонент реакционной среды в
процессах нитрования, в частности, при
получении взрывчатых ве-в. Виды:
1. башенная серная ки-та 75% 2. Купоросное
масло 92-94% 3. Олеум 18-24% SO3. Сырье
-
*Рядовой
колчедан. Состоит из пирита FeS2, в котором
52,6% S и примесей, которые
снижают
содержание серы до 30 - 35%.*Флотационный
колчедан.
Флотационный
пирит содержит до 40% серы.
*Углистый
пирит.
*Самородная
сера.
*Отходы
пр-ва SO2, H2S получаются при переработке
жидкого или тв
топлива,
на электростанциях при сжигании
серы.
Методы
производства серной кислоты:1.
Контактный метод (получается более
чистая серная
кислота и олеум 20-60%)
2.
Нитрозный способ
3.
Способ прямого сжигания серы или
сероводорода.
Отходы
производств.
Способ
прямого сжигания серы или H2S.
Сера
–
легкоплавкое ве-во: tпл.
113°С. Перед сжиганием ее расплавляют,
используя пар, полученный при утилизации
тепла ее сжигания. Расплавленная сера
отстаивается и фильтруется для удаления
имеющихся в природной сере примесей и
насосом подается в печь сжигания. Сера
горит в основном в парофазном состоянии.
В циклонной печи жидкая сера и воздух
подаются тангенциально и за счет
вихревого движения достигается
диспергирование жидкости и перемешивание
двух потоков. Мелкие капли быстро
испаряются и сера в парообразном
состоянии сгорает. Горение протекает
адиабатически, и t
зависит от концентрации образующегося
SO2: Sж+O2=SO2+319кДж
Р1.
За счет высокой теплоты сгорания серы
t в печи составляет более 1000°СКонтактный
способ производства
включает
в себя:
1.
Обжиг пирита
2.
Очистка и специальная очистка обжигового
газа
3.
контактное окисление SO2
в SO3
4.
поглощение серного ангидрида SO3
и получение товарной серной кислоты
или олеума.Стадия
1 -
Обжиг
серного колчедана или серы:
4FeS2+11O2=2Fe2O3(огарок)+8SO2;
S2+202=2SO2.Образующиеся
в процессе горения примеси
(SO3
и оксиды азота) приводят
или к коррозии аппаратуры или к
отравлению
катализатора, а также ухудшают качество
серной кислоты. Их удаляют в промывном
отделении.
Колчедан,
который поступает на обжиг, поступает
в виде кусков диаметром от 50 до 400 мм.
Он
дробиться с помощью щековых или валковых
дробилок и на грохотах отсеивается
фракция менее 10 мл. После крупные фракции
поступают на повторное дробление, а
более мелкая измельчается в пыль. На
основное пр-во идут частицы 10 мм.
Р2.
Исходные
вещества -
минеральное сырье - содержит примеси,
поэтому выходящие со стадии
обжига
газы подвергаются очистке. Первая стадия
- обжиг, специфичен для каждого вида
сырья.Обжиг
серосодержащего сырья.
Термическая
диссоциация:
2FeS2
=2FeS
+ S2.
Газофазное
горение серы:
S2 + 2О2= 2SO2.
Горение
пирротина: 4FeS2+7О2=
2Fe2O3+4SO2.
Или
суммарно: 4FeS2+1102=2 Fe2O3+8SO2.
При
небольшом избытке или недостатке
кислорода образуется смешанный оксид
железа
3FeS2
+ 8O2= Fe3O4+6SO2.
Обжиг
пирита осуществляется в обжиговых печах
различной конструкции.Обоснование
роли параметров обжига серосодержащего
сырья.Температура.
Термическое
разложение пирита начинается уже при
t около 200°C
и
одновременно воспламеняется сера. При
t выше 680°С интенсивно протекают все
3
ре-ции.
B
промышленности обжиг ведут при 850 -
900°C. В общем виде движущая сила этого
процесса может быть выражена
уравнением:
r(D)=k(D)F∆
(1)
Из этого уравнения видно, что факторами
увеличения производительности процесса
явл.
интенсификация
массопереноса (k(D)),
увеличение
поверхности контакта фаз (F) и конц.
реагентов.
Коэф. массопереноса может быть увеличен
повышением t,
поверхность
контакта фаз существенно возрастает с
увеличением степени дисперсности тв
материала,
а высокие конц. реагентов обеспечиваются
использованием обогащенного
пирита
и увеличением содержания кислорода в
окислителе. Следует иметь в виду, что
чрезмерное
повышение t способствует размягчению
и слипанию частиц тв
сырья,
а применение концентрированных реагентов
удорожает их стоимость и приводит к
чрезмерному
росту t в условиях автотермического
режима.Давление.
В
соответствии с уравнением (1) давление
является фактором скорости процесса,
однако
его рост может привести в условиях
автотермического режима к чрезмерному
увеличению
температуры
и слипанию частиц твердого материала.
Кроме того, увеличение давления приводит
к
увеличению расходов на компремирование.
Предпочитают работать при атмосферном
давлении.
Соотношение
воздух-колчедан поддерживается на
уровне, обеспечивающем избыток
кислорода
по сравнению с его стехиометрическим
количеством.
Ранее
доминирующим типом реактора был
многополочный реактор, снабженный
скребками
для
выравнивания слоя на каждой полке и
организации транспорта твердой фазы с
верхних полок
на
нижние.1.
ВXЗ (Воскресенский химический завод)
Р3.
В таком реакторе степень дисперсности
колчедана невелика, что существенно
ограничивает
производительность
реактора. Кроме того, в таком реакторе
движущиеся скребки в
высокотемпературной
зоне усложняют его конструкцию, создается
неоднородный t
режим
по полкам, в нем трудно организовать
отвод тепла из зоны реакции. Трудности
теплосъема
не
позволяют получить обжиговый газ с
концентрацией SO2 более 8-9%. Основное
ограничение
невозможность
использования мелких частиц, в то время
как для гетерогенного процесса
основной
способ ускорения реакции - дробление
частиц. По этой причине более перспективными
явл.
аппараты к «кипящем слоем» тв частиц.
Их степень дисперсности позволяет на
порядок
ускорить процесс.2.
Печь сжигания пирита в кипящем слое и
во взвешенном состоянии.Р4
Пылевидный колчедан подается через
питатель в реактор. Окислитель (воздух)
подается снизу через распределительную
решетку. Их витание в слое предотвращает
слипание и способствует хорошему
контакту с газом, выравнивает температурный
режим по всему слою, обеспечивает
подвижность тв. материала и его переток
в выходной патрубок для вывода огарка
из реактора. В слое подвижных частиц
можно расположить теплообменные
элементы. Благодаря увеличению глубины
обжига содержащие SO2
в обжиговом газе
увеличивается до 13-15%. Основной недостаток
печей кипящего слоя-повышенная
запыленность обжигового газа из-за
механической эрозии подвижных тв.
частиц. Это требует более тщательной
очистки газа от пыли в циклоне и
электрофильтре.Стадия
2:
Р5-Обжиговый
газ может непосредственно направляться
на пр-во серной кислоты
нитрозным
способом после очистки от пыли. На
производстве серной кислоты требуется
специальная очистка обж. газа.
Очистка включает в себя в основном
избавление от пыли. Применяют
циклон-аппараты.Обжиговый газ с высокой
скоростью входит в первую трубу
циклон-аппарата, выходит через ее
наибольшее сечение с большой скоростью
и закручивается вокруг вертикальной
трубы. Центробежная сила отбрасывает
пылинки к стенкам, которые ссыпаются в
его нижнюю часть и утилизируются с
огарком. Очищенный обжиговый газ уходит
сверху циклон-аппарата. Р6-Далее
обжиговый газ попадает в электрофильтры,
в центре которых находится коронный
электрофильтр, на который попадает
высокое напряжение (1000 В). Молекулы
воздуха ионизируются, притягиваются к
частичкам пыли, укрупняя их. При снятии
напряжения они ссыпаются в нижнюю часть
электрофильтра, а очищенный газ выходит
вверх. Р7Для
контактного способа требуется
дополнительная очистка (96/98 Н2SO4).
Обжиговый газ поступает в первую
поглотительную колонну, орошаемую 70%
H2SO4.
Колонна изнутри выложена огнеупорным
кирпичом. Затем, выйдя из первой, газ
входит во вторую орошаемую 30% серной
кислотой. Эта колонна внутри выложена
свинцовыми пластинами. Для увеличения
поверхности контакта все или некоторые
колонны этой схемы содержат насадку
колец Раминга. Поглощаются оксиды
железа, огарки, остаток и пыль поступают
в электрофильтр ЭФ, где очищаются от
тумана, влаги и частично унесенных тв
частиц («мокрый ЭФ»). Следующая колонна
– увлажнительная. Окончательно очищает.
Затем опять в «мокрый ЭФ» и в 4 колонну.
Здесь обжиговый газ очищается от паров
влаги и может направляться в стадию 3.
Внизу стекает отработанная кислота.
Здесь ее концентрируют, очищают от
примесей и возвращают в пр-во. Шлак,
содержащий селен, теллур, отправляют в
пр-во.Стадия
3.
Контактное
окисление SO2 в SO3
Реакция: 2SO2+O2= 2SO3+94,2кДж(обратимая).
Является обратимой и экзотермической
и протекает с уменьшением объема. Она
осуществляется на катализаторах, основой
которых является V2O5 с добавлением
оксидов щелочных металлов, нанесенных
на оксид кремния.Обоснование
роли параметров и их выбор.
Температура.
Выбор
температуры определяется верхним и
нижним пределом этого параметра. При
Т<400 0C активность катализаторов весьма
мала, а выше 600 0С происходит их термическая
дезактивация. Оптимальными являются
температуры, лежащие внутри этих
пределов. Давление
является фактором скорости процесса и
фактором смещения равновесия, однако
на практике предпочитают работать при
давлениях, близких к атмосферном. Время
контакта
выбирается,
исходя из максимально достижимой
конверсии. Поэтому за вр. реакц. выбир.
то min
t
при котором степень превращения конверсии
практически близка равновесной. Обычно
эта величина равна 90-95 %, а соответствующее
ей время – несколько секунд. Катализаторы:
1.
пентооксид ванадия V2O5,
модифицированный оксидами щелочных
Ме, нанесенных на оксид кремния (старый).
2.
сульфованадат натрия, нанесенный на
диатомид (СВД-новый). 3.
бария алюмованадат – новый. Катализатор
не активен до t
400. Выше 600С происходит термическая
дезактивация катализ. Оптимальные t
работы катализ. 400-600С. Соотношение
O2:SO2
является
фактором смещения равновесия, а также
фактором скорости процесса в соответствии
с уравнением Борескова:r
= k
* [
В этом случае сопротивлением в фазе
Ф(у) можно пренебречь.
При
большом коэффициенте (х)
член
m/(х)
мал
и как видно из уравнения, . В этом случае
сопротивлением Ф(х) можно пренебречь.
При выводе уравнения было принято
условие линейной зависимости между
равновесными концентрациями у*
и
х.
В
случае отсутствия такой зависимости
линия равновесия не будет прямой и
константу равновесия надо брать как
тангенс угла наклона касательной к
линии равновесия в данной точке. При
этом величины m
и
К(у)
будут
изменяться по длине аппарата. При
расчетах берут среднее значение
коэффициента распределения m.
Пр-во
H2SO4. По
объему производ. и потреб. занимает 1-ое
место. Из-за: ее дешевизны по сравн. со
всеми др. ки-тами, и ее св-в. H2SO4
не дымит, в конц. виде не разрушает черные
Ме, явл. одной из сильных кислот, в широком
диапазоне температур находится в жидком
состоянии. Безводная H2SO4
(моногидрат)-тяжелая маслянистая
жидкость, которая смешивается с водой
с выделением большого кол-ва
теплоты.Тривиальное название «купоросное
масло». Крупн. потреб.: пр-ва минер.
употреб.: суперфосфата, сульфата аммония.
Многие ки-ты и соли производ. в знач.
части при помощи серной ки-ты. Применение:
*в производстве цветных и редких Ме, а
также Ме-обраб. промышлен. *получение
ряда красителей (для тканей), лаков и
красок (для зданий и машин) *производ.
лекарств. ве-в *производ. пластмасс. При
помощи H2SO4
производят этиловый и др. спирты,
некоторые эфиры, ПАВ, ряд ядохимикатов
для борьбы с вредителями сельского
хозяйства и сорными травами. Разбавленные
ра-ры H2SO4
и её солей применяют в текстильной, а
также др. отраслях легкой промышленности.
В пищевой промышленности применяется
для получения крахмала, патоки и ряда
др. продуктов. В электротехнике она исп.
В качестве электролита в аккумуляторах.
Серную ки-ту исп. Для осушки газов и при
концентрации ки-т. Так же её применяют
как компонент реакционной среды в
процессах нитрования, в частности, при
получении взрывчатых ве-в. Виды:
1. башенная серная ки-та 75% 2. Купоросное
масло 92-94% 3. Олеум 18-24% SO3. Сырье
-
*Рядовой
колчедан. Состоит из пирита FeS2, в котором
52,6% S и примесей, которые
снижают
содержание серы до 30 - 35%.*Флотационный
колчедан.
Флотационный
пирит содержит до 40% серы.
*Углистый
пирит.
*Самородная
сера.
*Отходы
пр-ва SO2, H2S получаются при переработке
жидкого или тв
топлива,
на электростанциях при сжигании
серы.
Методы
производства серной кислоты:1.
Контактный метод (получается более
чистая серная
кислота и олеум 20-60%)
2.
Нитрозный способ
3.
Способ прямого сжигания серы или
сероводорода.
Отходы
производств.
Способ
прямого сжигания серы или H2S.
Сера
–
легкоплавкое ве-во: tпл.
113°С. Перед сжиганием ее расплавляют,
используя пар, полученный при утилизации
тепла ее сжигания. Расплавленная сера
отстаивается и фильтруется для удаления
имеющихся в природной сере примесей и
насосом подается в печь сжигания. Сера
горит в основном в парофазном состоянии.
В циклонной печи жидкая сера и воздух
подаются тангенциально и за счет
вихревого движения достигается
диспергирование жидкости и перемешивание
двух потоков. Мелкие капли быстро
испаряются и сера в парообразном
состоянии сгорает. Горение протекает
адиабатически, и t
зависит от концентрации образующегося
SO2: Sж+O2=SO2+319кДж
Р1.
За счет высокой теплоты сгорания серы
t в печи составляет более 1000°СКонтактный
способ производства
включает
в себя:
1.
Обжиг пирита
2.
Очистка и специальная очистка обжигового
газа
3.
контактное окисление SO2
в SO3
4.
поглощение серного ангидрида SO3
и получение товарной серной кислоты
или олеума.Стадия
1 -
Обжиг
серного колчедана или серы:
4FeS2+11O2=2Fe2O3(огарок)+8SO2;
S2+202=2SO2.Образующиеся
в процессе горения примеси
(SO3
и оксиды азота) приводят
или к коррозии аппаратуры или к
отравлению
катализатора, а также ухудшают качество
серной кислоты. Их удаляют в промывном
отделении.
Колчедан,
который поступает на обжиг, поступает
в виде кусков диаметром от 50 до 400 мм.
Он
дробиться с помощью щековых или валковых
дробилок и на грохотах отсеивается
фракция менее 10 мл. После крупные фракции
поступают на повторное дробление, а
более мелкая измельчается в пыль. На
основное пр-во идут частицы 10 мм.
Р2.
Исходные
вещества -
минеральное сырье - содержит примеси,
поэтому выходящие со стадии
обжига
газы подвергаются очистке. Первая стадия
- обжиг, специфичен для каждого вида
сырья.Обжиг
серосодержащего сырья.
Термическая
диссоциация:
2FeS2
=2FeS
+ S2.
Газофазное
горение серы:
S2 + 2О2= 2SO2.
Горение
пирротина: 4FeS2+7О2=
2Fe2O3+4SO2.
Или
суммарно: 4FeS2+1102=2 Fe2O3+8SO2.
При
небольшом избытке или недостатке
кислорода образуется смешанный оксид
железа
3FeS2
+ 8O2= Fe3O4+6SO2.
Обжиг
пирита осуществляется в обжиговых печах
различной конструкции.Обоснование
роли параметров обжига серосодержащего
сырья.Температура.
Термическое
разложение пирита начинается уже при
t около 200°C
и
одновременно воспламеняется сера. При
t выше 680°С интенсивно протекают все
3
ре-ции.
B
промышленности обжиг ведут при 850 -
900°C. В общем виде движущая сила этого
процесса может быть выражена
уравнением:
r(D)=k(D)F∆
(1)
Из этого уравнения видно, что факторами
увеличения производительности процесса
явл.
интенсификация
массопереноса (k(D)),
увеличение
поверхности контакта фаз (F) и конц.
реагентов.
Коэф. массопереноса может быть увеличен
повышением t,
поверхность
контакта фаз существенно возрастает с
увеличением степени дисперсности тв
материала,
а высокие конц. реагентов обеспечиваются
использованием обогащенного
пирита
и увеличением содержания кислорода в
окислителе. Следует иметь в виду, что
чрезмерное
повышение t способствует размягчению
и слипанию частиц тв
сырья,
а применение концентрированных реагентов
удорожает их стоимость и приводит к
чрезмерному
росту t в условиях автотермического
режима.Давление.
В
соответствии с уравнением (1) давление
является фактором скорости процесса,
однако
его рост может привести в условиях
автотермического режима к чрезмерному
увеличению
температуры
и слипанию частиц твердого материала.
Кроме того, увеличение давления приводит
к
увеличению расходов на компремирование.
Предпочитают работать при атмосферном
давлении.
Соотношение
воздух-колчедан поддерживается на
уровне, обеспечивающем избыток
кислорода
по сравнению с его стехиометрическим
количеством.
Ранее
доминирующим типом реактора был
многополочный реактор, снабженный
скребками
для
выравнивания слоя на каждой полке и
организации транспорта твердой фазы с
верхних полок
на
нижние.1.
ВXЗ (Воскресенский химический завод)
Р3.
В таком реакторе степень дисперсности
колчедана невелика, что существенно
ограничивает
производительность
реактора. Кроме того, в таком реакторе
движущиеся скребки в
высокотемпературной
зоне усложняют его конструкцию, создается
неоднородный t
режим
по полкам, в нем трудно организовать
отвод тепла из зоны реакции. Трудности
теплосъема
не
позволяют получить обжиговый газ с
концентрацией SO2 более 8-9%. Основное
ограничение
невозможность
использования мелких частиц, в то время
как для гетерогенного процесса
основной
способ ускорения реакции - дробление
частиц. По этой причине более перспективными
явл.
аппараты к «кипящем слоем» тв частиц.
Их степень дисперсности позволяет на
порядок
ускорить процесс.2.
Печь сжигания пирита в кипящем слое и
во взвешенном состоянии.Р4
Пылевидный колчедан подается через
питатель в реактор. Окислитель (воздух)
подается снизу через распределительную
решетку. Их витание в слое предотвращает
слипание и способствует хорошему
контакту с газом, выравнивает температурный
режим по всему слою, обеспечивает
подвижность тв. материала и его переток
в выходной патрубок для вывода огарка
из реактора. В слое подвижных частиц
можно расположить теплообменные
элементы. Благодаря увеличению глубины
обжига содержащие SO2
в обжиговом газе
увеличивается до 13-15%. Основной недостаток
печей кипящего слоя-повышенная
запыленность обжигового газа из-за
механической эрозии подвижных тв.
частиц. Это требует более тщательной
очистки газа от пыли в циклоне и
электрофильтре.Стадия
2:
Р5-Обжиговый
газ может непосредственно направляться
на пр-во серной кислоты
нитрозным
способом после очистки от пыли. На
производстве серной кислоты требуется
специальная очистка обж. газа.
Очистка включает в себя в основном
избавление от пыли. Применяют
циклон-аппараты.Обжиговый газ с высокой
скоростью входит в первую трубу
циклон-аппарата, выходит через ее
наибольшее сечение с большой скоростью
и закручивается вокруг вертикальной
трубы. Центробежная сила отбрасывает
пылинки к стенкам, которые ссыпаются в
его нижнюю часть и утилизируются с
огарком. Очищенный обжиговый газ уходит
сверху циклон-аппарата. Р6-Далее
обжиговый газ попадает в электрофильтры,
в центре которых находится коронный
электрофильтр, на который попадает
высокое напряжение (1000 В). Молекулы
воздуха ионизируются, притягиваются к
частичкам пыли, укрупняя их. При снятии
напряжения они ссыпаются в нижнюю часть
электрофильтра, а очищенный газ выходит
вверх. Р7Для
контактного способа требуется
дополнительная очистка (96/98 Н2SO4).
Обжиговый газ поступает в первую
поглотительную колонну, орошаемую 70%
H2SO4.
Колонна изнутри выложена огнеупорным
кирпичом. Затем, выйдя из первой, газ
входит во вторую орошаемую 30% серной
кислотой. Эта колонна внутри выложена
свинцовыми пластинами. Для увеличения
поверхности контакта все или некоторые
колонны этой схемы содержат насадку
колец Раминга. Поглощаются оксиды
железа, огарки, остаток и пыль поступают
в электрофильтр ЭФ, где очищаются от
тумана, влаги и частично унесенных тв
частиц («мокрый ЭФ»). Следующая колонна
– увлажнительная. Окончательно очищает.
Затем опять в «мокрый ЭФ» и в 4 колонну.
Здесь обжиговый газ очищается от паров
влаги и может направляться в стадию 3.
Внизу стекает отработанная кислота.
Здесь ее концентрируют, очищают от
примесей и возвращают в пр-во. Шлак,
содержащий селен, теллур, отправляют в
пр-во.Стадия
3.
Контактное
окисление SO2 в SO3
Реакция: 2SO2+O2= 2SO3+94,2кДж(обратимая).
Является обратимой и экзотермической
и протекает с уменьшением объема. Она
осуществляется на катализаторах, основой
которых является V2O5 с добавлением
оксидов щелочных металлов, нанесенных
на оксид кремния.Обоснование
роли параметров и их выбор.
Температура.
Выбор
температуры определяется верхним и
нижним пределом этого параметра. При
Т<400 0C активность катализаторов весьма
мала, а выше 600 0С происходит их термическая
дезактивация. Оптимальными являются
температуры, лежащие внутри этих
пределов. Давление
является фактором скорости процесса и
фактором смещения равновесия, однако
на практике предпочитают работать при
давлениях, близких к атмосферном. Время
контакта
выбирается,
исходя из максимально достижимой
конверсии. Поэтому за вр. реакц. выбир.
то min
t
при котором степень превращения конверсии
практически близка равновесной. Обычно
эта величина равна 90-95 %, а соответствующее
ей время – несколько секунд. Катализаторы:
1.
пентооксид ванадия V2O5,
модифицированный оксидами щелочных
Ме, нанесенных на оксид кремния (старый).
2.
сульфованадат натрия, нанесенный на
диатомид (СВД-новый). 3.
бария алюмованадат – новый. Катализатор
не активен до t
400. Выше 600С происходит термическая
дезактивация катализ. Оптимальные t
работы катализ. 400-600С. Соотношение
O2:SO2
является
фактором смещения равновесия, а также
фактором скорости процесса в соответствии
с уравнением Борескова:r
= k
* [
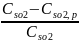 ]0,8
*
Сso2.
Технология
контактного окисления SO2Полочный
контактный аппарат:Р8Обжиговый
газ поступает в нижнюю часть. Кварц –
для равномерного распределения потока.
Газ последовательно проходит полки со
слоями катализатора, где потом идет
окисление SO2→SO3 и одновременно
разогревается от 300 0С внизу до 800С–900С
вверху за счет экзотермичности. Степень
окисления SO2→SO3 в полочном аппарате
достигает 90%. Для доокисления SO2→SO3
используется второй контактные аппарат,
включенный в схему. Недостаток-невозможность
терморегуляции. Схема с двумя контактными
аппаратами позволяет частично
минимизировать недостаток полочного
аппарат.Важнейшая
задача произ-ва H2SO4-увеличение
степени превращения диоксида серы и
снижения его выбросов в атмосф. Один из
наиболее рациональных методов решения
этой задачи-метод двойного контактирования
и двойной абсорбции (ДКДА): реакционную
смесь, в которой степень превращения
SO2 составляет 90 – 95%, охлаждают и направляют
в промежуточный абсорбер для выделения
SO3; в оставшемся реакционном газе
соотношение O2 : SO2 существенно повышается,
что приводит к смещению равновесия
реакции вправо. Вновь нагретый реакционный
газ опять подают в контактный аппарат,
где на достигается 95% степень превращения
оставшегося SO2. Суммарная степень
превращения SO2 в таком процессе достигает
99,5 – 99,8%. Адсорбция
триоксида серы. H2SO4+H2O=H2SO4+Q.
Абсорбция триоксида серы – последняя
стадия процесса на которой образуется
серная кислота. Взаимодействие SO3
с водой протекает достаточно интенсивно
как в жидкой, так и в газовой фазе. Кроме
того, H2SO4
может растворять в себе SO3,
образуя олеум. Этот продукт удобен для
транспортировки, поскольку он не вызывает
коррозии даже обычных сталей. В то же
время растворы серной кислоты чрезвычайно
агрессивны.Р9
Схема
отделения абсорбции в производстве
серной кислоты.
1-олеумный
абсорбер;
2-моногидратный
абсорбер;
3-холодильник;
4-сборники.
Газ, содержащий SO3
после реактора проходит последовательно
олеумый (1) и моногидратный (2) абсорберы.
Другой компонент реакции (H2O)
подается противотоком через сборник в
моногидратный абсорбер. Кислота оттуда
поступает в олеумный. В нем циркулирует
20% раствор Н2SO4,
который частично отбирается как конечный
продукт – олеум. Кислота из предыдущего
абсорбера – моногидрат – также может
быть продуктом. В системе циркуляции
предусмотрены холодильники для съема
тепла реакции и обеспечения более
эффективной абсорбции. При температурах
меньше 100°С SO3
поглощается практически полностью,
диоксид серы – практически не поглощается.
Нитрозный
способ (башенный) производства серной
кислоты Р101.Башня-денитратор
2.Башня-концентратор
3.Продукционная
башня 4.Окисленная
башня 5-7.
Асборбционные
башни 8.Сборники
9.Электрофильтр
10.Трубы
для отходящих газов 11.Холодильники
12.Компрессор.
Около
1/3 обжигового газа поступает в 1, остальные
2/3 в 2. На выходе из башни 1 и 2 газовые
потоки объединяются и идут в 3. Затем в
4 и последовательно в 5, 6, 7. Между 6 и 7
поставлен трубокомпрессор. В 9 происходит
отделение тумана мелких капелек серной
кислоты и газ идет в трубу на выброс в
атм. В денитраторе происходит гидролиз
нитрозилсерной кислоты с образованием
серной и азотистой кислот:
HNSO5+H2O↔H2SO4+HNO2–Q (эндо) Одновременно: SO2 +
H2O↔H2SO3+Q (экзо) Окисление сернистого газа
протекают преимущ. в жидкой фазе:
H2SO3(ж.)+HNO3(ж.)↔H2SO4(ж.)+NO(г.)+Q Недостаток:
недостаточная концентр.2NO+O2↔2NO2;
NO:
NO2=1:1.
Азотистый ангидрид NO+NO2=N2O3.
Растворяясь в H2SO4,
орошается в абсорбционные колонные
5-7, образуя нитрозил, который прекрасно
растворяется в серной кислоте , давая
нитрозу, поступающую в 1.N2O3+H2SO4↔2HNSO5+H2O+Q.
Недостаток
метода:разбавленная
кислота. Достоинство: нет очистки
обжигового газа.
Пр-во
серной кислоты по методу «мокрого
катализа» Позволяет
утилизировать газы сероочистки,
получаемые при очистке коксового газа,
газов нефтепереработке, которые очень
богаты H2S
(85 – 95%). Это экологически выгодно. Около
10% серной кислоты получают по этому
методу. Процесс состоит из 3х стадий:
Стадия
1: Сжигание
H2S до SО2 :2H2S+3O2=2H2O+1038 кДж. Стадия
2: Контактное
окисление SO2→SO3 (любой контактный
аппарат)Стадия 3: Улавливание SO3 с
образованием H2SО4. Достоинства: получается
высокочистая серная кислота и используются
отходы производства.
Перспективы развития сернокислотных
пр-в.
Мощным средством повышения производительности
сернокислотных производств является
увеличение концентрации диоксида серы.
Высококонцентрированные газы, содержащие
до 80% SO2
уже начали получать в производствах
цветных Ме из их сульфидных руд с
применением технического кислорода.
Получение высококонц. сернистого газа
позволяет создать энерготехнологические
циклические производства серной кислоты
из серы и колчедана. Диоксид серы,
полученный с применением технического
кислорода окисляют на 90% в контактном
аппарате с «кипящем слоем» катализатора.
При абсорбции SO3
получают высококонцентрированный олеум
и моногидрат. Газ после абсорбции
возвращают на контактирование. В реакторе
общая степень превращения составляет
99,995%. Для отвода накапливающегося в
результате многократного рецикла азота
часть газа после абсорбции пропускают
через малогабаритную сернокислотную
установку, из которой азот выбрасывается
в атмосферу. Интенсивность работы
циклической системы, работающей под
давлением около 1 МПа, с применением
кислорода в десятки раз превышает
интенсивность обычных систем. Потери
серы с отходящими газами и соответственно
выбросы SO2
и SO3
в окружающую среду также снижены в
десятки раз. Схемы предусматривают
генерирование водяного пара (4 Мпа) за
счет тепла газов обжига, который может
быть использован как в самой установке
для компенсации затрат энергии на работу
компрессоров и насосов, так и в других
цехах завода. Тепло сернистых газов
после прохождения очередного
каталитического слоя можно использовать
для предварительного подогрева реагентов
на входе в контактный аппарат. Тепло
сорбции используется для бытовых нужд.
Важнейшие
направления развития пр-ва серной ки-ты.
1.Увеличение
мощности аппаратуры при одновременной
комплексной автоматизации производства.
2.Интенсификация
процессов путем применения реакторов
«кипящего слоя» (как на стадии обжига,
так и при контактном окислении SO2),
более активных катализаторов, повышенных
давлений и использование технического
кислорода в процессе окисления.
3.Разработка
энерготехнологических схем с максимальным
использованием теплоты экзотермических
реакций, в том числе циклических систем
под давлением. 4.Увеличение
степеней превращения на всех стадиях
производства для снижения расходных
коэффициентов по сырью и материалам и
снижение вредных выбросов.
5.Утилизация
вредных выбросов из отходящих газов, а
также тв отходов (огарок). Пр-во
HNO3-
представляет собой жидкость с p=1,52 г/см3,
tкип.=86ºС, tзам.= -46ºС. При нагревании
разлагается: 4HNO3 = NO2 +4NO + 2H2O. В
промышленности выпускают 2 сорта HNO3:
Разбавленную с концентрацией 45-60%;
Концентрир.-96-98,5%. Меланж-концент. HNO3
+10% концентр. H2SO4.
Технологические
схемы производства делятся на 2 группы:
1. Пр-во при атмосферном давлении –
разбав-ая. HNO3;
2.В ряде предусмотрено ее концентрирование
– концентр. HNO3.
Пр-во
разбавл. HNO3.
Физико-химические основы метода.1.
4NН3+ 5О2→ 4NO+6H2O+907 кДж; 2. NО2 + H2O +1100 кДж;
3. N2 + H2O +1210 кДж; 4. NН3+ NО →N2+H2O+1870 кДж
диспропорц. Непревзойденным по
селективности катализатором для 1-ой
ре-ции явл. катализатор на основе платины
или ее сплавов. Селективность повышается
до 98%. Др. катализаторами явл. оксидные
катализаторы на основе Fe,
Ni,
Cr,
Co.
Эти катализаторы, обладая несомненными
+: более низкой стоимостью, уступают Pt
по селективности. С точки зрения
удешевления процесса пользуются
двухступенчатой схемой: 1
стадия–катализатор–платина; 2
стадия–катализатор–оксид металла.
Каталитическое
окисление аммиака на платине.
Р11Процесс
каталит. окисл. начинается с активирования
адсорбции О2
на поверхности kt.
После этого следует адсорбция NH3
с образованием промежуточного активного
комплекса с перераспределением связи
с высвобождением NO и воды. Чистая Pt
быстро разрушается при высокой
температуре. Используется Pt-Pj –kt, в
котором содержание полония 5-10%. По мере
эксплуатации гладкая поверхность kt
сетки становится матовой пористой, т.е.
увеличивается поверхность kt
и его активность. Pt
отравляется необратимо примесями
воздуха, аммиаком, фосфином и сероводородом.
Срок службы 1,5 года. Кроме хим. отравлений
поверхность изнашивается механически,
и мелкая пыль kt
уносится с потоком газа. Из-за дороговизны
kt
ставят пылеуловители. Исп. двухступенчатых
kt
позволяет достичь на 1 ступени степени
конверсии 87-90%, а после 2-ух ступеней, где
вторая – MeO kt,
степень конверсии 97%. Сам процесс
окисления аммиака О2
на практике начинается уже при 140ºС, но
при этом образуется N2,
N2O,
кол-во NO, наоборот, незначительно. С
ростом t
процесса увеличивается и выход NO. Окисл.
NO в NO2. 2NO
+ O2 ↔ 2NO2;
]0,8
*
Сso2.
Технология
контактного окисления SO2Полочный
контактный аппарат:Р8Обжиговый
газ поступает в нижнюю часть. Кварц –
для равномерного распределения потока.
Газ последовательно проходит полки со
слоями катализатора, где потом идет
окисление SO2→SO3 и одновременно
разогревается от 300 0С внизу до 800С–900С
вверху за счет экзотермичности. Степень
окисления SO2→SO3 в полочном аппарате
достигает 90%. Для доокисления SO2→SO3
используется второй контактные аппарат,
включенный в схему. Недостаток-невозможность
терморегуляции. Схема с двумя контактными
аппаратами позволяет частично
минимизировать недостаток полочного
аппарат.Важнейшая
задача произ-ва H2SO4-увеличение
степени превращения диоксида серы и
снижения его выбросов в атмосф. Один из
наиболее рациональных методов решения
этой задачи-метод двойного контактирования
и двойной абсорбции (ДКДА): реакционную
смесь, в которой степень превращения
SO2 составляет 90 – 95%, охлаждают и направляют
в промежуточный абсорбер для выделения
SO3; в оставшемся реакционном газе
соотношение O2 : SO2 существенно повышается,
что приводит к смещению равновесия
реакции вправо. Вновь нагретый реакционный
газ опять подают в контактный аппарат,
где на достигается 95% степень превращения
оставшегося SO2. Суммарная степень
превращения SO2 в таком процессе достигает
99,5 – 99,8%. Адсорбция
триоксида серы. H2SO4+H2O=H2SO4+Q.
Абсорбция триоксида серы – последняя
стадия процесса на которой образуется
серная кислота. Взаимодействие SO3
с водой протекает достаточно интенсивно
как в жидкой, так и в газовой фазе. Кроме
того, H2SO4
может растворять в себе SO3,
образуя олеум. Этот продукт удобен для
транспортировки, поскольку он не вызывает
коррозии даже обычных сталей. В то же
время растворы серной кислоты чрезвычайно
агрессивны.Р9
Схема
отделения абсорбции в производстве
серной кислоты.
1-олеумный
абсорбер;
2-моногидратный
абсорбер;
3-холодильник;
4-сборники.
Газ, содержащий SO3
после реактора проходит последовательно
олеумый (1) и моногидратный (2) абсорберы.
Другой компонент реакции (H2O)
подается противотоком через сборник в
моногидратный абсорбер. Кислота оттуда
поступает в олеумный. В нем циркулирует
20% раствор Н2SO4,
который частично отбирается как конечный
продукт – олеум. Кислота из предыдущего
абсорбера – моногидрат – также может
быть продуктом. В системе циркуляции
предусмотрены холодильники для съема
тепла реакции и обеспечения более
эффективной абсорбции. При температурах
меньше 100°С SO3
поглощается практически полностью,
диоксид серы – практически не поглощается.
Нитрозный
способ (башенный) производства серной
кислоты Р101.Башня-денитратор
2.Башня-концентратор
3.Продукционная
башня 4.Окисленная
башня 5-7.
Асборбционные
башни 8.Сборники
9.Электрофильтр
10.Трубы
для отходящих газов 11.Холодильники
12.Компрессор.
Около
1/3 обжигового газа поступает в 1, остальные
2/3 в 2. На выходе из башни 1 и 2 газовые
потоки объединяются и идут в 3. Затем в
4 и последовательно в 5, 6, 7. Между 6 и 7
поставлен трубокомпрессор. В 9 происходит
отделение тумана мелких капелек серной
кислоты и газ идет в трубу на выброс в
атм. В денитраторе происходит гидролиз
нитрозилсерной кислоты с образованием
серной и азотистой кислот:
HNSO5+H2O↔H2SO4+HNO2–Q (эндо) Одновременно: SO2 +
H2O↔H2SO3+Q (экзо) Окисление сернистого газа
протекают преимущ. в жидкой фазе:
H2SO3(ж.)+HNO3(ж.)↔H2SO4(ж.)+NO(г.)+Q Недостаток:
недостаточная концентр.2NO+O2↔2NO2;
NO:
NO2=1:1.
Азотистый ангидрид NO+NO2=N2O3.
Растворяясь в H2SO4,
орошается в абсорбционные колонные
5-7, образуя нитрозил, который прекрасно
растворяется в серной кислоте , давая
нитрозу, поступающую в 1.N2O3+H2SO4↔2HNSO5+H2O+Q.
Недостаток
метода:разбавленная
кислота. Достоинство: нет очистки
обжигового газа.
Пр-во
серной кислоты по методу «мокрого
катализа» Позволяет
утилизировать газы сероочистки,
получаемые при очистке коксового газа,
газов нефтепереработке, которые очень
богаты H2S
(85 – 95%). Это экологически выгодно. Около
10% серной кислоты получают по этому
методу. Процесс состоит из 3х стадий:
Стадия
1: Сжигание
H2S до SО2 :2H2S+3O2=2H2O+1038 кДж. Стадия
2: Контактное
окисление SO2→SO3 (любой контактный
аппарат)Стадия 3: Улавливание SO3 с
образованием H2SО4. Достоинства: получается
высокочистая серная кислота и используются
отходы производства.
Перспективы развития сернокислотных
пр-в.
Мощным средством повышения производительности
сернокислотных производств является
увеличение концентрации диоксида серы.
Высококонцентрированные газы, содержащие
до 80% SO2
уже начали получать в производствах
цветных Ме из их сульфидных руд с
применением технического кислорода.
Получение высококонц. сернистого газа
позволяет создать энерготехнологические
циклические производства серной кислоты
из серы и колчедана. Диоксид серы,
полученный с применением технического
кислорода окисляют на 90% в контактном
аппарате с «кипящем слоем» катализатора.
При абсорбции SO3
получают высококонцентрированный олеум
и моногидрат. Газ после абсорбции
возвращают на контактирование. В реакторе
общая степень превращения составляет
99,995%. Для отвода накапливающегося в
результате многократного рецикла азота
часть газа после абсорбции пропускают
через малогабаритную сернокислотную
установку, из которой азот выбрасывается
в атмосферу. Интенсивность работы
циклической системы, работающей под
давлением около 1 МПа, с применением
кислорода в десятки раз превышает
интенсивность обычных систем. Потери
серы с отходящими газами и соответственно
выбросы SO2
и SO3
в окружающую среду также снижены в
десятки раз. Схемы предусматривают
генерирование водяного пара (4 Мпа) за
счет тепла газов обжига, который может
быть использован как в самой установке
для компенсации затрат энергии на работу
компрессоров и насосов, так и в других
цехах завода. Тепло сернистых газов
после прохождения очередного
каталитического слоя можно использовать
для предварительного подогрева реагентов
на входе в контактный аппарат. Тепло
сорбции используется для бытовых нужд.
Важнейшие
направления развития пр-ва серной ки-ты.
1.Увеличение
мощности аппаратуры при одновременной
комплексной автоматизации производства.
2.Интенсификация
процессов путем применения реакторов
«кипящего слоя» (как на стадии обжига,
так и при контактном окислении SO2),
более активных катализаторов, повышенных
давлений и использование технического
кислорода в процессе окисления.
3.Разработка
энерготехнологических схем с максимальным
использованием теплоты экзотермических
реакций, в том числе циклических систем
под давлением. 4.Увеличение
степеней превращения на всех стадиях
производства для снижения расходных
коэффициентов по сырью и материалам и
снижение вредных выбросов.
5.Утилизация
вредных выбросов из отходящих газов, а
также тв отходов (огарок). Пр-во
HNO3-
представляет собой жидкость с p=1,52 г/см3,
tкип.=86ºС, tзам.= -46ºС. При нагревании
разлагается: 4HNO3 = NO2 +4NO + 2H2O. В
промышленности выпускают 2 сорта HNO3:
Разбавленную с концентрацией 45-60%;
Концентрир.-96-98,5%. Меланж-концент. HNO3
+10% концентр. H2SO4.
Технологические
схемы производства делятся на 2 группы:
1. Пр-во при атмосферном давлении –
разбав-ая. HNO3;
2.В ряде предусмотрено ее концентрирование
– концентр. HNO3.
Пр-во
разбавл. HNO3.
Физико-химические основы метода.1.
4NН3+ 5О2→ 4NO+6H2O+907 кДж; 2. NО2 + H2O +1100 кДж;
3. N2 + H2O +1210 кДж; 4. NН3+ NО →N2+H2O+1870 кДж
диспропорц. Непревзойденным по
селективности катализатором для 1-ой
ре-ции явл. катализатор на основе платины
или ее сплавов. Селективность повышается
до 98%. Др. катализаторами явл. оксидные
катализаторы на основе Fe,
Ni,
Cr,
Co.
Эти катализаторы, обладая несомненными
+: более низкой стоимостью, уступают Pt
по селективности. С точки зрения
удешевления процесса пользуются
двухступенчатой схемой: 1
стадия–катализатор–платина; 2
стадия–катализатор–оксид металла.
Каталитическое
окисление аммиака на платине.
Р11Процесс
каталит. окисл. начинается с активирования
адсорбции О2
на поверхности kt.
После этого следует адсорбция NH3
с образованием промежуточного активного
комплекса с перераспределением связи
с высвобождением NO и воды. Чистая Pt
быстро разрушается при высокой
температуре. Используется Pt-Pj –kt, в
котором содержание полония 5-10%. По мере
эксплуатации гладкая поверхность kt
сетки становится матовой пористой, т.е.
увеличивается поверхность kt
и его активность. Pt
отравляется необратимо примесями
воздуха, аммиаком, фосфином и сероводородом.
Срок службы 1,5 года. Кроме хим. отравлений
поверхность изнашивается механически,
и мелкая пыль kt
уносится с потоком газа. Из-за дороговизны
kt
ставят пылеуловители. Исп. двухступенчатых
kt
позволяет достичь на 1 ступени степени
конверсии 87-90%, а после 2-ух ступеней, где
вторая – MeO kt,
степень конверсии 97%. Сам процесс
окисления аммиака О2
на практике начинается уже при 140ºС, но
при этом образуется N2,
N2O,
кол-во NO, наоборот, незначительно. С
ростом t
процесса увеличивается и выход NO. Окисл.
NO в NO2. 2NO
+ O2 ↔ 2NO2;
![]() ,
𝐾𝑝
сильно
зависит от t.
Скорость понижается при повышении t.
Скорость окислен. NO до NO2 явл. самой
низкой из всех стадий. При охлажд. может
протекать димеризация NO2 : 2NO2↔ N2O4 +57
кДж. Данная ре-ция протекает с высокой
скоростью. Со снижением t
степень димеризации снижается. Абсорбция
NO2: Если
суммировать: 3NO2 + H2O →2HNO3+NO + 136 кДж. Механизм
ре-ции:
Сначала NO2 из газовой фазы через
пограничный слой диффундирует у
поверхности жидкости и абсорбируется
ею. Затем идет взаимодействие NO2 с Н2О.
HNO2 разлагается сравнительно медленно,
NO плохо растворим в водных растворах
HNO3, поэтому выходит в газовую фазу,
покидая ра-р. Концентр. получающейся
HNO3 определяется условием равновесия
NO2 над ра-ром HNO3. С ростом t,
растет
и
одновременно снижается разница, т.е.
движущаяся сила процесса, т.о. понижается
конц. продукц. HNO3 и наоборот. Получение
разбавленной азотной кислоты при
атмосферном давлении.Р12
Воздух через заборную трубу проходит
через водяной скруббер 1, суконный фильтр
2, где очищается от пыли и аэрозолей.
Очищенный воздух и аммиак направляются
в вентилятор 3, где смешиваются так, что
конц. NH3
достигает 5-10% объема и направляется на
картонный фильтр 4, где окончательно
очищается от пыли и капельной влаги и
затем направляется в реактор 5, назыв.
конвертором. В верхней части конвертора
находятся Pt-Po сетки, где протекает окисл.
NH3
O2
воздуха, так что степень превращения
NH3→NO достигает 97-98%/. На выходе из
конвертора t
отходящих газов достигает 800ºС. Конвертор
смонтирован на котле-утилизаторе, где
происходит в результате исп. тепла от
отходящих газов получение водяного
пара с р=4МПа и t=450ºС,
после котла-утилизатора t
отходящих газов снижается до 160 ºС и
дальнейшее их охлаждение происходит в
водяных холодильниках 7 и 8. Здесь частично
конденсируются водяные пары. В
незначительной степени протекает
окисление NO в NO2 и может получаться ≈5%
HNO3. В холодильнике 8 за счет более низкой
t
получается более конц. HNO3 ≈30%. Окончательно
охлажденный газ вентилятором 9 направл.
в абсорбционную систему, сост. из 6-8
поглотительных башен 10, орошаемых HNO3,
где продолжается превращение NO в NO2,
одновременно NO2 взаимодействует с
раствором HNO3, повышает ее концентрацию.
Для поглощения NO2 в последнюю башню
подают на орошение воду, а образовавшуюся
ки-ту, противотоком направляют от
последних башен в первую. Газ (NO)
направляется в окислительную башню 11
для завершения окисления в NO2 и далее
в башню щелочной абсорбции 12, орошаемую
ра-ром соды, либо известковой водой. Там
происходит поглощение нитрозных газов.
NO2+NO+ Na2CO3→2NaNO2 =CO2↑; NO2+NaCO3→NaNO2+NaNO3+CO2↑
Очищен. газы идут на выброс в атмосферу,
а нитрит-нитратные ра-ры на переработку.
Получение
концентрированной HNO3.
Концентрирование. Мах
tкип.=121ºС достигается при конц. 68,4% HNO3.
В этой точке (азиотроп) состав паров
совпадает с составом жидкой фазы. Для
конц. выше 68,4% необходимо исп. перегонку
разбавл. HNO3
с добавлением конц. H2SO4.
В этом случае получ. гидраты H2SO4
кипят при более высокой t,
чем конц. HNO3.
Перегонку разбавл. HNO3
ведут с 92-94%H2SO4.
Процесс осущ. в тарельчатых барабанных
колоннах или колоннах с насадками из
колец (Раминга), изготовленных из
кислотоупорного чугуна (ферросилида),
с содержанием Si-12-18%. Р131-Тарельчатая
барботажная колонна;2-Испаритель разбавл.
HNO3; 3-Холодильник продукции конц. HNO3;
4-Конденсатор для охлаждения нитрозных
газов. Пары HNO3 с незначительным содержанием
паров H2O и отжига N из колонны направляются
в конденсатор, где конденсируется HNO3,
а нитрозные газы идут на дальнейшее
улавливание. Часть нитрозных ↑
растворяется в HNO3, поэтому из конденсатора
HNO3 возвращается на верхние участки
колонны, где она отдувается от оксидов
и как продукция отходится в холодильник.
Отработанная H2SO4 (≈70%) отходит в нижней
части колонный и без охлаждения
направляется на упаривание.
Концентрирование
с Mg(NO3)2.
В отпарную колонну барабанного типа
сверху подается 80% р-р Mg(NO3)2, нагретый до
160 – 180ºС, а ниже в колонну подается
разбл. 55% HNO3. В части колонны с помощью
кипятильника поддерживается t=160-180ºС.
Выходящие из колонны газы 97% HNO3 направляются
в дистилляционную колонну, оттуда пары
конц. HNO3 с t=86
ºС поступ. в конденсатор, а далее на
охлаждение. Выделен. в нижней части
колонны (≈70%) HNO3 возвращ. в отпарную
колонну, а разбавл. ра-р Mg(NO3)2 (≈55-60%) из
нижней части отпарной колонны отправл.
на концентрирование после предварительного
нагрева в вакуум испарители, откуда 80%
Mg(NO3)2 направляется на орошение отпарной
колонны.Прямой
синтез конц. HNO3.
Из нитрозных газов получают NO2 абсорбцией
конц. ки-той. При низких t=
-10ºС, с получение нитроолеума: НNO3 + n NO2
→HNO3 ∙ n NO2. Из нитроолеума в отбелочных
колоннах при t=80ºС,
высвобождается газообразный NO2,
направляется на конденсацию охлаждения
до -8 ºС. При этом происходит димеризация
NO2 с получением N2O4. Жидкий N2O4 смешивают
со слабой HNO3
(55-65%) и направляют в автоклав, под p, куда
доп. подается O2.
В автоклаве при t=90ºС
и p=5
Мпа протекает ре-ция: 2N2O4(ж.) + 2H2O(ж.) +
O2(г.) → HNO3(ж.) + 59,5 кДж. Для смещения
равновесия вправо необходим избыток
N2O4 – по стехим. – 100% (в 2 раза). Из автоклава
конц. HNO3, содерж. еще и 25% NO2 и вместе с
олиумом подается в отбелочную колонну.
Там ки-та освобождается от растворенного
NO2 и частично выводится в виде продукционной
97-98%, а частично для получ. нитролиума.
Пр-во
фосфорной кислоты. Функциональная
схема производства экстракционной
H3PO4.
Хорошо
исследованным явл. сернокислотное
разложение апатитового концентрата,
получ. обогащением хибинской
апатито-нефелиновой руды и содерж. ≈39%
Р2О5. Его разложение – гетерогенная
ре-ция “ж–тв тело”. Ca5F(PO4)3+5H2SO4+5nH2О=3H3PO4+
5CaSO4*nH2O(↓)+HF
(↑). Кальциевая составляющая выпадает
в ↓ в виде гипса (CaSO4), вторая уходит в ↑
в виде HF.
Функциональная схема
производства ЭФК включ. разложение
измельченного сырья в реакторе
(экстракторе), фильтрацию тв ↓, упаривание
H3PO4
до товарной конц. и очистку отходящих
↑. Образ. HF
взаимод. с пустой породой сырья.
SiO2+4HF=SiF4(газ)+2H2O. Поэтому при промывке
отходящих ↑ H2O
оба летучих продукта улавливаются в
виде кремнефтористоводородной ки-ты
H2SiF4. SiF4+2HF=H2SiF6. Р14Функциональная
схема производства экстракционной
H3PO4:
1 – реактор разложения апатита
(экстрактор); 2 – вакуум фильтр; 3 –
сборник фильтратов; 4 – колона выпарки
H3PO4;
5 – система очистки ↑.
Сернокислотное разложение апатита. С
растворением апатита происходит образ.
тв CaSO4, который плохо растворим. Связывание
Са2+ в CaSO4 должно увеличивать скорость
растворения апатита. К конц. Н2SO4 в
реакторе предъявляются жесткие требов.:
строгое поддержание этой конц. на
оптимальном уровне во всем V
реакционной зоны. Реакционный
узел -
последовательность секций с интенсивным
перемешиванием реагентов в каждой из
них и перетоком между ними для организации
движения реакционной массы по каскаду.
В первых секциях происходит растворение
апатита. Остальные секции работают как
дозреватели, в них формируются кристаллы
CaSO4. Между последней и 1-ой секциями
организован интенсивный рецикл.
Образование
продукционной
H3PO4.
Включает
отделение ↓ CaSO4 (фосфогипса) и
концентрирование кис-ты. В зависимости
от условий разложения образ. кристаллы
CaSO4·2Н2О (дигидрат) или CaSO4·0,5Н2О (полугидрат).
В дигидратном процессе t 343-353 К и конц.
H3PO4
25-35 %. В полугидратном процессе эти
показатели составляют 358-378 К и 35 мас.%.
Фосфогипс отделяют на вакуум-фильтре.
Фильтрат направляют на выпаривание,
где конц. ки-ты увелич. до 52-54%. Это и есть
продукционная ки-та. При выпаривании
из ки-ты удаляется 80-90% F
в виде HF. Отходящие из реактора и из узла
выпаривания фторсодержащие ↑ в системах
очистки поглощаются с образов.
кремнефтористоводородной ки-ты.
2Ca3(PO4)2+
10CO+6SiO2=P4+10CO2+6CaSiO3; P4+5O2=P2O5; P2O5+3H2O= 2H3PO4. Пр-во
минеральных удобрений (МУ).
Классификация
МУ.
МУ классифиц. по 3 главным признакам –
агрохим. назначению, составу и св-вам.
МУ: 1.по агрохимич.
назначению:1.1прямые(простые:(а)азотные,
б)фосфорные,в)калийные);комплексные:а)двойные,б)тройные,в)смешанные,г)сложные);1.2косвенные;2.по
св-вам;3.по составу (по агрегатному
состоянию):тв и ж. Фосфорные
удобрения (ФУ).
Один из методов механической обработки
- измельчение фосфатов. Полученная
фосфористая мука при исп. в кислых почвах
медленно растворяется в почвенных Н2О
и становится долговременно действующим
удобрением. ФУ могут быть получены
термическим разложением фосфатов при
t=1200
– 1800ºС. Пр-во
простого суперфосфата.
2Ca3F(PO4)3+7H2SO4+6,5H2O= 3[Ca(H2PO4)2*
H2O]+7[CaSO4*0,5H2O]+2HF+227,4кДж.(1) Разложение
протекает в 2 стадии.
На
1-ой стадии ≈70%
апатита реагирует с H2SO4.
При этом образ. H3PO4
и полугидрат CaSO4
(1). Эта стадия характер. образов.
поверхностных пленок CaSO4 на зернах
фосфата. 1-ая стадия заканчивается через
20–40 минут после смешения фосфата с
H2SO4.
После
полного исчезновения H2SO4
начинается 2-ая стадия разложения, в
которой оставшийся апатит (~30%) разлагается
H3PO4:
Ca5F(PO4)3+7
H3PO4+5H2O=5[Ca(H2PO4)2*H2O]+HF (2). Образ. монокальцийфосфат
в отличие от CaSO4
не сразу выпадает в↓. Он постепенно
насыщает р-р H3PO4,
после чего начинает выкристаллизовываться
в виде Ca(H2PO4)2*H2O и протекает значительно
медленнее, чем реакция (1). Она начинается
в суперфосфатных камерах и длится в
течение 5–20 суток хранения суперфосфата
на складе. Пр-во
двойного суперфосфата.
Двойной суперфосфат – конц. фосфорное
удобрение. Получ. разложением природных
фосфатов H3PO4.
Оно содержит 42 – 50% усвояемого P2O5. В
отличие от последнего двойной суперфосфат
почти не содержит балласта – сульфата
кальция. Процесс протекает по уравнению
(2). Азотнокислое
разложение фосфатов. Получение сложных
удобрений.Ca5F(PO4)3+10HNO3=
3H3PO4+5Ca(NO3)2+HF (3). В результате образуется
азотнокислая вытяжка, р-р, содержащий
Ca(NO3)2 и свободную H3PO4.
Сущ. ряд методов дальнейшей переработки
азотнокислой вытяжки. Во многих процессах
вытяжку нейтрализуют NH3
получая фосфаты аммония (NP-удобрения).
Если перед гранулированием нейтрализованной
пульпы к ней добавляют соли калия (KCl,
K2SO4), то получают тройное NPK-удобрение –
нитроаммофоску. Азотнокислый метод
разложения фосфатов позволяет наряду
с получением NPK-удобрений попутно
выделять из сырья стронций, редкоземельные
элементы и др., находящие применение в
различных отраслях промышленности.
Пр-во
азотных удобрений. Аммиачная
селитра, карбамид, сульфат аммония,
водные растворы аммиака и др. Азотные
удобрения отличаются друг от друга по
содержанию азота, по форме соединений
азота (нитратные, аммонийные, амидные),
фазовому состоянию (твердые и жидкие).
Пр-во
аммиачной селитры.Р15
Реактор-нейтрализатор
ИНТ
1-реакционный
стакан; 2-барбатер
NH3;
3-барбатер
HNO3;
4-колпачковые
тарелки; 5-каплеотбойник.
Аммиачная селитра или нитрат аммония
(NH4NO3) – кристаллическое ве-во белого
цвета, содерж. 35% N
в аммонийной и нитратной формах, обе
формы легко усваиваются растениями.
Гранулированную аммиачную селитру
применяют в больших масштабах перед
посевом и для всех видов подкормок. В
меньших масштабах ее исп. для производства
взрывчатых ве-в. Для получения аммиачной
селитры применяют ряд технологических
приемов. Во-первых, гранулирование. В
основе процесса производства аммиачной
селитры лежит гетерофазная ре-ция
взаимод. NH3↑
с ра-ром HNO3.
В настоящее время наиболее распространены
схемы с частичным упариванием ра-ра за
счет тепла нейтрализации. Нейтрализацию
осущ. в аппарате ИТН (использование
тепла нейтрализации). Аппарат состоит
из 2 цилиндров (внешний и внутренний).
Во внутренний подается NH3↑
и разбрызгивается HNO3.
Внутренняя часть представляет собой
реакционное пространство, внешняя –
зона испарения. Отвод тепла из зоны
ре-ции необходим не только для выпаривания
ра-ра, но и во избежание перегрева и
разложения HNO3
и аммиачной селитры. Соковый пар,
образующийся как результат испарения
H2O
из реакционного ра-ра исп. для подогрева
реагентов и упаривания реакционного
ра-ра. Р16Схема
производства аммиачной селитры с
частичным упариванием ра-ра за счет Q
нейтрализатора.
1-теплообменник
подогрева HNO3;
2-теплообменник
подогрева NH3;
3-сборник;
4-реактор
ИНТ; 5-донейтрализатор;
6-выпарной
аппарат;7-сепаратор;
8-грануляционная
башня;
9-барометрический
конденсатор; 10-транспортер.
Ра-р аммиачной селитры (60-80%) поступает
в донейтрализатор(5). Сюда добавляют NH3
как нейтрализующий агент и ве-ва
уменьшающие слеживаемость удобрений.
Выпарку проводят в 2 или 3 ступени с исп.
в качестве греющих агентов сокового
пара из аппарата ИТН, вторичного пара
выпарки и свежего насыщенного пара. Для
простоты на схеме показан один выпарной
аппарат (6). В выпарной установке плав
доводят до содержания в нем NH4NO3 98 – 99%.
Гранулирование производят путем
разбрызгивания плава в полой железобетонной
башне (8) высотой 30–35 м. Падающие капли
застывают в гранулы в потоке холодного
воздуха, поступающего противотоком с
помощью вентиляторов. Окончательная
сушка осущ. горячим воздухом во вращающемся
сушильном барабане.
Электрохим.
Пр-ва. Теоретические основы электролиза
водных растворов и расплавленных сред.
Технология электролиза раствора хлорида
натрия.
Основные
направления применения электрохимических
производств. При
электролизе водных ра-ров и расплавленных
сред могут быть получены хим. продукты:
H2, CI2,KOH
и NaOH.
Методом электролиза водных ра-ров осущ.
энергохим. синтез многих неорган. и
орган. ве-в – гипохлоритов, хлоратов,
перхлоратов, HCI,
перманганатов, диоксида марганца, а
также адипонитрила, антидетонаторов.
На электролизе водных ра-ров основаны
гидроэлектрометаллургия и гальванотехника.
Гидроэлектрометалургия
–извлечение Ме из водных ра-ров их солей
путем электролиза. Важнейшая
область электрохимии–получение
и эксплуатация хим. источников тока.
Хим. источники тока подразделяются на
первичные и вторичные. Первичные:
источники энергии, которые могут быть
исп. лишь однократно: батареи и
гальванические элементы. Вторичные:
аккумуляторы, работоспособность которых
восстанавливается пропусканием
электрического тока. Электрохим. методы
все более широко внедряются в технику
очистки сточных вод, Преимущества
электрохим. методов.
1.
В
электрохим. процессах аппаратура во
многих случаях проще и компактнее по
сравнению с альтернативными хим.
процессами; 2.
Электрохим.
методы получения ряда продуктов
характеризуются меньшим числом
производственных стадий и операций,
более дешевым сырьем и большей глубиной
его превращения, одновременным
образованием (причем в раздельном виде)
ценных продуктов.
3.
Чистота получаемых продуктов. Основной
– электрохим. процессов – высокая
энергоемкость, поэтому энергетические
затраты составляют главную статью
себестоимости продуктов. Электролиз
раствора хлорида натрия. Электролиз
ра-ра NaCl – наиболее простой и экономичный
метод одновременного получения 3
важнейших хим. продуктов-CI2,
H2 и NaOH
с использованием дешевого и доступного
природного сырья. CI2
применяют как сырье для производства
хлорорганических растворителей и
пластмасс, синтетических каучуков,
химических волокон, ядохимикатов. В
металлургии: для хлорирующего обжига
руд, в текстильной и целлюлозно-бумажной
промышленности – для очистки и отбеливания
целлюлозы, бумажной массы и тканей.
Большие кол-ва Cl2
идут на очистку и стерилизацию сточных
вод и питьевой H2O.
2NaCl+2H2O=2H2+2NaOH+Cl2. Гидроксид натрия
используется в производстве многих
химических продуктов, прежде всего в
промышленном органическом синтезе, в
целлюлозно-бумажном производстве, в
производстве искусственных волокон, в
металлургии (пр-во алюминия), в
нефтехимической промышленности и др.
Электролиз
раствора NaCl осуществляется двумя
методами:1)электролиз
с твердым катодом и фильтрующей
диафрагмой;2) электролиз без диафрагмы
с жидким ртутным катодом. Электролиз
раствора NaCl с твердым катодом и фильтрующей
диафрагмой.
К:
2H2O+2e=2OH(-)+2H(адс)(1);
2Н(адс)=Н2.
Материалами для катода служит сталь,
на которой водород выделяется с
относительно невысоким перенапряжением
(0,3 В). Снижение потенциала до 0,3 – 0,4 В
можно достичь применением пористых
графитовых катодов, для упрочнения и
гидрофобизации пропитанных
политетрафторэтиленом и активированных
солями меди или серебра. Накапливающиеся
в катодном пространстве гидроксид-ионы
образуют нейтральные молекулы гидроксида
натрия: OH-+Na+=NaOH(3). На аноде выделяется
хлор: A:2Cl(-)-2e=Cl2 (4). Материалом анода служит
оксидно-рутениевая система, обладающая
прочностью и химической инертность по
отношению к кислороду являющемуся
побочным продуктом, образующемся на
аноде: A:4OH(-)-4e=O2+2H2O (5). В объеме электролита
анодного пространства в результате
гидролиза хлора идут побочные химические
реакции: Cl2+H2O↔HOCl+HCl(6),
Cl2+OH(-)↔HOCl+Cl(-)(7),
HOCl+OH(-)=OCl+H2O(8).
Образующийся в результате этой
последовательной реакции гипохлорит
анион претерпевает анодное окисление:
A:6OCl(-)+3H2O+6e
=2OCl3(-)+4Cl(-)+3O2+6H(+)
(9). Электролиз реализуют в электролизерах
непрерывного действия с вертикальными
фильтрующими диафрагмами при противотоке
движения электролита и OH
–ионов.
Р17Схема
электролизитера с фильтрующей диафрагмой:
1-пористая
асбестовая диафрагма; 2-перфорированный
стальной катод; 3-катодное
пространство; 4-оксидно-рутениевый
анод; 5-анодное
пространство. Корпус ванны разделен на
катодное и анодное пространства пористой
диафрагмой из асбеста. Диафрагма плотно
прилегает к перфорированному стеклянному
катоду. В анодном пространстве расположен
оксидно-рутениевый анод. Очищенный
рассол подают в анодное пространство
и вследствие гидростатического давления
он фильтруется через диафрагму и катод
в катодное пространство. Из катодного
пространства непрерывно отводят водород
и раствор гидроксида натрия, а из анодного
– хлор. В образующемся хлор-газе
содержится 95-96% Cl2. Хлор-газ охлаждают
до 20С (при этом конденсируется вода) и
дополнительно сушат промывкой
концентрированной серной кислотой.
Катодный продукт-раствор NaOH
содержит NaOH и неразложившийся NaCl. Раствор
выпаривают, при этом NaCl переходит в
твердую фазу, т.к. его растворимость
резко снижается с увеличением концентрации
NaOH. После выпарки и плавки щелоков
получают безводный гидроксид натрия,
содержащий 92 – 95% NaOH и 2 – 4% NaCl. Электролиз
раствора хлорида натрия с ртутным
катодом. На
перфорированном графитовом (или
оксидно-рутениевом) аноде выделяется
хлор. K: Na(+)+nHg+e=NaHg(n), A: 2Cl(-)-2e=Cl2. Амальгаму
натрия, содержащую 0,1 – 0,3% Na, выводят из
электролизера и разлагают нагретой
водой в отдельном реакторе-разлагателе.
В разлагателе идет электрохимическая
реакция, соответствующая процессу в
короткозамкнутом гальваническом
элементе NaHg n [NaOH] С в котором амальгама
служит катодом. Na+H2O +2e=Na(+)+OH(-)+1/2H2;
Na(+)+OH(-)=NaOH. Схема
электролизера с ртутным катодом.
Р18Глубоко
очищенный концентрированный раствор
NaCl подают в наклонный удлиненный
электролизер, по дну которого самотеком,
противотоком рассолу, движется ртуть,
служащая катодом. Над ртутью расположен
горизонтальный оксидно-рутениевый
анод, погруженный в рассол. Анодная
жидкость, содержащая непрореагировавший
NaCl, выводится из электролизера совместно
с хлор-газом, от которого отделяется в
сепараторах и продувочных колоннах.
Хлор подают на осушку, а обесхлоренный
рассол после очистки от ртути и примесей
насыщается каменной солью и возвращается
в электролизер. Амальгама натрия из
электролизера перетекает в наклонный
реактор-разлагатель, где движется
противотоком дистиллированной воде,
подаваемой в количестве, обеспечивающем
получение 45%-ого раствора NaOH. На дне
разлагателя размещены гребенчатые
графитовые плиты, образующие с амальгамой
короткозамкнутый гальванический элемент
NaHg
n
[NaOH] С. Отводимый гидроксид натрия
отделяют в сепараторах от водорода и
передают потребителям. Ртуть, вытекающую
из разлагателя, ртутным насосом
перекачивают в электролизер.
Технология
связанного азота Под
связанным азотом понимают азот,
находящийся в виде различных соединений
– аммиака, азотной ки-ты и её солей,
карбамида, аминов, аминоки-т, белков и
т.д. Вовлечение азота в био-, гео- и хим
циклы осуществляется путем ее фиксации
некоторыми почвенными микроорганизмами.
Теоретические
основы синтеза аммиака. Основания выбора
параметров процесса. Синтез
аммиака протекает в соответствии со
стехиометрическим уравнением:
N2+3H2↔2NH3+103,6кДж.
Эта реакция обратимая, экзотермическая
и сопровождается уменьшением объема.
Реакция протекает на катализаторе –
пористом железе с добавлением
стабилизирующих и промотирующих добавок
(Al2O3, K2O, CaO, SiO2 и др.). Он активен и термически
устойчив в области температур 650 – 830
К. Соединения серы отравляют катализатор
необратимо, а кислород и некоторые его
соединения, особенно СО-обратимо. Факторы
смещения равновесия. 1)
На практике диапазон оптимальных
температур составляет 400-500°С в зависимости
от совокупности других параметров. 2)
На практике работают при давлениях 30 –
32 МПа. 3) Соотношение реагентов выбирают
исходя из требований стехиометрии. 4)
Высокие объемные скорости в сочетании
с соблюдением оптимального температурного
режима, применение азото-водородной
смеси высокой степени чистоты и
использованием достаточно активных
контактных масс должны обеспечить
большую производительность цехов
синтеза аммиака при высоких экономических
показателях процесса. Стадии
процесса:
1)
Диффузия
N2
и H2
из газового V
в поверхности порами катализатора. 2)
Хемосорбция газов на поверхности
катализ. 3)
Взаимодействие азота и водорода на
поверхности катализатора. Важно: азот
принимает е от катализ., отдает е и на
поверхности могут образов. NH3
амидные группы. 4)
Десорбция.
Оптимальные
условия для синтеза аммиака:
1)
АВС-высокая степень очистки; 2)
Оптимальное соотношение азота к водороду
=1:3; 3)
оптимальный температурный профиль по
длине слоя катализатора; 4)
снижение
содержания аммиака на входе в контактный
аппарат; 5)
оптимальная конструкция для колоны
синтеза.Технологическая
схема производства азота: в зависимости
от давления 3 схемы синтеза аммиака:
1. при низком давлении 100-500 атм; 2. при
среднем давлении 250-360 атм; 3. при высоком
давлении 450-1000 атм.Содержание аммиака
на выходе из контактного аппарата от
14 до 20% (один проход). Газовая смесь из
контактного аппарата охлаждается.
Аммиак конденсируется и отделяется от
газа, а непрореагирующая АВС содержит
аммиак, К отработанной реакционной
смеси добавляется свежая АВС, по
возможности более чистая, поскольку
при многократной циркуляции постепенно
накапливаются примеси, которые частично
растворяются в жидком аммиаке и выводятся
из цикла, но их накопление снижает
производительность колонны синтеза.
Для снижения примесей прибегают к
выбросу части циркулирующей газовой
смеси (поддувочные газы). Р19
1-паровой котел, 2-колона синтеза, 3-водяной
холодильник, 4-сепаратор, 5-трубоциркулирующий
компрессор, 6-конденсационная колона,
7-испаритель, 8-сборники жидкого аммиака.
Подготовленная АВС подается сверху в
колону синтеза, где и происходит синтез
аммиака. Реакционная смесь, содержащая
аммиак и непрореагировавшие АВ выводится
из колоны синтеза (нижняя часть) при
температуре 400 °С. И направляется в котел
утилизатор, где отдает тепло реакции
воде, а полученный пар используется в
технологических целях (для обогрева
цехов). Из котла утилизатора газовая
смесь вновь идет в колону синтеза, где
проходит теплообменник, дополнительно
охлаждается до 90-100С и направляется в
водяной холодильник и сепаратор. В
водяном холодильнике при р=30*10(6) Па.
Конденсируется часть аммиака. Далее
газ с помощью циркуляционного насоса
подается в 6 и 7. За счет испарения аммиака
жидкого получают холод для более полного
выделения аммиака из газовой смеси. Из
6 газ снова направляется в 2, при этом
аммиака здесь не более 2-3%. Свежую азотно
водородную смесь подают в нижнюю часть
6, где она промывается проходя через
аммиак жид. от влаги СО и масел.. р20
Основные
направления в развитии производства
аммиака. 1.Кооперирование
азотной промышленности с промышленностью
основного органического синтеза на
базе использования природного газа и
газов нефтепереработки в качестве
сырья; 2.
Укрупнение всего производства в целом,
и отдельных его подсистем;
3. Разработка
процессов на основе более активных
каталитических систем и снижения за
счет этого давления в процессе. 4.
Применение колонн синтеза с «кипящем
слоем» катализатора; 5.
Дальнейшее совершенствование систем
рационального использования тепла. Для
последней проблемы можно использовать
энерготехнологическую систему в
производстве аммиака. Сжатие вначале
природного газа на стадии конверсии до
4,5 МПа, а затем азото-водородной смеси
до 30 – 32 МПа, ее циркуляция в подсистеме
синтеза осуществляется помощью мощных
турбокомпрессоров. Используется
энергоноситель – пар с высокими
параметрами: давлением до 10 МПа и
температурой 720 – 740 К. Для привода других
компрессоров используют также паровые
турбины на энергоносителях с меньшими
параметрами. В пр-ве аммиака исп.
высокопотенциальные технологические
потоки: конвертированный газ и дымовые
газы после конверсии метана. Но их
энергии и потенциала недостаточно для
образования пара с высокими параметрами.
Необходим дополнительный высокотемпературный
источник энергии. Им является
вспомогательный котел с огневым
обогревом, установленный в газоходе
после трубчатой печи. Пар, получаемый
в котлах, утилизаторах, в линиях
технологических потоков и в дополнительном
котле, собирается в паросборнике и
оттуда распределяется на паровые
турбины-приводы компрессоров.
Пр-во
уксусной кислоты микробиологическим
синтезом. В
химич. промышленности техническая
уксусная кислота производится окислением
ацетальлегида. Пищевую же уксусную
кислоту получаю окислением этилового
спирта под действием фермента
алкогольоксидаза, вырабатываемого
уксусно-кислыми бактериями:
C2H5OH+O2=CH3COOH+H2O.
Процесс протекает с выделением теплоты
в аэробных условиях (с подачей воздуха
для жизнедеятельности микроорганизмов)
при температуре 301-308 К в кислой среде
(рН = 3). Сырьем служит очищенный разбавленный
спирт (примеси угнетают уксусно-кислые
бактерии). Реакция (брожение) осуществляется
непрерывно или переодически в реакторе
— ферментере. Технологическая схема
производства приведена на рис.
Технологическая
схема производства уксусной кислоты
из спирта: Р211-емкость
для приготовления питательной среды;
2-компрессор; 3-фильтр бактерицидный;
4-ферментеры; 5-пастерезатор; 6-фильтр-пресс;
7-емкость для купажирования; 8-сборник
готового продукта. Непрерывный процесс
проводят в каскаде ферментеров 4, каждый
из которых отвечает за определенную
стадию процесса. В соответствие со
стадиями процесса в каждом ферментаторе
поддерживаются заданные условия
культивирования. Подаваемый в ферментер
воздух проходит бактерицидный фильтр
3 для сохранения чистоты бактериальной
культуры. Из последнего ферментера
выводится сырой уксус, содержащий около
9% СН3СООН и поступает на тепловую
обработку в аппарат 5 на пастеризацию.
При Т 360К происходит инактивация
бактериальной массы и коагуляция
коллоидных частии. После фильтра 6 в
емкости 7 уксус доводят до нужной конц.
и направляют в сборник готового продукта
8. Выход уксусной кислоты достигает до
90 кг на 100 л безводного спирта.
Биохимические
производства-основаны
на получении продуктов при помощи живых
организмов. Основным «производителем»
продукции являются микроорганизмы
(бактерии, дрожжи) и продукты их
жизнедеятельности. Микробиологический
синтез происходит в клетках микроорганизмов.
Особое внимание в биотехнологических
производствах уделяется интенсификации
процесса наращивания биомассы, отвечающей
определенным требованиям по составу и
свойствам и чувствительной к условиям
своего развития (составу питательной
среды — субстрату, кислороду, температуре,
давлению, рН и др.). Кинетика биохимических
процессов также специфична-микроорганизмы
могут «задыхаться» от недостатка
кислорода, жиреть от избытка питания и
терять от этого свою активность, не
развиваться в тесноте, отравляться
продуктами обмена (метаболизма).
Биотехнологические процессы привлекательны
тем, что протекают с высокой степенью
специфичности при t
и давлениях, близких к нормальным, и не
требуют больших энергетических затрат.
По
виду продуктов биохимические производства
можно разделить на несколько групп.
1.Пр-во
продуктов, содержащих жизнеспособные
микроорганизмы: биопрепараты, повышающие
плодородность почвы, закваски для
получения кисломолочных продуктов и
силосования кормов, биологические
средства защиты растений. 2.
Пр-во биопрепаратов, в состав которых
входит инактивированная биомасса клеток
и продуктов ее переработки: кормовые
дрожжи, белково-витаминные концентраты
и т.д. 3.
Пр-во очищенных продуктов жизнедеятельности
организмов: витамины, гормоны, ферменты,
антибиотики, аминокислоты и др. Также
биохимические процессы используются
для производства продуктов химической
технологии, для очистки сточных вод,
для извлечения металлов из руд в
металлургии и иных целей в других
отраслях промышленности. По содержанию
биохимические процессы резко отличаются
от химических, но по использованию и
организации технолог. процессов аналог.
процессам хим. тех. Пр-во
пищевых белков. В
отличие от сложных белков, белки
одноклеточных организмов (БОО) используются
как пищевая добавка. Обогащением
белковыми добавками на основе БОО
улучшают качество растительного белка.
Эти добавки повышают содержание
витаминов, микроэлементов, а главное —
аминокислот, несинтезирусмых многими
растениями. Микробиологический
синтез белка, продукт которого представляет
собой инактивированную массу клеток,
— основной промышленный способ его
получения. Его принципиальная схема
включает:
Р221.
подготовку
культуры микроорганизма (продуцента),
например, определенного вида дрожжей
— одноклеточного гриба, размножающегося
почкованием;2.
приготовление питательной среды
(субстрата), включающего вещества сырья,
подвергающегося превращению под
действием фер-ментов, и минеральные
соли, регулирующие жизнедеятельность
клеток;3.культивирование
(выращивание) продуцента, в ходе которого
увеличивается его биомасса (эта стадия
обычно называется ферментацией);4.отделение
биомассы; отработанная культурная
жидкость направляется на очистку;5.тепловая
обработка готовой биомассы, ее сушка и
очистка; в ходе этих операций происходит
инактивация массы и ее подготовка в
виде товарной продукции. Здесь обратим
внимание только на ферментер, обеспечивающий
собственно пр-во продукта — биомассы.
Кроме того, ферментер также должен
обеспечить «комфортные» условия
жизнедеятельности микроорганизмов —
их физиолого-биохимическую активность.
Необходимо обеспечить однородность
среды по всему объему реактора, иначе
локальные неоднородности, застойные
зоны существенно изменят ход размножения
клеток микроорганизма. Было предложено
много разных вариантов конструкций
ферментеров (рис. 6.61). В колонном ферментере
однородность среды обеспечивается
барбатажем воздуха (рис. 6.61, а), а также
специальными вставками, встроенными
диффузором с турбиной или механическими
смесителями. В фермептере, показанном
на рис. 6.61, б, перемешивание жидкости
осуществляется высоким эрлифтом —
барбатаж воздуха вызывает циркуляцию
жидкости. Циркуляция может быть обеспечена
также принудительно с помощью встроенного
перемешивающего устройства. В этих
конструкциях необходимо исключить
образование застойных зон в углах
аппарата. В ферментере горизонтального
типа (рис. 6.61, в) поток принудительно
циркулирует по объему аппарата, похожего
на тороил. Однако скорость движения
потока должна быть достаточной, чтобы
предотвратить расслаивание жидкости.
Схема
ферментеров:Р23
а-коллоного(1.
реакционная зона, 2.
пеногаситель, 3.охлаждающаяя
рубашка, 4.
распределитель воздуха),
б-циркуляционного (1.реакционная
колонна, 2.зона
сепарации, 3.циркуляционная
труба, 4.теплообменник,
5.барбатер),
в-горизонтального
(1.винтовое
перемешивающее устройство, 2.горизонтальный
реактор,3.сепарационная
емкость, 4.теплообменник);
А-питание,Б-газ на аэрацию,В-дрожжевая
суспензия,Г-отработанный газ. В
представленных ферментерах обеспечивается
интенсивное перемешивание жидкости, и
процесс описывается моделью идеального
смешения. На выбор схемы и аппаратуры
в малотоннажных производствах наиболее
сильно будет сказываться «рецептурная»
составляющая процессов. Химико-технологич.
методы защиты окруж. среды. Деятельность
человека приводит к образованию большого
числа отходов. Основ.
направления предохранения окр. среды:1.
собственно уменьшение отходов; 2. получ.
из отходов полезных продуктов с возвратом
последних в сферу производства или
потребления; 3. очистка отходящих потоков
от примесей для уменьшения нагрузки на
окружающую среду; 4. обезвреживание
токсичных отходов для предотвращения
катастрофических последствий; 5.
захоронение отходов, т.е. их локализация
в определенных местах без контакта с
окружающей средой. Большое разнообразие
отходов по составу образующих ве-в
обуславливает первостепенное значение
методов хим. тех. в их обработке. Ввиду
большого многообразия отходов предложить
обобщенную схему их утилизации и
обезвреживания не представляется
возможным, поэтому рассмотрим некоторые
из них. В основу выбора положены методы
обработки отходов из разных источников
их образования.
Утилизация
и обезвреживание тв отходов. Тв
отходы могут содержать разнообразные
по природе компоненты-Ме, неорганические
и орган. ве-ва, поэтому сначала их
сепарируют, Фракции с ве-вами орган.
природы подвергают, как правило,
термической переработке. Утилизация
пластмасс и эластомеров.
Отходы пластических масс и эластомеров
образуются в отраслях, связанных с их
синтезом и переработкой. В эту же группу
отходов также попадают бытовые изделия
из полимерных материалов при окончания
их срока эксплуатации. Длительное время
захоронение в почву и сжигание были
наиболее распространенными способами
уничтожения отходов пластмасс и
эластомеров. Теплота, выделяющаяся при
их сжигании, исп. для генерирования
водяного пара. Однако термическое
разложение приводит к образованию сажи,
выделению токсичных газов и повторному
загрязнению воздушного бассейна. К
основным способам утилизации отходов
пластических масс относятся: термическое
разложение путем пиролиза; деполимеризация
с получением исх. низкомолекулярных
продуктов (мономеров, олигомеров);
вторичная переработка. Пиролиз полимеров
осущ. при t
1100—1400 К без доступа воздуха и позволяет
получить высококалорийное топливо,
сырье и полупродукты, исп. в различных
технологических процессах, а также
мономеры для синтеза полимеров. При
пиролизе отходов полиэтилена образуются
следующие полезные продукты (в %): этилен
— 25, метан — 16, бензол — 12, пропилен —
10. Установка термического пиролиза
включает дробилку, шнековый питатель,
печь пиролиза, скруббер ля промывки
пирогаза, холодильник, ректификационную
колонку разделения углеводородов и
камеру сжигания отходящих ↑, В случае
переработки поливинилхлорида
предусматривается скруббер для поглощения
НСТ. Печь пиролиза отходов представляет
обогреваемую вертикальную цилиндрическую
камеру, в кот. измельченные пластмассовые
отходы перемещаются под действием силы
тяжести вниз, а продукты пиролиза,
выходящие через верх печи, направляются
па переработку. Процессу деполимеризации
с получ. мономеров подвергают только
те вилы пластмасс, кот. распадаются при
сравнительно низких t
(570—710 К). К таким полимерам относятся
полистирол и его сополимеры, полиакрилаты.
Пиролиз полистирола сопровождается
получением 50-70% исх. стирола; при
термическом разложении полиметилметакрилата
выход газообразного метилакрилата
достигает 91-96%. Наиб. эффективным способом
утилизации отходов полимерных материалов
явл. их вторичная переработка.
Утилизация
и обезвреживание жидких отходов. Основ.
массу жидких отходов составляют сточные
воды. Применение конкретного метода
очистки сточной вода зависит от характера
примесей. Наиб.
часто употребляемые приемы очистки
сточных вод объединяют в группы:
1)
для суспензионных и эмульгированных
примесей — отстаивание, флотация,
фильтрация, осветление, центрифугирование);
коагуляция, флотация, электрические
методы осаждения (для мелкодисперсных
и коллоидных частиц; 2)
для очистки от неорган. соед.-дистилляция,
ионообмен, обратный осмос, ультрафильтрация,
реагентное осаждение, методы охлаждения,
электрические методы; 3)
для очистки от орган. соед.-экстракция,
абсорбция, флотация, ионообмен, реагентные
методы; биохим. окисление, жидкофазное
окисление, парофазное окисление,
озонирование, хлорирование, электрохим.
окисление; 4)
для очистки от ↑ и паров-отдувка, нагрев,
реагентные методы; 5)
для уничтожения вредных ве-в-термическое
разложение. Очистку сточных вод от соед.
P
и N
осущ. с помощью специальных Ме. Метод
биолог. очистки сточных вод основан на
исп. способности гетеротрофных
микроорганизмов питаться разнообразными
орган., подвергая последние биохим.
превращениям. Исп. св-в адаптации бактерий
активного ила позволяет успешно решать
вопросы биолог. очистки стоков воды
хим. производств, содерж. сложные орг.
соед. неприродного происхождения.Контактируя
с орган. ве-вами микроорганизмы частично
разрушают их, превращая в воду, диоксид
углерода, нитрит- и сульфат-ионы и др.
Др. часть ве-ва идет на образование их
биомассы. Разрушение орган. соед. назыв.
биохим. окислением, оно происходит
избирательно, поэтому некоторые соед.
разрушаются легко, др.-медленно или
совсем не окисляются. Методы
биохим. очистки сточных вод делятся на
аэробные и анаэробные.
Первые методы основ. на исп. аэробных
групп микроорганизмов, для жизнедеятельности
кот. треб. постоянный приток O2
и t
293-313 К. При изменении кислородного и
температурного режимов состав и число
микроорганизмов меняется, также меняется
эффективность очистки стоков. В случае
анаэробной очистки микроорганизмы
культивируются в активном иле или
биопленке, где биохим. процессы протекают
без доступа O2.
Этот метод исп. для обезвреживания
осадков. Биохим. очистка не может
считаться универсальной, т. к. поступ.
со сточными водами ценные орган. ве-ва
не извлекаются из них, а перерабатываются
в избыточный ил, также требующий
обезвреживания. Пока это единствен.
метод, кот. обеспеч. очистку сточных вод
до показателей, позвол. исп. их в оборотных
системах охлаждения.Сооружения биолог.
очистки могут иметь различные
технологические схемы, кот. выбирают в
зависимости от характеристик, поступ.
в них сточных вод. Схема
биолог. очистки сточных вод:Р241-усреднительные
емкости; 2-накопитель
хоз.-бытовых сточных вод; 3-смесительная
камера; 4-аэротенк;
5-вторич.
отстойник. Сточные воды первоначально
поступ. в усреднительные емкости 1, а
затем в смесительную камеру 3. Сюда же
поступают хоз.-бытовые сточные воды из
накопителя 2, а также условно-чистая
вода. Накопитель хоз.-бытовых сточных
вод играет роль усреднителя, поскольку
эти воды имеют переменный характер
загрязнения. Смешанный сток поступает
в аэротенк 4, куда подастся воздух. Иловая
смесь из аэротенка направляется во
вторичный отстойник 5, где очищен. вода
отделяется от актин. ила и направляется
в водоем или на подпитку водооборотных
систем. Активный ил из вторич. отстойников
возвращ. в аэротенк в виде возвратного
ила, а частично в виде избыточ. ила
направл. на обработку. Термоокислительные
методы обезвреживания сточных вод.
В термоокислительных методах обезвреживания
орган. примеси в сточных водах окисляются
O2
воздуха при повышенной t
до безвредных продуктов. Это пламенный
(«огневой») метод, жидкофазное окисление
и парофазное каталитич. окисление.Выбор
метода обезвреживания зависит от объема
сточных вод, их теплотворной способности,
экономичности процесса и требований к
очищенным стокам. По теплотворной
способности загрязненные стоки делятся
на сточные воды, способные гореть
самостоятельно за счет содержащихся в
них органических ве-в и воды,
термоокислительное обезвреживание
которых возможно только с вводом
топлива.Огневой
метод обезвреживания
сточных вод явл. универсальным и обладает
высокой степенью очистки (98—99,9%). В этом
методе сточная вода вводится в распыленном
состоянии в высокотемпературные продукты
сгорания топлива (1200-1300 К). Вода испаряется,
а ортанические примеси сгорают, образуя
СО2 и Н2О. Минеральные примеси при этом
образуют тв или расплавлен. частицы,
кот. выводятся из камеры печи. В связи
с этим огневой метод рекомендуется исп.
в след. случаях: *при большом кол-ве
сточных вод, содерж. высокотоксич. орган.
примеси, обезвреживание кот. др. Ме
невозможно или экономически невыгодно;
*при
наличии горючих вторич. энергоресурсов,
кот. могут быть исп. вместо
топлива.Р251-нагнетательный
насос; 2-циклонный
реактор; 3-парогенератор
(котел-утилизатор); 4-струйный
аппарат; 5-циркуляционный
насос; 6-емкость;
7-каплеотделитель:
8-дымовая
труба. На рис. представлена схема
установки огневого обезвреживания
сточных вод, включ. циклонный реактор
2, парогенератор (котел-утилизатор) 3 и
струйный аппарат 4. Из реактора продукты
сгорания поступ. в камеру охлаждения
парогенератора. Наличие эжектора позвол.
исключ. дымосос. Циркуляционный насос
5 исп. для подачи ра-ра минеральных ве-в
из емкости б в реактор 2 и в струйный
аппарат 4. Пройдя каплеотделитель 7,
очищ. ↑ поступ. в дымовую трубу 8 и из
нее-в атмосферу. На некоторых установках
для утилизации теплоты исп. водогрейные
котлы, водоаммиачные абсорбционные
холодильные машины и циклоны для сухой
очистки газа. В процессе обезвреживания
сточных вод, содерж. орган. соед. S,
CI2,
нитросоединения, образуются SO2, SO3, Р2О5,
НСl. Эти ве-ва могут взаимод. с образов.
новых более токсичных соед., что необходим.
иметь в виду при удалении отходящих ↑
в окр. среду.
Метод жидкофазного
окисления основан на окислении орган.
ве-в, растворенных в сточной воде, O2
воздуха при t
370-620К и p
2-28 МПа. Повышение давления ускоряет
процесс и глубину окисления вследствие
увелич. растворимости в воде O2.
Жидкофазное окисление осущ. как на
катализаторах, так и без них. В качестве
kt
исп. Ме (Рt, Рd, Сu, Zn, Мn), нанесенные на
оксид алюминия или актив. уголь.
В
методе парофазного каталитического
окисления исп. гетерогенное каталитическое
окисление O2
воздуха летучих орган. соед., находящихся
в сточных волах. Процесс окисления
интенсивно протекает в присутствии
медно- хромовых, цинковых, марганцевых
катализаторов. При высокой t
350-400 большинство орган. ве-в подвергается
полному окислению (98,5—99,9%). В данном
процессе могут быть исп. конструкции
реакторов, характерные для
гетерогенно-каталит. процессов.
Обезвреживание
газообразных отходов.Очистка
выбрасываемых в атмосферу ↑ от орган.
примесей может быть достигнута их
сжиганием при высоких t
1200-1300 К, но такой способ требует больших
затрат первичного топлива, что
нерентабельно при обезвреживании ↑ с
малым содерж. вредных ве-в. В связи с
этим получ. применение каталит. очистка,
осущ. при более низкой t
до 600-700 К. Каталит. очистка от орган. ве-в
основана на каталит. окислении или
восстановлении примесей. Активные
компоненты катализаторов, исп. для
очистки отходящих ↑, можно разделить
на 3 группы: благородные
Ме; сплавы; оксидные системы.
Они должны окислять более 90% СО и
углеводородов в широком интервале t
250-800 в присутствии воды и не должны
отравляться соед. серы. Наиб. распространены
платиновые катализаторы вследствие
способности ускорять самые различные
ре-ции превращения орган. соед. в
окислительных и восстановительных
средах. Для обезвреживания ↑ исп. и
более дешевые kt
на основе оксидов неплатиновых Ме (Ni,
Сu, Сr, Мn). Очистка-дорогостоящий процесс
в плане затрат на оборудование и энергию,
огромная часть кот. расходуется на
прокачку ↑ через систему. Т.к. зернистый
слой kt
облад. большим гидравлическим
сопротивлением, в последнее время
получили распространение катализаторы
сотовой формы. Их сопротивление на
порядок ниже зернистого слоя и в них
лучше перерабатывать запыленный газ-в
прямых каналах меньше задерживается
пыль. Принципиальные
схемы каталит. очистки ↑ с репродуктивным
теплообменником (а) и доп. подогревом
(б):Р261-реактор
2-теплообменник 3-горелка. Наиб. часто
применяется след. принципиальная схема
каталит. очистки (рис. 6.65, а). Очищаемые
↑, пройдя отбойники и циклоны для
отделения конденсата и взвешенных
частиц, захваченных тазовым потоком,
нагреваются в рекуперативном теплообменнике
2 до t
ре-ии и направляют в реактор 1. Очищенные
↑ охлаждаются в теплообменнике 1 и
выбрасывают в атмосферу. Автотермическое
проведение процесса возможно при содерж.
горючих примесей 5-10 г/м3.При меньшем
содержании окисляемых примесей разогрев
в реакторе будет небольшой, что приведет
к уменьшению разности t
(движущей силы) в теплообменнике, равной
ΔТ(ад), т. е. к возрастанию капитальных
затрат на увеличение поверхности
теплообменника. Доп. подвод теплоты,
осущ. сжиганием топлива (природного
газа-рис. 6.65, б), позволяет сэкономить
на теплообменнике. Абсорбционно-каталитическая
очистка.
Абсорбция остается эф. способом извлечения
малых кол-в примесей, но накопившиеся
на сорбенте примеси надо удалять. Обычно
адсорбент заменяют на новый, а отработ.
становится отходом, состоящ. из накопленной
примеси и собственно сорбента, что тоже
приход. утилизир. Абсорбционно-каталит.
метод позвол. избежать доп. отходов.
Загрязненный газ пропуск. через
абсорбционный аппарат. После завершения
цикла абсорбции поглощенную примесь
десорбируют, для чего насыщенный сорбент
продувают нагретым воздухом. Выходящий
газ направляют в реактор каталитической
очистки. Если примесь имеет органич.
природу, происходит се глубокое окисл.
Очистка небольшого V
газа, содержащего большое кол-во примеси,
— процесс более эффективный, нежели
удаление малых количеств примеси из
большого V
загрязненного газа, те. из первоначальной
смеси.Р27
Схема
адсорбционно-каталитической очистки
газа от оксидов
N:1-
абсорбер; 2-реактор: 3-подогреватель;
4-смеситель; 5-дымовая труба.
Технологическая схема подобного процесса
для очистки отходящего газа от оксидов
N
приведена па рис. Абсорбер загружен
неолитом, на котором хорошо сорбируются
оксиды азота при t
295 К и p 0,1 МПа. После цикла адсорбции
производится переключение адсорбера
на режим прогрева и регенерации, который
проводится обратной продувкой слоя
цеолита воздухом при t
495К и давлении 0,1 МПа. Разогрев адсорбера
осуществляется циркулирующим горячим
воздухом. Газы регенерации через
подогреватель 3 с температурой 570К
направляются в реактор 2. В смесителе 4
к газу добавляется подогретый аммиак,
и на kat.
оксиды N
восстанавливаются до азота. После
каталит. очистки ↑, содерж. не более
0,005% оксидов N,
выбрасывается в атмосф.
Пр-во
метанола.
Раньше
получали сухой перегонкой древесины.
Сейчас потребность в метаноле возраста.
СН3ОН-это бесцветная жидкость, исх.
сырьем для неё служит синтез газ, который
получают конверсией метана:СО+Н2О
,
𝐾𝑝
сильно
зависит от t.
Скорость понижается при повышении t.
Скорость окислен. NO до NO2 явл. самой
низкой из всех стадий. При охлажд. может
протекать димеризация NO2 : 2NO2↔ N2O4 +57
кДж. Данная ре-ция протекает с высокой
скоростью. Со снижением t
степень димеризации снижается. Абсорбция
NO2: Если
суммировать: 3NO2 + H2O →2HNO3+NO + 136 кДж. Механизм
ре-ции:
Сначала NO2 из газовой фазы через
пограничный слой диффундирует у
поверхности жидкости и абсорбируется
ею. Затем идет взаимодействие NO2 с Н2О.
HNO2 разлагается сравнительно медленно,
NO плохо растворим в водных растворах
HNO3, поэтому выходит в газовую фазу,
покидая ра-р. Концентр. получающейся
HNO3 определяется условием равновесия
NO2 над ра-ром HNO3. С ростом t,
растет
и
одновременно снижается разница, т.е.
движущаяся сила процесса, т.о. понижается
конц. продукц. HNO3 и наоборот. Получение
разбавленной азотной кислоты при
атмосферном давлении.Р12
Воздух через заборную трубу проходит
через водяной скруббер 1, суконный фильтр
2, где очищается от пыли и аэрозолей.
Очищенный воздух и аммиак направляются
в вентилятор 3, где смешиваются так, что
конц. NH3
достигает 5-10% объема и направляется на
картонный фильтр 4, где окончательно
очищается от пыли и капельной влаги и
затем направляется в реактор 5, назыв.
конвертором. В верхней части конвертора
находятся Pt-Po сетки, где протекает окисл.
NH3
O2
воздуха, так что степень превращения
NH3→NO достигает 97-98%/. На выходе из
конвертора t
отходящих газов достигает 800ºС. Конвертор
смонтирован на котле-утилизаторе, где
происходит в результате исп. тепла от
отходящих газов получение водяного
пара с р=4МПа и t=450ºС,
после котла-утилизатора t
отходящих газов снижается до 160 ºС и
дальнейшее их охлаждение происходит в
водяных холодильниках 7 и 8. Здесь частично
конденсируются водяные пары. В
незначительной степени протекает
окисление NO в NO2 и может получаться ≈5%
HNO3. В холодильнике 8 за счет более низкой
t
получается более конц. HNO3 ≈30%. Окончательно
охлажденный газ вентилятором 9 направл.
в абсорбционную систему, сост. из 6-8
поглотительных башен 10, орошаемых HNO3,
где продолжается превращение NO в NO2,
одновременно NO2 взаимодействует с
раствором HNO3, повышает ее концентрацию.
Для поглощения NO2 в последнюю башню
подают на орошение воду, а образовавшуюся
ки-ту, противотоком направляют от
последних башен в первую. Газ (NO)
направляется в окислительную башню 11
для завершения окисления в NO2 и далее
в башню щелочной абсорбции 12, орошаемую
ра-ром соды, либо известковой водой. Там
происходит поглощение нитрозных газов.
NO2+NO+ Na2CO3→2NaNO2 =CO2↑; NO2+NaCO3→NaNO2+NaNO3+CO2↑
Очищен. газы идут на выброс в атмосферу,
а нитрит-нитратные ра-ры на переработку.
Получение
концентрированной HNO3.
Концентрирование. Мах
tкип.=121ºС достигается при конц. 68,4% HNO3.
В этой точке (азиотроп) состав паров
совпадает с составом жидкой фазы. Для
конц. выше 68,4% необходимо исп. перегонку
разбавл. HNO3
с добавлением конц. H2SO4.
В этом случае получ. гидраты H2SO4
кипят при более высокой t,
чем конц. HNO3.
Перегонку разбавл. HNO3
ведут с 92-94%H2SO4.
Процесс осущ. в тарельчатых барабанных
колоннах или колоннах с насадками из
колец (Раминга), изготовленных из
кислотоупорного чугуна (ферросилида),
с содержанием Si-12-18%. Р131-Тарельчатая
барботажная колонна;2-Испаритель разбавл.
HNO3; 3-Холодильник продукции конц. HNO3;
4-Конденсатор для охлаждения нитрозных
газов. Пары HNO3 с незначительным содержанием
паров H2O и отжига N из колонны направляются
в конденсатор, где конденсируется HNO3,
а нитрозные газы идут на дальнейшее
улавливание. Часть нитрозных ↑
растворяется в HNO3, поэтому из конденсатора
HNO3 возвращается на верхние участки
колонны, где она отдувается от оксидов
и как продукция отходится в холодильник.
Отработанная H2SO4 (≈70%) отходит в нижней
части колонный и без охлаждения
направляется на упаривание.
Концентрирование
с Mg(NO3)2.
В отпарную колонну барабанного типа
сверху подается 80% р-р Mg(NO3)2, нагретый до
160 – 180ºС, а ниже в колонну подается
разбл. 55% HNO3. В части колонны с помощью
кипятильника поддерживается t=160-180ºС.
Выходящие из колонны газы 97% HNO3 направляются
в дистилляционную колонну, оттуда пары
конц. HNO3 с t=86
ºС поступ. в конденсатор, а далее на
охлаждение. Выделен. в нижней части
колонны (≈70%) HNO3 возвращ. в отпарную
колонну, а разбавл. ра-р Mg(NO3)2 (≈55-60%) из
нижней части отпарной колонны отправл.
на концентрирование после предварительного
нагрева в вакуум испарители, откуда 80%
Mg(NO3)2 направляется на орошение отпарной
колонны.Прямой
синтез конц. HNO3.
Из нитрозных газов получают NO2 абсорбцией
конц. ки-той. При низких t=
-10ºС, с получение нитроолеума: НNO3 + n NO2
→HNO3 ∙ n NO2. Из нитроолеума в отбелочных
колоннах при t=80ºС,
высвобождается газообразный NO2,
направляется на конденсацию охлаждения
до -8 ºС. При этом происходит димеризация
NO2 с получением N2O4. Жидкий N2O4 смешивают
со слабой HNO3
(55-65%) и направляют в автоклав, под p, куда
доп. подается O2.
В автоклаве при t=90ºС
и p=5
Мпа протекает ре-ция: 2N2O4(ж.) + 2H2O(ж.) +
O2(г.) → HNO3(ж.) + 59,5 кДж. Для смещения
равновесия вправо необходим избыток
N2O4 – по стехим. – 100% (в 2 раза). Из автоклава
конц. HNO3, содерж. еще и 25% NO2 и вместе с
олиумом подается в отбелочную колонну.
Там ки-та освобождается от растворенного
NO2 и частично выводится в виде продукционной
97-98%, а частично для получ. нитролиума.
Пр-во
фосфорной кислоты. Функциональная
схема производства экстракционной
H3PO4.
Хорошо
исследованным явл. сернокислотное
разложение апатитового концентрата,
получ. обогащением хибинской
апатито-нефелиновой руды и содерж. ≈39%
Р2О5. Его разложение – гетерогенная
ре-ция “ж–тв тело”. Ca5F(PO4)3+5H2SO4+5nH2О=3H3PO4+
5CaSO4*nH2O(↓)+HF
(↑). Кальциевая составляющая выпадает
в ↓ в виде гипса (CaSO4), вторая уходит в ↑
в виде HF.
Функциональная схема
производства ЭФК включ. разложение
измельченного сырья в реакторе
(экстракторе), фильтрацию тв ↓, упаривание
H3PO4
до товарной конц. и очистку отходящих
↑. Образ. HF
взаимод. с пустой породой сырья.
SiO2+4HF=SiF4(газ)+2H2O. Поэтому при промывке
отходящих ↑ H2O
оба летучих продукта улавливаются в
виде кремнефтористоводородной ки-ты
H2SiF4. SiF4+2HF=H2SiF6. Р14Функциональная
схема производства экстракционной
H3PO4:
1 – реактор разложения апатита
(экстрактор); 2 – вакуум фильтр; 3 –
сборник фильтратов; 4 – колона выпарки
H3PO4;
5 – система очистки ↑.
Сернокислотное разложение апатита. С
растворением апатита происходит образ.
тв CaSO4, который плохо растворим. Связывание
Са2+ в CaSO4 должно увеличивать скорость
растворения апатита. К конц. Н2SO4 в
реакторе предъявляются жесткие требов.:
строгое поддержание этой конц. на
оптимальном уровне во всем V
реакционной зоны. Реакционный
узел -
последовательность секций с интенсивным
перемешиванием реагентов в каждой из
них и перетоком между ними для организации
движения реакционной массы по каскаду.
В первых секциях происходит растворение
апатита. Остальные секции работают как
дозреватели, в них формируются кристаллы
CaSO4. Между последней и 1-ой секциями
организован интенсивный рецикл.
Образование
продукционной
H3PO4.
Включает
отделение ↓ CaSO4 (фосфогипса) и
концентрирование кис-ты. В зависимости
от условий разложения образ. кристаллы
CaSO4·2Н2О (дигидрат) или CaSO4·0,5Н2О (полугидрат).
В дигидратном процессе t 343-353 К и конц.
H3PO4
25-35 %. В полугидратном процессе эти
показатели составляют 358-378 К и 35 мас.%.
Фосфогипс отделяют на вакуум-фильтре.
Фильтрат направляют на выпаривание,
где конц. ки-ты увелич. до 52-54%. Это и есть
продукционная ки-та. При выпаривании
из ки-ты удаляется 80-90% F
в виде HF. Отходящие из реактора и из узла
выпаривания фторсодержащие ↑ в системах
очистки поглощаются с образов.
кремнефтористоводородной ки-ты.
2Ca3(PO4)2+
10CO+6SiO2=P4+10CO2+6CaSiO3; P4+5O2=P2O5; P2O5+3H2O= 2H3PO4. Пр-во
минеральных удобрений (МУ).
Классификация
МУ.
МУ классифиц. по 3 главным признакам –
агрохим. назначению, составу и св-вам.
МУ: 1.по агрохимич.
назначению:1.1прямые(простые:(а)азотные,
б)фосфорные,в)калийные);комплексные:а)двойные,б)тройные,в)смешанные,г)сложные);1.2косвенные;2.по
св-вам;3.по составу (по агрегатному
состоянию):тв и ж. Фосфорные
удобрения (ФУ).
Один из методов механической обработки
- измельчение фосфатов. Полученная
фосфористая мука при исп. в кислых почвах
медленно растворяется в почвенных Н2О
и становится долговременно действующим
удобрением. ФУ могут быть получены
термическим разложением фосфатов при
t=1200
– 1800ºС. Пр-во
простого суперфосфата.
2Ca3F(PO4)3+7H2SO4+6,5H2O= 3[Ca(H2PO4)2*
H2O]+7[CaSO4*0,5H2O]+2HF+227,4кДж.(1) Разложение
протекает в 2 стадии.
На
1-ой стадии ≈70%
апатита реагирует с H2SO4.
При этом образ. H3PO4
и полугидрат CaSO4
(1). Эта стадия характер. образов.
поверхностных пленок CaSO4 на зернах
фосфата. 1-ая стадия заканчивается через
20–40 минут после смешения фосфата с
H2SO4.
После
полного исчезновения H2SO4
начинается 2-ая стадия разложения, в
которой оставшийся апатит (~30%) разлагается
H3PO4:
Ca5F(PO4)3+7
H3PO4+5H2O=5[Ca(H2PO4)2*H2O]+HF (2). Образ. монокальцийфосфат
в отличие от CaSO4
не сразу выпадает в↓. Он постепенно
насыщает р-р H3PO4,
после чего начинает выкристаллизовываться
в виде Ca(H2PO4)2*H2O и протекает значительно
медленнее, чем реакция (1). Она начинается
в суперфосфатных камерах и длится в
течение 5–20 суток хранения суперфосфата
на складе. Пр-во
двойного суперфосфата.
Двойной суперфосфат – конц. фосфорное
удобрение. Получ. разложением природных
фосфатов H3PO4.
Оно содержит 42 – 50% усвояемого P2O5. В
отличие от последнего двойной суперфосфат
почти не содержит балласта – сульфата
кальция. Процесс протекает по уравнению
(2). Азотнокислое
разложение фосфатов. Получение сложных
удобрений.Ca5F(PO4)3+10HNO3=
3H3PO4+5Ca(NO3)2+HF (3). В результате образуется
азотнокислая вытяжка, р-р, содержащий
Ca(NO3)2 и свободную H3PO4.
Сущ. ряд методов дальнейшей переработки
азотнокислой вытяжки. Во многих процессах
вытяжку нейтрализуют NH3
получая фосфаты аммония (NP-удобрения).
Если перед гранулированием нейтрализованной
пульпы к ней добавляют соли калия (KCl,
K2SO4), то получают тройное NPK-удобрение –
нитроаммофоску. Азотнокислый метод
разложения фосфатов позволяет наряду
с получением NPK-удобрений попутно
выделять из сырья стронций, редкоземельные
элементы и др., находящие применение в
различных отраслях промышленности.
Пр-во
азотных удобрений. Аммиачная
селитра, карбамид, сульфат аммония,
водные растворы аммиака и др. Азотные
удобрения отличаются друг от друга по
содержанию азота, по форме соединений
азота (нитратные, аммонийные, амидные),
фазовому состоянию (твердые и жидкие).
Пр-во
аммиачной селитры.Р15
Реактор-нейтрализатор
ИНТ
1-реакционный
стакан; 2-барбатер
NH3;
3-барбатер
HNO3;
4-колпачковые
тарелки; 5-каплеотбойник.
Аммиачная селитра или нитрат аммония
(NH4NO3) – кристаллическое ве-во белого
цвета, содерж. 35% N
в аммонийной и нитратной формах, обе
формы легко усваиваются растениями.
Гранулированную аммиачную селитру
применяют в больших масштабах перед
посевом и для всех видов подкормок. В
меньших масштабах ее исп. для производства
взрывчатых ве-в. Для получения аммиачной
селитры применяют ряд технологических
приемов. Во-первых, гранулирование. В
основе процесса производства аммиачной
селитры лежит гетерофазная ре-ция
взаимод. NH3↑
с ра-ром HNO3.
В настоящее время наиболее распространены
схемы с частичным упариванием ра-ра за
счет тепла нейтрализации. Нейтрализацию
осущ. в аппарате ИТН (использование
тепла нейтрализации). Аппарат состоит
из 2 цилиндров (внешний и внутренний).
Во внутренний подается NH3↑
и разбрызгивается HNO3.
Внутренняя часть представляет собой
реакционное пространство, внешняя –
зона испарения. Отвод тепла из зоны
ре-ции необходим не только для выпаривания
ра-ра, но и во избежание перегрева и
разложения HNO3
и аммиачной селитры. Соковый пар,
образующийся как результат испарения
H2O
из реакционного ра-ра исп. для подогрева
реагентов и упаривания реакционного
ра-ра. Р16Схема
производства аммиачной селитры с
частичным упариванием ра-ра за счет Q
нейтрализатора.
1-теплообменник
подогрева HNO3;
2-теплообменник
подогрева NH3;
3-сборник;
4-реактор
ИНТ; 5-донейтрализатор;
6-выпарной
аппарат;7-сепаратор;
8-грануляционная
башня;
9-барометрический
конденсатор; 10-транспортер.
Ра-р аммиачной селитры (60-80%) поступает
в донейтрализатор(5). Сюда добавляют NH3
как нейтрализующий агент и ве-ва
уменьшающие слеживаемость удобрений.
Выпарку проводят в 2 или 3 ступени с исп.
в качестве греющих агентов сокового
пара из аппарата ИТН, вторичного пара
выпарки и свежего насыщенного пара. Для
простоты на схеме показан один выпарной
аппарат (6). В выпарной установке плав
доводят до содержания в нем NH4NO3 98 – 99%.
Гранулирование производят путем
разбрызгивания плава в полой железобетонной
башне (8) высотой 30–35 м. Падающие капли
застывают в гранулы в потоке холодного
воздуха, поступающего противотоком с
помощью вентиляторов. Окончательная
сушка осущ. горячим воздухом во вращающемся
сушильном барабане.
Электрохим.
Пр-ва. Теоретические основы электролиза
водных растворов и расплавленных сред.
Технология электролиза раствора хлорида
натрия.
Основные
направления применения электрохимических
производств. При
электролизе водных ра-ров и расплавленных
сред могут быть получены хим. продукты:
H2, CI2,KOH
и NaOH.
Методом электролиза водных ра-ров осущ.
энергохим. синтез многих неорган. и
орган. ве-в – гипохлоритов, хлоратов,
перхлоратов, HCI,
перманганатов, диоксида марганца, а
также адипонитрила, антидетонаторов.
На электролизе водных ра-ров основаны
гидроэлектрометаллургия и гальванотехника.
Гидроэлектрометалургия
–извлечение Ме из водных ра-ров их солей
путем электролиза. Важнейшая
область электрохимии–получение
и эксплуатация хим. источников тока.
Хим. источники тока подразделяются на
первичные и вторичные. Первичные:
источники энергии, которые могут быть
исп. лишь однократно: батареи и
гальванические элементы. Вторичные:
аккумуляторы, работоспособность которых
восстанавливается пропусканием
электрического тока. Электрохим. методы
все более широко внедряются в технику
очистки сточных вод, Преимущества
электрохим. методов.
1.
В
электрохим. процессах аппаратура во
многих случаях проще и компактнее по
сравнению с альтернативными хим.
процессами; 2.
Электрохим.
методы получения ряда продуктов
характеризуются меньшим числом
производственных стадий и операций,
более дешевым сырьем и большей глубиной
его превращения, одновременным
образованием (причем в раздельном виде)
ценных продуктов.
3.
Чистота получаемых продуктов. Основной
– электрохим. процессов – высокая
энергоемкость, поэтому энергетические
затраты составляют главную статью
себестоимости продуктов. Электролиз
раствора хлорида натрия. Электролиз
ра-ра NaCl – наиболее простой и экономичный
метод одновременного получения 3
важнейших хим. продуктов-CI2,
H2 и NaOH
с использованием дешевого и доступного
природного сырья. CI2
применяют как сырье для производства
хлорорганических растворителей и
пластмасс, синтетических каучуков,
химических волокон, ядохимикатов. В
металлургии: для хлорирующего обжига
руд, в текстильной и целлюлозно-бумажной
промышленности – для очистки и отбеливания
целлюлозы, бумажной массы и тканей.
Большие кол-ва Cl2
идут на очистку и стерилизацию сточных
вод и питьевой H2O.
2NaCl+2H2O=2H2+2NaOH+Cl2. Гидроксид натрия
используется в производстве многих
химических продуктов, прежде всего в
промышленном органическом синтезе, в
целлюлозно-бумажном производстве, в
производстве искусственных волокон, в
металлургии (пр-во алюминия), в
нефтехимической промышленности и др.
Электролиз
раствора NaCl осуществляется двумя
методами:1)электролиз
с твердым катодом и фильтрующей
диафрагмой;2) электролиз без диафрагмы
с жидким ртутным катодом. Электролиз
раствора NaCl с твердым катодом и фильтрующей
диафрагмой.
К:
2H2O+2e=2OH(-)+2H(адс)(1);
2Н(адс)=Н2.
Материалами для катода служит сталь,
на которой водород выделяется с
относительно невысоким перенапряжением
(0,3 В). Снижение потенциала до 0,3 – 0,4 В
можно достичь применением пористых
графитовых катодов, для упрочнения и
гидрофобизации пропитанных
политетрафторэтиленом и активированных
солями меди или серебра. Накапливающиеся
в катодном пространстве гидроксид-ионы
образуют нейтральные молекулы гидроксида
натрия: OH-+Na+=NaOH(3). На аноде выделяется
хлор: A:2Cl(-)-2e=Cl2 (4). Материалом анода служит
оксидно-рутениевая система, обладающая
прочностью и химической инертность по
отношению к кислороду являющемуся
побочным продуктом, образующемся на
аноде: A:4OH(-)-4e=O2+2H2O (5). В объеме электролита
анодного пространства в результате
гидролиза хлора идут побочные химические
реакции: Cl2+H2O↔HOCl+HCl(6),
Cl2+OH(-)↔HOCl+Cl(-)(7),
HOCl+OH(-)=OCl+H2O(8).
Образующийся в результате этой
последовательной реакции гипохлорит
анион претерпевает анодное окисление:
A:6OCl(-)+3H2O+6e
=2OCl3(-)+4Cl(-)+3O2+6H(+)
(9). Электролиз реализуют в электролизерах
непрерывного действия с вертикальными
фильтрующими диафрагмами при противотоке
движения электролита и OH
–ионов.
Р17Схема
электролизитера с фильтрующей диафрагмой:
1-пористая
асбестовая диафрагма; 2-перфорированный
стальной катод; 3-катодное
пространство; 4-оксидно-рутениевый
анод; 5-анодное
пространство. Корпус ванны разделен на
катодное и анодное пространства пористой
диафрагмой из асбеста. Диафрагма плотно
прилегает к перфорированному стеклянному
катоду. В анодном пространстве расположен
оксидно-рутениевый анод. Очищенный
рассол подают в анодное пространство
и вследствие гидростатического давления
он фильтруется через диафрагму и катод
в катодное пространство. Из катодного
пространства непрерывно отводят водород
и раствор гидроксида натрия, а из анодного
– хлор. В образующемся хлор-газе
содержится 95-96% Cl2. Хлор-газ охлаждают
до 20С (при этом конденсируется вода) и
дополнительно сушат промывкой
концентрированной серной кислотой.
Катодный продукт-раствор NaOH
содержит NaOH и неразложившийся NaCl. Раствор
выпаривают, при этом NaCl переходит в
твердую фазу, т.к. его растворимость
резко снижается с увеличением концентрации
NaOH. После выпарки и плавки щелоков
получают безводный гидроксид натрия,
содержащий 92 – 95% NaOH и 2 – 4% NaCl. Электролиз
раствора хлорида натрия с ртутным
катодом. На
перфорированном графитовом (или
оксидно-рутениевом) аноде выделяется
хлор. K: Na(+)+nHg+e=NaHg(n), A: 2Cl(-)-2e=Cl2. Амальгаму
натрия, содержащую 0,1 – 0,3% Na, выводят из
электролизера и разлагают нагретой
водой в отдельном реакторе-разлагателе.
В разлагателе идет электрохимическая
реакция, соответствующая процессу в
короткозамкнутом гальваническом
элементе NaHg n [NaOH] С в котором амальгама
служит катодом. Na+H2O +2e=Na(+)+OH(-)+1/2H2;
Na(+)+OH(-)=NaOH. Схема
электролизера с ртутным катодом.
Р18Глубоко
очищенный концентрированный раствор
NaCl подают в наклонный удлиненный
электролизер, по дну которого самотеком,
противотоком рассолу, движется ртуть,
служащая катодом. Над ртутью расположен
горизонтальный оксидно-рутениевый
анод, погруженный в рассол. Анодная
жидкость, содержащая непрореагировавший
NaCl, выводится из электролизера совместно
с хлор-газом, от которого отделяется в
сепараторах и продувочных колоннах.
Хлор подают на осушку, а обесхлоренный
рассол после очистки от ртути и примесей
насыщается каменной солью и возвращается
в электролизер. Амальгама натрия из
электролизера перетекает в наклонный
реактор-разлагатель, где движется
противотоком дистиллированной воде,
подаваемой в количестве, обеспечивающем
получение 45%-ого раствора NaOH. На дне
разлагателя размещены гребенчатые
графитовые плиты, образующие с амальгамой
короткозамкнутый гальванический элемент
NaHg
n
[NaOH] С. Отводимый гидроксид натрия
отделяют в сепараторах от водорода и
передают потребителям. Ртуть, вытекающую
из разлагателя, ртутным насосом
перекачивают в электролизер.
Технология
связанного азота Под
связанным азотом понимают азот,
находящийся в виде различных соединений
– аммиака, азотной ки-ты и её солей,
карбамида, аминов, аминоки-т, белков и
т.д. Вовлечение азота в био-, гео- и хим
циклы осуществляется путем ее фиксации
некоторыми почвенными микроорганизмами.
Теоретические
основы синтеза аммиака. Основания выбора
параметров процесса. Синтез
аммиака протекает в соответствии со
стехиометрическим уравнением:
N2+3H2↔2NH3+103,6кДж.
Эта реакция обратимая, экзотермическая
и сопровождается уменьшением объема.
Реакция протекает на катализаторе –
пористом железе с добавлением
стабилизирующих и промотирующих добавок
(Al2O3, K2O, CaO, SiO2 и др.). Он активен и термически
устойчив в области температур 650 – 830
К. Соединения серы отравляют катализатор
необратимо, а кислород и некоторые его
соединения, особенно СО-обратимо. Факторы
смещения равновесия. 1)
На практике диапазон оптимальных
температур составляет 400-500°С в зависимости
от совокупности других параметров. 2)
На практике работают при давлениях 30 –
32 МПа. 3) Соотношение реагентов выбирают
исходя из требований стехиометрии. 4)
Высокие объемные скорости в сочетании
с соблюдением оптимального температурного
режима, применение азото-водородной
смеси высокой степени чистоты и
использованием достаточно активных
контактных масс должны обеспечить
большую производительность цехов
синтеза аммиака при высоких экономических
показателях процесса. Стадии
процесса:
1)
Диффузия
N2
и H2
из газового V
в поверхности порами катализатора. 2)
Хемосорбция газов на поверхности
катализ. 3)
Взаимодействие азота и водорода на
поверхности катализатора. Важно: азот
принимает е от катализ., отдает е и на
поверхности могут образов. NH3
амидные группы. 4)
Десорбция.
Оптимальные
условия для синтеза аммиака:
1)
АВС-высокая степень очистки; 2)
Оптимальное соотношение азота к водороду
=1:3; 3)
оптимальный температурный профиль по
длине слоя катализатора; 4)
снижение
содержания аммиака на входе в контактный
аппарат; 5)
оптимальная конструкция для колоны
синтеза.Технологическая
схема производства азота: в зависимости
от давления 3 схемы синтеза аммиака:
1. при низком давлении 100-500 атм; 2. при
среднем давлении 250-360 атм; 3. при высоком
давлении 450-1000 атм.Содержание аммиака
на выходе из контактного аппарата от
14 до 20% (один проход). Газовая смесь из
контактного аппарата охлаждается.
Аммиак конденсируется и отделяется от
газа, а непрореагирующая АВС содержит
аммиак, К отработанной реакционной
смеси добавляется свежая АВС, по
возможности более чистая, поскольку
при многократной циркуляции постепенно
накапливаются примеси, которые частично
растворяются в жидком аммиаке и выводятся
из цикла, но их накопление снижает
производительность колонны синтеза.
Для снижения примесей прибегают к
выбросу части циркулирующей газовой
смеси (поддувочные газы). Р19
1-паровой котел, 2-колона синтеза, 3-водяной
холодильник, 4-сепаратор, 5-трубоциркулирующий
компрессор, 6-конденсационная колона,
7-испаритель, 8-сборники жидкого аммиака.
Подготовленная АВС подается сверху в
колону синтеза, где и происходит синтез
аммиака. Реакционная смесь, содержащая
аммиак и непрореагировавшие АВ выводится
из колоны синтеза (нижняя часть) при
температуре 400 °С. И направляется в котел
утилизатор, где отдает тепло реакции
воде, а полученный пар используется в
технологических целях (для обогрева
цехов). Из котла утилизатора газовая
смесь вновь идет в колону синтеза, где
проходит теплообменник, дополнительно
охлаждается до 90-100С и направляется в
водяной холодильник и сепаратор. В
водяном холодильнике при р=30*10(6) Па.
Конденсируется часть аммиака. Далее
газ с помощью циркуляционного насоса
подается в 6 и 7. За счет испарения аммиака
жидкого получают холод для более полного
выделения аммиака из газовой смеси. Из
6 газ снова направляется в 2, при этом
аммиака здесь не более 2-3%. Свежую азотно
водородную смесь подают в нижнюю часть
6, где она промывается проходя через
аммиак жид. от влаги СО и масел.. р20
Основные
направления в развитии производства
аммиака. 1.Кооперирование
азотной промышленности с промышленностью
основного органического синтеза на
базе использования природного газа и
газов нефтепереработки в качестве
сырья; 2.
Укрупнение всего производства в целом,
и отдельных его подсистем;
3. Разработка
процессов на основе более активных
каталитических систем и снижения за
счет этого давления в процессе. 4.
Применение колонн синтеза с «кипящем
слоем» катализатора; 5.
Дальнейшее совершенствование систем
рационального использования тепла. Для
последней проблемы можно использовать
энерготехнологическую систему в
производстве аммиака. Сжатие вначале
природного газа на стадии конверсии до
4,5 МПа, а затем азото-водородной смеси
до 30 – 32 МПа, ее циркуляция в подсистеме
синтеза осуществляется помощью мощных
турбокомпрессоров. Используется
энергоноситель – пар с высокими
параметрами: давлением до 10 МПа и
температурой 720 – 740 К. Для привода других
компрессоров используют также паровые
турбины на энергоносителях с меньшими
параметрами. В пр-ве аммиака исп.
высокопотенциальные технологические
потоки: конвертированный газ и дымовые
газы после конверсии метана. Но их
энергии и потенциала недостаточно для
образования пара с высокими параметрами.
Необходим дополнительный высокотемпературный
источник энергии. Им является
вспомогательный котел с огневым
обогревом, установленный в газоходе
после трубчатой печи. Пар, получаемый
в котлах, утилизаторах, в линиях
технологических потоков и в дополнительном
котле, собирается в паросборнике и
оттуда распределяется на паровые
турбины-приводы компрессоров.
Пр-во
уксусной кислоты микробиологическим
синтезом. В
химич. промышленности техническая
уксусная кислота производится окислением
ацетальлегида. Пищевую же уксусную
кислоту получаю окислением этилового
спирта под действием фермента
алкогольоксидаза, вырабатываемого
уксусно-кислыми бактериями:
C2H5OH+O2=CH3COOH+H2O.
Процесс протекает с выделением теплоты
в аэробных условиях (с подачей воздуха
для жизнедеятельности микроорганизмов)
при температуре 301-308 К в кислой среде
(рН = 3). Сырьем служит очищенный разбавленный
спирт (примеси угнетают уксусно-кислые
бактерии). Реакция (брожение) осуществляется
непрерывно или переодически в реакторе
— ферментере. Технологическая схема
производства приведена на рис.
Технологическая
схема производства уксусной кислоты
из спирта: Р211-емкость
для приготовления питательной среды;
2-компрессор; 3-фильтр бактерицидный;
4-ферментеры; 5-пастерезатор; 6-фильтр-пресс;
7-емкость для купажирования; 8-сборник
готового продукта. Непрерывный процесс
проводят в каскаде ферментеров 4, каждый
из которых отвечает за определенную
стадию процесса. В соответствие со
стадиями процесса в каждом ферментаторе
поддерживаются заданные условия
культивирования. Подаваемый в ферментер
воздух проходит бактерицидный фильтр
3 для сохранения чистоты бактериальной
культуры. Из последнего ферментера
выводится сырой уксус, содержащий около
9% СН3СООН и поступает на тепловую
обработку в аппарат 5 на пастеризацию.
При Т 360К происходит инактивация
бактериальной массы и коагуляция
коллоидных частии. После фильтра 6 в
емкости 7 уксус доводят до нужной конц.
и направляют в сборник готового продукта
8. Выход уксусной кислоты достигает до
90 кг на 100 л безводного спирта.
Биохимические
производства-основаны
на получении продуктов при помощи живых
организмов. Основным «производителем»
продукции являются микроорганизмы
(бактерии, дрожжи) и продукты их
жизнедеятельности. Микробиологический
синтез происходит в клетках микроорганизмов.
Особое внимание в биотехнологических
производствах уделяется интенсификации
процесса наращивания биомассы, отвечающей
определенным требованиям по составу и
свойствам и чувствительной к условиям
своего развития (составу питательной
среды — субстрату, кислороду, температуре,
давлению, рН и др.). Кинетика биохимических
процессов также специфична-микроорганизмы
могут «задыхаться» от недостатка
кислорода, жиреть от избытка питания и
терять от этого свою активность, не
развиваться в тесноте, отравляться
продуктами обмена (метаболизма).
Биотехнологические процессы привлекательны
тем, что протекают с высокой степенью
специфичности при t
и давлениях, близких к нормальным, и не
требуют больших энергетических затрат.
По
виду продуктов биохимические производства
можно разделить на несколько групп.
1.Пр-во
продуктов, содержащих жизнеспособные
микроорганизмы: биопрепараты, повышающие
плодородность почвы, закваски для
получения кисломолочных продуктов и
силосования кормов, биологические
средства защиты растений. 2.
Пр-во биопрепаратов, в состав которых
входит инактивированная биомасса клеток
и продуктов ее переработки: кормовые
дрожжи, белково-витаминные концентраты
и т.д. 3.
Пр-во очищенных продуктов жизнедеятельности
организмов: витамины, гормоны, ферменты,
антибиотики, аминокислоты и др. Также
биохимические процессы используются
для производства продуктов химической
технологии, для очистки сточных вод,
для извлечения металлов из руд в
металлургии и иных целей в других
отраслях промышленности. По содержанию
биохимические процессы резко отличаются
от химических, но по использованию и
организации технолог. процессов аналог.
процессам хим. тех. Пр-во
пищевых белков. В
отличие от сложных белков, белки
одноклеточных организмов (БОО) используются
как пищевая добавка. Обогащением
белковыми добавками на основе БОО
улучшают качество растительного белка.
Эти добавки повышают содержание
витаминов, микроэлементов, а главное —
аминокислот, несинтезирусмых многими
растениями. Микробиологический
синтез белка, продукт которого представляет
собой инактивированную массу клеток,
— основной промышленный способ его
получения. Его принципиальная схема
включает:
Р221.
подготовку
культуры микроорганизма (продуцента),
например, определенного вида дрожжей
— одноклеточного гриба, размножающегося
почкованием;2.
приготовление питательной среды
(субстрата), включающего вещества сырья,
подвергающегося превращению под
действием фер-ментов, и минеральные
соли, регулирующие жизнедеятельность
клеток;3.культивирование
(выращивание) продуцента, в ходе которого
увеличивается его биомасса (эта стадия
обычно называется ферментацией);4.отделение
биомассы; отработанная культурная
жидкость направляется на очистку;5.тепловая
обработка готовой биомассы, ее сушка и
очистка; в ходе этих операций происходит
инактивация массы и ее подготовка в
виде товарной продукции. Здесь обратим
внимание только на ферментер, обеспечивающий
собственно пр-во продукта — биомассы.
Кроме того, ферментер также должен
обеспечить «комфортные» условия
жизнедеятельности микроорганизмов —
их физиолого-биохимическую активность.
Необходимо обеспечить однородность
среды по всему объему реактора, иначе
локальные неоднородности, застойные
зоны существенно изменят ход размножения
клеток микроорганизма. Было предложено
много разных вариантов конструкций
ферментеров (рис. 6.61). В колонном ферментере
однородность среды обеспечивается
барбатажем воздуха (рис. 6.61, а), а также
специальными вставками, встроенными
диффузором с турбиной или механическими
смесителями. В фермептере, показанном
на рис. 6.61, б, перемешивание жидкости
осуществляется высоким эрлифтом —
барбатаж воздуха вызывает циркуляцию
жидкости. Циркуляция может быть обеспечена
также принудительно с помощью встроенного
перемешивающего устройства. В этих
конструкциях необходимо исключить
образование застойных зон в углах
аппарата. В ферментере горизонтального
типа (рис. 6.61, в) поток принудительно
циркулирует по объему аппарата, похожего
на тороил. Однако скорость движения
потока должна быть достаточной, чтобы
предотвратить расслаивание жидкости.
Схема
ферментеров:Р23
а-коллоного(1.
реакционная зона, 2.
пеногаситель, 3.охлаждающаяя
рубашка, 4.
распределитель воздуха),
б-циркуляционного (1.реакционная
колонна, 2.зона
сепарации, 3.циркуляционная
труба, 4.теплообменник,
5.барбатер),
в-горизонтального
(1.винтовое
перемешивающее устройство, 2.горизонтальный
реактор,3.сепарационная
емкость, 4.теплообменник);
А-питание,Б-газ на аэрацию,В-дрожжевая
суспензия,Г-отработанный газ. В
представленных ферментерах обеспечивается
интенсивное перемешивание жидкости, и
процесс описывается моделью идеального
смешения. На выбор схемы и аппаратуры
в малотоннажных производствах наиболее
сильно будет сказываться «рецептурная»
составляющая процессов. Химико-технологич.
методы защиты окруж. среды. Деятельность
человека приводит к образованию большого
числа отходов. Основ.
направления предохранения окр. среды:1.
собственно уменьшение отходов; 2. получ.
из отходов полезных продуктов с возвратом
последних в сферу производства или
потребления; 3. очистка отходящих потоков
от примесей для уменьшения нагрузки на
окружающую среду; 4. обезвреживание
токсичных отходов для предотвращения
катастрофических последствий; 5.
захоронение отходов, т.е. их локализация
в определенных местах без контакта с
окружающей средой. Большое разнообразие
отходов по составу образующих ве-в
обуславливает первостепенное значение
методов хим. тех. в их обработке. Ввиду
большого многообразия отходов предложить
обобщенную схему их утилизации и
обезвреживания не представляется
возможным, поэтому рассмотрим некоторые
из них. В основу выбора положены методы
обработки отходов из разных источников
их образования.
Утилизация
и обезвреживание тв отходов. Тв
отходы могут содержать разнообразные
по природе компоненты-Ме, неорганические
и орган. ве-ва, поэтому сначала их
сепарируют, Фракции с ве-вами орган.
природы подвергают, как правило,
термической переработке. Утилизация
пластмасс и эластомеров.
Отходы пластических масс и эластомеров
образуются в отраслях, связанных с их
синтезом и переработкой. В эту же группу
отходов также попадают бытовые изделия
из полимерных материалов при окончания
их срока эксплуатации. Длительное время
захоронение в почву и сжигание были
наиболее распространенными способами
уничтожения отходов пластмасс и
эластомеров. Теплота, выделяющаяся при
их сжигании, исп. для генерирования
водяного пара. Однако термическое
разложение приводит к образованию сажи,
выделению токсичных газов и повторному
загрязнению воздушного бассейна. К
основным способам утилизации отходов
пластических масс относятся: термическое
разложение путем пиролиза; деполимеризация
с получением исх. низкомолекулярных
продуктов (мономеров, олигомеров);
вторичная переработка. Пиролиз полимеров
осущ. при t
1100—1400 К без доступа воздуха и позволяет
получить высококалорийное топливо,
сырье и полупродукты, исп. в различных
технологических процессах, а также
мономеры для синтеза полимеров. При
пиролизе отходов полиэтилена образуются
следующие полезные продукты (в %): этилен
— 25, метан — 16, бензол — 12, пропилен —
10. Установка термического пиролиза
включает дробилку, шнековый питатель,
печь пиролиза, скруббер ля промывки
пирогаза, холодильник, ректификационную
колонку разделения углеводородов и
камеру сжигания отходящих ↑, В случае
переработки поливинилхлорида
предусматривается скруббер для поглощения
НСТ. Печь пиролиза отходов представляет
обогреваемую вертикальную цилиндрическую
камеру, в кот. измельченные пластмассовые
отходы перемещаются под действием силы
тяжести вниз, а продукты пиролиза,
выходящие через верх печи, направляются
па переработку. Процессу деполимеризации
с получ. мономеров подвергают только
те вилы пластмасс, кот. распадаются при
сравнительно низких t
(570—710 К). К таким полимерам относятся
полистирол и его сополимеры, полиакрилаты.
Пиролиз полистирола сопровождается
получением 50-70% исх. стирола; при
термическом разложении полиметилметакрилата
выход газообразного метилакрилата
достигает 91-96%. Наиб. эффективным способом
утилизации отходов полимерных материалов
явл. их вторичная переработка.
Утилизация
и обезвреживание жидких отходов. Основ.
массу жидких отходов составляют сточные
воды. Применение конкретного метода
очистки сточной вода зависит от характера
примесей. Наиб.
часто употребляемые приемы очистки
сточных вод объединяют в группы:
1)
для суспензионных и эмульгированных
примесей — отстаивание, флотация,
фильтрация, осветление, центрифугирование);
коагуляция, флотация, электрические
методы осаждения (для мелкодисперсных
и коллоидных частиц; 2)
для очистки от неорган. соед.-дистилляция,
ионообмен, обратный осмос, ультрафильтрация,
реагентное осаждение, методы охлаждения,
электрические методы; 3)
для очистки от орган. соед.-экстракция,
абсорбция, флотация, ионообмен, реагентные
методы; биохим. окисление, жидкофазное
окисление, парофазное окисление,
озонирование, хлорирование, электрохим.
окисление; 4)
для очистки от ↑ и паров-отдувка, нагрев,
реагентные методы; 5)
для уничтожения вредных ве-в-термическое
разложение. Очистку сточных вод от соед.
P
и N
осущ. с помощью специальных Ме. Метод
биолог. очистки сточных вод основан на
исп. способности гетеротрофных
микроорганизмов питаться разнообразными
орган., подвергая последние биохим.
превращениям. Исп. св-в адаптации бактерий
активного ила позволяет успешно решать
вопросы биолог. очистки стоков воды
хим. производств, содерж. сложные орг.
соед. неприродного происхождения.Контактируя
с орган. ве-вами микроорганизмы частично
разрушают их, превращая в воду, диоксид
углерода, нитрит- и сульфат-ионы и др.
Др. часть ве-ва идет на образование их
биомассы. Разрушение орган. соед. назыв.
биохим. окислением, оно происходит
избирательно, поэтому некоторые соед.
разрушаются легко, др.-медленно или
совсем не окисляются. Методы
биохим. очистки сточных вод делятся на
аэробные и анаэробные.
Первые методы основ. на исп. аэробных
групп микроорганизмов, для жизнедеятельности
кот. треб. постоянный приток O2
и t
293-313 К. При изменении кислородного и
температурного режимов состав и число
микроорганизмов меняется, также меняется
эффективность очистки стоков. В случае
анаэробной очистки микроорганизмы
культивируются в активном иле или
биопленке, где биохим. процессы протекают
без доступа O2.
Этот метод исп. для обезвреживания
осадков. Биохим. очистка не может
считаться универсальной, т. к. поступ.
со сточными водами ценные орган. ве-ва
не извлекаются из них, а перерабатываются
в избыточный ил, также требующий
обезвреживания. Пока это единствен.
метод, кот. обеспеч. очистку сточных вод
до показателей, позвол. исп. их в оборотных
системах охлаждения.Сооружения биолог.
очистки могут иметь различные
технологические схемы, кот. выбирают в
зависимости от характеристик, поступ.
в них сточных вод. Схема
биолог. очистки сточных вод:Р241-усреднительные
емкости; 2-накопитель
хоз.-бытовых сточных вод; 3-смесительная
камера; 4-аэротенк;
5-вторич.
отстойник. Сточные воды первоначально
поступ. в усреднительные емкости 1, а
затем в смесительную камеру 3. Сюда же
поступают хоз.-бытовые сточные воды из
накопителя 2, а также условно-чистая
вода. Накопитель хоз.-бытовых сточных
вод играет роль усреднителя, поскольку
эти воды имеют переменный характер
загрязнения. Смешанный сток поступает
в аэротенк 4, куда подастся воздух. Иловая
смесь из аэротенка направляется во
вторичный отстойник 5, где очищен. вода
отделяется от актин. ила и направляется
в водоем или на подпитку водооборотных
систем. Активный ил из вторич. отстойников
возвращ. в аэротенк в виде возвратного
ила, а частично в виде избыточ. ила
направл. на обработку. Термоокислительные
методы обезвреживания сточных вод.
В термоокислительных методах обезвреживания
орган. примеси в сточных водах окисляются
O2
воздуха при повышенной t
до безвредных продуктов. Это пламенный
(«огневой») метод, жидкофазное окисление
и парофазное каталитич. окисление.Выбор
метода обезвреживания зависит от объема
сточных вод, их теплотворной способности,
экономичности процесса и требований к
очищенным стокам. По теплотворной
способности загрязненные стоки делятся
на сточные воды, способные гореть
самостоятельно за счет содержащихся в
них органических ве-в и воды,
термоокислительное обезвреживание
которых возможно только с вводом
топлива.Огневой
метод обезвреживания
сточных вод явл. универсальным и обладает
высокой степенью очистки (98—99,9%). В этом
методе сточная вода вводится в распыленном
состоянии в высокотемпературные продукты
сгорания топлива (1200-1300 К). Вода испаряется,
а ортанические примеси сгорают, образуя
СО2 и Н2О. Минеральные примеси при этом
образуют тв или расплавлен. частицы,
кот. выводятся из камеры печи. В связи
с этим огневой метод рекомендуется исп.
в след. случаях: *при большом кол-ве
сточных вод, содерж. высокотоксич. орган.
примеси, обезвреживание кот. др. Ме
невозможно или экономически невыгодно;
*при
наличии горючих вторич. энергоресурсов,
кот. могут быть исп. вместо
топлива.Р251-нагнетательный
насос; 2-циклонный
реактор; 3-парогенератор
(котел-утилизатор); 4-струйный
аппарат; 5-циркуляционный
насос; 6-емкость;
7-каплеотделитель:
8-дымовая
труба. На рис. представлена схема
установки огневого обезвреживания
сточных вод, включ. циклонный реактор
2, парогенератор (котел-утилизатор) 3 и
струйный аппарат 4. Из реактора продукты
сгорания поступ. в камеру охлаждения
парогенератора. Наличие эжектора позвол.
исключ. дымосос. Циркуляционный насос
5 исп. для подачи ра-ра минеральных ве-в
из емкости б в реактор 2 и в струйный
аппарат 4. Пройдя каплеотделитель 7,
очищ. ↑ поступ. в дымовую трубу 8 и из
нее-в атмосферу. На некоторых установках
для утилизации теплоты исп. водогрейные
котлы, водоаммиачные абсорбционные
холодильные машины и циклоны для сухой
очистки газа. В процессе обезвреживания
сточных вод, содерж. орган. соед. S,
CI2,
нитросоединения, образуются SO2, SO3, Р2О5,
НСl. Эти ве-ва могут взаимод. с образов.
новых более токсичных соед., что необходим.
иметь в виду при удалении отходящих ↑
в окр. среду.
Метод жидкофазного
окисления основан на окислении орган.
ве-в, растворенных в сточной воде, O2
воздуха при t
370-620К и p
2-28 МПа. Повышение давления ускоряет
процесс и глубину окисления вследствие
увелич. растворимости в воде O2.
Жидкофазное окисление осущ. как на
катализаторах, так и без них. В качестве
kt
исп. Ме (Рt, Рd, Сu, Zn, Мn), нанесенные на
оксид алюминия или актив. уголь.
В
методе парофазного каталитического
окисления исп. гетерогенное каталитическое
окисление O2
воздуха летучих орган. соед., находящихся
в сточных волах. Процесс окисления
интенсивно протекает в присутствии
медно- хромовых, цинковых, марганцевых
катализаторов. При высокой t
350-400 большинство орган. ве-в подвергается
полному окислению (98,5—99,9%). В данном
процессе могут быть исп. конструкции
реакторов, характерные для
гетерогенно-каталит. процессов.
Обезвреживание
газообразных отходов.Очистка
выбрасываемых в атмосферу ↑ от орган.
примесей может быть достигнута их
сжиганием при высоких t
1200-1300 К, но такой способ требует больших
затрат первичного топлива, что
нерентабельно при обезвреживании ↑ с
малым содерж. вредных ве-в. В связи с
этим получ. применение каталит. очистка,
осущ. при более низкой t
до 600-700 К. Каталит. очистка от орган. ве-в
основана на каталит. окислении или
восстановлении примесей. Активные
компоненты катализаторов, исп. для
очистки отходящих ↑, можно разделить
на 3 группы: благородные
Ме; сплавы; оксидные системы.
Они должны окислять более 90% СО и
углеводородов в широком интервале t
250-800 в присутствии воды и не должны
отравляться соед. серы. Наиб. распространены
платиновые катализаторы вследствие
способности ускорять самые различные
ре-ции превращения орган. соед. в
окислительных и восстановительных
средах. Для обезвреживания ↑ исп. и
более дешевые kt
на основе оксидов неплатиновых Ме (Ni,
Сu, Сr, Мn). Очистка-дорогостоящий процесс
в плане затрат на оборудование и энергию,
огромная часть кот. расходуется на
прокачку ↑ через систему. Т.к. зернистый
слой kt
облад. большим гидравлическим
сопротивлением, в последнее время
получили распространение катализаторы
сотовой формы. Их сопротивление на
порядок ниже зернистого слоя и в них
лучше перерабатывать запыленный газ-в
прямых каналах меньше задерживается
пыль. Принципиальные
схемы каталит. очистки ↑ с репродуктивным
теплообменником (а) и доп. подогревом
(б):Р261-реактор
2-теплообменник 3-горелка. Наиб. часто
применяется след. принципиальная схема
каталит. очистки (рис. 6.65, а). Очищаемые
↑, пройдя отбойники и циклоны для
отделения конденсата и взвешенных
частиц, захваченных тазовым потоком,
нагреваются в рекуперативном теплообменнике
2 до t
ре-ии и направляют в реактор 1. Очищенные
↑ охлаждаются в теплообменнике 1 и
выбрасывают в атмосферу. Автотермическое
проведение процесса возможно при содерж.
горючих примесей 5-10 г/м3.При меньшем
содержании окисляемых примесей разогрев
в реакторе будет небольшой, что приведет
к уменьшению разности t
(движущей силы) в теплообменнике, равной
ΔТ(ад), т. е. к возрастанию капитальных
затрат на увеличение поверхности
теплообменника. Доп. подвод теплоты,
осущ. сжиганием топлива (природного
газа-рис. 6.65, б), позволяет сэкономить
на теплообменнике. Абсорбционно-каталитическая
очистка.
Абсорбция остается эф. способом извлечения
малых кол-в примесей, но накопившиеся
на сорбенте примеси надо удалять. Обычно
адсорбент заменяют на новый, а отработ.
становится отходом, состоящ. из накопленной
примеси и собственно сорбента, что тоже
приход. утилизир. Абсорбционно-каталит.
метод позвол. избежать доп. отходов.
Загрязненный газ пропуск. через
абсорбционный аппарат. После завершения
цикла абсорбции поглощенную примесь
десорбируют, для чего насыщенный сорбент
продувают нагретым воздухом. Выходящий
газ направляют в реактор каталитической
очистки. Если примесь имеет органич.
природу, происходит се глубокое окисл.
Очистка небольшого V
газа, содержащего большое кол-во примеси,
— процесс более эффективный, нежели
удаление малых количеств примеси из
большого V
загрязненного газа, те. из первоначальной
смеси.Р27
Схема
адсорбционно-каталитической очистки
газа от оксидов
N:1-
абсорбер; 2-реактор: 3-подогреватель;
4-смеситель; 5-дымовая труба.
Технологическая схема подобного процесса
для очистки отходящего газа от оксидов
N
приведена па рис. Абсорбер загружен
неолитом, на котором хорошо сорбируются
оксиды азота при t
295 К и p 0,1 МПа. После цикла адсорбции
производится переключение адсорбера
на режим прогрева и регенерации, который
проводится обратной продувкой слоя
цеолита воздухом при t
495К и давлении 0,1 МПа. Разогрев адсорбера
осуществляется циркулирующим горячим
воздухом. Газы регенерации через
подогреватель 3 с температурой 570К
направляются в реактор 2. В смесителе 4
к газу добавляется подогретый аммиак,
и на kat.
оксиды N
восстанавливаются до азота. После
каталит. очистки ↑, содерж. не более
0,005% оксидов N,
выбрасывается в атмосф.
Пр-во
метанола.
Раньше
получали сухой перегонкой древесины.
Сейчас потребность в метаноле возраста.
СН3ОН-это бесцветная жидкость, исх.
сырьем для неё служит синтез газ, который
получают конверсией метана:СО+Н2О
 СО2+Н2;
СН4+2Н2О=СО+Н2.
Катализатор
для СН3ОН-вместо
8тчастейиnO и 1 массовая часть Р2О3. Степень
конверсии исх. смеси в метаноле за один
проход под действием катализатора
составляет 5%, поэтому исп. циркуляционную
схему с промежуточным выделением
образующегося СН3ОН, ре-ция экзотермической
и идёт с уменьшением общего объёма
системы, t=430-450С, p=300-320 атм.С1
Свежая смесь СО и Н2 сжим. компрессором
1
до 399 атм. и поступает в смеситель 2,
в который поступ. возвратный синтез
газ, газ перемещ. и поступает в конденсатор,
где задерж. пары воды и механические
примеси. После синтеза газ поступает в
теплообменник 4
обогрев контакт. газами из 5,
нагрев до 250С и идёт в колону синтеза 5,
её конструкция соответсвует конструкции
для синтеза NH3, газ проходит трубки с
гранулир. kt, образ. до 5% метанола и мало
высших спиртов. Контактные газы проходят
теплообменник 4,
где отдают тепло на нагрев смеси, затем
водой охлаждают до 20-30С. Метанол конден.
в жидком состояние, переходящий в воды
и высшие спирты. Эти продукты поступают
в сепаратор 7,
где отдел синтез газ поступить 8
сжим. до 300 атм. и идёт в 2.
Жидкие продукты собираются в общем
сборнике 9
и подверг. ректификации в 10.
Пары метанола конденс. в холодильнике
11и
жидкий метанол сгорают в приемнике 12.
Часть спирта исп. для орошения колон-
спиртов. флегма. В колонке остаются
высшие спирты, которые используются в
фармацевтике и для др. целей.Был предложен
новый катализатор на основе СuO и MnO2,
p=50атм., а степень конверсии 20-25%, но при
этом образов. много высших спиртов.Пр-во
формальдегида.
Явл. ценным сырьем для органического
синтеза (получение дивинила, изопрена,
ФФС, формальдегида и др. продуктов).
Получение его основано на каталитическом
окислении метанола на поверхности
Ag-cat t=700-750C. Давление-атмосферное. Саt
исп. в виде сетки или Ag-напоситца на
поверхности пемзы.С2.
CH3OH+1/2O2=CH2OH+H2O,
CH3OH
СО2+Н2;
СН4+2Н2О=СО+Н2.
Катализатор
для СН3ОН-вместо
8тчастейиnO и 1 массовая часть Р2О3. Степень
конверсии исх. смеси в метаноле за один
проход под действием катализатора
составляет 5%, поэтому исп. циркуляционную
схему с промежуточным выделением
образующегося СН3ОН, ре-ция экзотермической
и идёт с уменьшением общего объёма
системы, t=430-450С, p=300-320 атм.С1
Свежая смесь СО и Н2 сжим. компрессором
1
до 399 атм. и поступает в смеситель 2,
в который поступ. возвратный синтез
газ, газ перемещ. и поступает в конденсатор,
где задерж. пары воды и механические
примеси. После синтеза газ поступает в
теплообменник 4
обогрев контакт. газами из 5,
нагрев до 250С и идёт в колону синтеза 5,
её конструкция соответсвует конструкции
для синтеза NH3, газ проходит трубки с
гранулир. kt, образ. до 5% метанола и мало
высших спиртов. Контактные газы проходят
теплообменник 4,
где отдают тепло на нагрев смеси, затем
водой охлаждают до 20-30С. Метанол конден.
в жидком состояние, переходящий в воды
и высшие спирты. Эти продукты поступают
в сепаратор 7,
где отдел синтез газ поступить 8
сжим. до 300 атм. и идёт в 2.
Жидкие продукты собираются в общем
сборнике 9
и подверг. ректификации в 10.
Пары метанола конденс. в холодильнике
11и
жидкий метанол сгорают в приемнике 12.
Часть спирта исп. для орошения колон-
спиртов. флегма. В колонке остаются
высшие спирты, которые используются в
фармацевтике и для др. целей.Был предложен
новый катализатор на основе СuO и MnO2,
p=50атм., а степень конверсии 20-25%, но при
этом образов. много высших спиртов.Пр-во
формальдегида.
Явл. ценным сырьем для органического
синтеза (получение дивинила, изопрена,
ФФС, формальдегида и др. продуктов).
Получение его основано на каталитическом
окислении метанола на поверхности
Ag-cat t=700-750C. Давление-атмосферное. Саt
исп. в виде сетки или Ag-напоситца на
поверхности пемзы.С2.
CH3OH+1/2O2=CH2OH+H2O,
CH3OH CO
+2H2, 2CO
CO
+2H2, 2CO CO2+
«С»,
2Ag+2C=Ag2C2. Жидкий
метанол поступает в кипятильник 2
внутри которого теплообменники обогрев.
горяч. водой. Снизу через слой метанола
пропускается очищенная азотоводородная
смесь, воздух насыщается парами метанола
и поступает в теплообменник 5.
Обогрев до t=800С. Метанол-азотоводородная
смесь нагревается до 750С и поступает. в
контактный аппарат 6,
внутри которого серебряная сетка или
слой пемзы, пропитанный серебром.
Требования:
отсутсвие следов железа (при их наличии:
CH3OH=CO2+H2,
CO=CO2+C(сажа),2Ag+
2C=Ag2C2).
При
этом cat отравляется необрат. Поэтому
ахотоводородная смесь и метанол очищают,
а контакт. аппарат изготавливают из
алюминия. При 750С метанол окисл. на 90%.
Газовая смесь проходит теплообменник
1
и отдаёт часть тепла на объём воды и газ
идёт в котёл 8
и охлаждается до 20С и поступает в общий
распределитель 9,
отчасти идёт в аппарат 10.
Здесь получается 30-40% формальдегида, в
котором 10% метанола, который явл.
ингибитором ре-ции полимеризации
формальдегида. Часть формальдегида
поступ. в рассольн. холодильник 11
и при 21С превращается в жидкость.Пр-во
этанола.В
настоящее время в России млрд тонн.
Основное кол-во-пр-во дивинила, эфиров,
растворителей, пищевых продуктов
топлива. Сущ.
4 метола получения:1.биохимический
способ;2.Серно-кислая
гидратациях этилена;3.Парообразная
гидратация этилена; 4.
Гидролиз древесины.1-исп.
картофель, фрукты, зерно.
Исх.
сырье разматывают, обрабатывают горяч.
водой до Клейстера, выдерж. несколько
часов до охлаждения 60С и добавл. солод
(проросшее зерно, в котором есть фермент
амилаза и под его действием происходит
гидролиз целлюлозы до мальтозы. Ра-р
охлаждают и добавляют дрожжи. Глюкоза
под действием дрожжей переходит в 2 моля
СО2 и 2 моля С6Н5ОН. Получ. бражка, которую
фильтрую и ректифицироваться по 2
ступеням. Сначала получ. спирт-81%, при
вторич. ректификации получают
спирт-ректификат (96,5-97,5%). 2-C3.
98% ацетилен сжим. компрессором до 15 атм
и пропускается через гидрататор 2
по всей высоте которого есть ситуативно
тарелки и водяной теплообменник для
поддержания 80-90С. Сверху аппарат орошает
98% H2SO4
и отрабатывают алкил-сульфатом. Содержащая
примеси смесь, промыв. в реакторе 3
водой, а в 4
NaOH,
и сжигают на факеле. Ра-р алкилсульфата
поступает в гидролизер 5,
где образуется спирт и диэтиловый эфир.
Образовавшуюся кислоту (50 %) подвергают
бараб. концентрирование в 6
и далее 98% H2SO4
собирают в приемнике 7
и далее может совращаться в пр-во. Пары
содержат спирт, эфир, воду и H2SO4.
Они охлаждаются в 8
и поступает в
9,
заполненный 29% Ca(OH)2.
Смесь пересушивают и H2SO4
удаляется. Гибс отделяют в центрифуге
10,
а водяной растворов спирта, эфира
поступают на 2-ух ступенчатый ректификатор
11.
Здесь отделяется эфир, затем спирт и с
конц. 83% его отправляют на дополнительную
ректификацию в аппарат 12.
Пары спирта концентрируют а холодильнике
13,
а жидкий спирт собирают в 14.
Часть спирта исп. на орошение башни 12.
Исходный ацетилен получают пиролизом
2
при 800С, при этом образуется пирогаз,
который содержит этилены и бутонных.
Пирогаз охлажд. и разделяют, пропускают
через колонку, орошают H2SO4
разной концентрации.
Раствора
газов обрабатывают в ситчатых колонках
оставшимся паром и газы отдув., затем
промывают водой и сушат.3-Использование
98% ацетилена. C2H4+H2O(c
H3PO4,
300ºC,
90атм)=C2H5OH+Q.
98%
ацетилен, пары воды и немного H3PO4
необходимо для восполнения её портрет
с поверхности kat. 1
сжигет до 70-89 атм., затем проходит
теплообменник 2
(t=110-150), за счёт постоянных газов из 4
в камеры печи 3
перегревают до 280С. С этой температурой
и давлением исходная смесь поступает
в полочной контакт
4,
внутр. часть которого из меди. За один
проход через катализатор образуется
до 5% этанола и немного эфира.C4.
Контактные
газы вначале проходят теплообменник 2
и водяной холодильник 5
и охлаждаются до 20-30С. При этом, спирты,
эфиры и вода конденсируются и отделяется
в аппарате 6,
а ацетилен приходит в 7
и промывается водой и возвращается в
процесс. Жидкие продукты из 6
и 7
поступают на мойку, которая проводит
Са(ОН)2:H3PO4+Ca(OH)2=
CaHPO4+2H2OВ
результате получается удобрение. Раствор
15% спирта и эфира выступают на
двух-ступенчатый ректификатор. На 1-ой
ступени в аппарате 8
отделяют эфиры, а растворы спирта
перегоняются в 12.
Жидкий спирт в 13.
Часть спирта-флегматик при орошении
11.
Общая степень конверсии 95%.4-При
переработке древесины образуется до
30% отходов/опилок.
В
составе древесины хвойных пород:
целлюлоза, гемицеллюлозы, пектоза,
лигнин, продукты не способные к гидролизу,
т.е. данное сырье является ценным для
производства глюкозы и фурфурола.
Процесс гидролиза при 15-20 атм, 2 часа, в
результате образуется глюкоза. Раствор
её нейтрализует оксидом кальция, далее
процесс брожения.C5.Древесные
отходы загружают в гидролизер, заливают
H2SO4
и гидролизирует, обогревают при 150°
выдерживают 2
часа. Протекает полном гидролиз с
образованием раствора глюкозы и
фурфурола. Через 2 часа жидкость продолжает
поступать в конденсатор 2
за счёт понижение давления от 20 до 1 атм.
Образуется раствор глюкозы и пары
фурфурола. Пары конденсируют в холодильник
3
и сливается в приёмник 4.
Из него получается пластмасса и смолы.
Водный раствор глюкоза поступает в
реактор 5,
заполненный гидроксидом кальция. Здесь
нейтрализуется кислота и образуется
гипс в реакторе 6.
Водный раствор глюкозы упаривается в
вакууме или подвергается брожению в
реакторе 7.
Через 12-14 суток брожение заканчивается,
всё фильтруют и ректифицируют. Пары
спирта кантеры в холодильнике 10.
Часть спирта идёт на орошение колонны,
а лигнин отмывают водой, сушиться и
используют для пиролиза для получения
фенола. При этом получается ароматич.
карбоновые кислоты.Пр-во
уксусной кислоты.
Она используется как сырье и среда для
проведения реакции окисления Ar УВ.
Сейчас укс.кисл получается kat окислением
ацетальдегида и мягкой бензиновой
фракцией или н-бутана. Если kat явл.
ацетальдегид Cu и добавляется этилметил
кетон то образуется только ангидрид
укс.кисл, тк кетоны образуют азиотропы
с водой, которая уходит из колоны и
реакция гидростанции не протекает.С6
Укс.альдегид и очищенная N-H смесь насосами
1
и 2
подаётся в окислительную колону 3 по
всей высоте катализатора-Стичатые
тарелки и по ним сверху стекает раствор
ацетата Mn в укс.кисл; есть в колоне водный
теплообменник и по нему циркулирует
горячая вода, тк промежуточный продукт
надуксусная кислота, которая взрываюоопасна.
Пр-во
уксусной кислоты окислением С4Н10.
С4Н10
получают термическим и kat. крекингом
нефти и высокотемпературного мазута,
гудрона и полугудрона. Окислен. С4Н10
приводит к образов. сложной смеси
продуктов. А CH3COOH
получ. в результате разрушения цепи. В
качестве примесей могут быть получены
сложные эфиры. Т.к. целевым яв пр-во
СН3СООН, то окисл. ведут таким обр., чтобы
степень превращения С4Н10-через слой
р-ра kat
не превышала 30-40%. Затем С4Н10 отделяют и
возвращают в пр-во.С7.С4Н10
сжим. в компрессоре 1 до 60атм. при сжигании
газы сильно разогрев. и поэтому после
помпрессора проходят Н2Охолодильник 3
и поступают в окислительную колонну 4.
Сюда же поступ. АВ смесь. Сверху по
ситчатым тарелкам стекает р-р (CH3COO)2Mn
в СН3СООН и дополн. продув. инертным
газом, тк при окислении образуется
пероксидные соединения, которые
взрывоопасны. Продукты окисления сначала
проходят в холодильник 5, t=20,
затем подверг. дрессимилированию 6до
р=1атм. В результате падения р происходит
конденсация всех компонентов за исключ.
С4Н10 и газов. В 7 собир. жидкость, а газовая
смесь проходит через 8 промыв. холодной
Н2О и газы идут в процесс (предварительно
мет. глубоко охлажд. С4Н10 отделяют от Н2
и N2).
Жидкость продолжает продуваться через
башню 7 и 8. подвергается ступенчатой
ректификации, отдел. пары и подаются в
10 и собир. в приемнике 11. Метод очень
дешевый, тк исходн. С4Н10 есть в больших
кол-вах, низкая цена.
Пр-во
высших карбоновых кислот(ВКК).
ВКК
получают каталитическим окислением
н-алканов, при этом в зависимости от
условий производится окисление с
разрушением цепи, так и без него. Можно
регулировать состав продуктов. Окисление
протекает под действием молекул О2 и
лимитируемая t, которая связана с длиной
цепи.
Высшие
парафины. Содержат
С15-С30. Kat-применение MnO2 или CrO3, либо
(CH3COO)2Mn или Со. Инициатором яв. НВr или
NO, NO2. Широко применяется каталит.
окисление-это фракция с t=375-425, соед.
С25-С35. При получении кислот обязательным
является отсутствие в них соединений
с разветвлённой цепью и ароматич. УВ,
тк они яв. сильными канцерогенам.
Биологическая среда водной артерии не
способна потреблять из алкана- Многие
вещества яв. не разлагаемыми(из ВКК).
Учитывая это из исходной фракции нефти,
выделяются изоалканы и ароматич. УВ и
затем обрабвтыв. парафинами или мочевиной.
Мочевина образует адукты с н-алканами,
а ароматич. УВ остаются в растворе.
Выпавшие в осадок адукты отделяются и
затем обрабатываются тёплой водой,
мочевина растворяется и всплывает
парафин с н-алканами, затем ещё раз
отмывается тёплой водой и только после
этого он используется как сырье в методе
Башкирова:C8.Чистый
парафин и KMnO4 загружают в ёмкость 1
снабжённую
водным теплообменником и мешалкой,
парафин расплавляется и KMnO4 в нем
равномерно распределяется, полученая
смесь подаётся в окислительную колонну
3,
продувается N-H смесью разбавленной в 5
раз N2. Данная смесь 24 часа перемешивается
сверху вниз в колоне и при t=105С алкан
окисляется. При этом образуются кислоты,
спирты, кретоны, эфиры, лактоны. Для
уменьшения побочных продуктов степень
окисления парафина за один переход
35-40%. Продукты окисления (оксиданты)
вместе с kat поступают в 4,
где он перемешивается и обрабатывается
конц. HCl: 2KMnO4+16HCl
=2KCl+2MnCl2+5Cl2+8H2O.
Таким образом kat разлагается и переходит
в водную реакционную форму а Cl2 уходит.
Верхний орган. слой идёт в 5,
орошается холодной водой, при этом
отмываются водорастворимые ВКК. Их
отделяют в 6 и подвергают ректификации.
В органическом слое соединеяются
карбоновые ки-ты, спирты, лактоны, эфиры
и оставшиеся парафин. Для отделения
продуктов окисления из необходимо
перевести в водорастворимую форму-
мыла. Для этого оксидант идёт в 7,
где обрабатывается 20% Na2СO3:2RCOOH
(Na2CO3)->2 RCOONa+CO2+H2O.
Для разрешения лактонов оксиданты идут
в 8 и обрабатываются 30% NaOH и все переходит
в мыла. Образуется
2 слоя: водные
растворимые мыла, твёрдый парафин.
Для
разделения компонентов идут в 9,
верхний слой парафина отбирается и
промывается водой, обезвоживается и
возвращается в процесс, а водный раствор
мыла содержит мало парафина - его сжим.
до 22 атм в 10
и нагревают до 400С в 11.
Это вызывает полное расслоение остатка
парафина, который отделяется в 12.
Для перевода мыла в кислоты его в 13
обрабатывают 10% H2SO4, образуется слой
карболовой кислоты который разделяется
в 15.
Затем вакуумную ректификации в 15
при p 20ммртст получают продукты:
С5-С7-шампунь, С7-С9-шампунь взрослый,
С10-С17- детское мыло, С18-С20-хоз.мыло,
С20-пластифинат. Пр-во
высших спиртов (ВС).
ВС
расходуются на синтез ПАВ, СМС, фарматив.
Исходное сырье-это керосиновая фракция
температурой плавления 270-320С, которая
перед окисление подвергается карбомиды
очистки с целью удаления ароматических
УВ. Окисление проводят при атм р,
t=130-140, 3 часа. Для остановленный реакции
на спиртах добавляют 4-5% H2B2O7, котор. С
образованными спиртами образуют эфиры,
а они уступ. к окислению:3ROH+B(OH)3=(RO)3B+
3H2O.
Образование кислот, сложных эфиров
лактон не значительно. Образованная
смесь подвергается ректификации.
C9Очищенная
парафин. фракция и тв. H3BO4 (5% от m парафина)
поступает в
1,
имеющий мешалку и водонагреватель.
Парафин расплавляется перемешиванием
с H3BO3 и смесь поступает в окислительную
колону 3,
продувается снизу N-H смесью с соединениями
О2 примерно 3,5-4%. Смесь при температуре
130С обрабатывается три часа и за это
время до 40% парафина окисляется в спирты
в небольшом количестве ки-т лактонов.
Продукт окисления из 3
поступает в центрифугу 5,
где отделяется H3BO3, затем вакуумная
ректификация осущ. при давлении 10 ммртс.
Не прореагировавший парафин конденсируют
в холодильник
7 и
поступает в 8
и обрабатывается 30% NaOH. Дальше отделяются
карбоновые кислоты, лактоны, эфиры в
виде мыла. Парафин и мыло разделяется
в 9,
затем парафин промывают водой, обезвреж.
и идёт в процесс, а борные эфиры поступают
на гидролиз в 10,
где обрабвтыв. H3BO3 продувной. Ее отделяют,
снизу поступают спирты, а мыла отдуваются
в башне 12.
Промышленный
органический синтез. Химическая
переработка нефти (Н.). Современные
пр-ва орган. ве-в базируется на ископаемом
органич. сырье: Н. и природный газ.
Доминирующим источником сырья является
Н. Н. различается месторождением, заметно
отличается по своему фракционному
составу, а именно содержание лёгких,
средних и тяжёлых фракций, которые
выкипают до 180С. Основные химич. элементы
нефти: С(82-87%) Н2(11-14%) S(0,1-7%) N2(до 3%) О2(1%).
Общее количество алкенов нефти достигает
30-50%, циклоалканов 25-75%, арены содержаться
в меньших количествах. Соотношение
между группами углеводородов придают
Н. различные свойства и влияют на
номенклатуру полученного продукта.Н.-основной
источник сырья для нефтеперерабатывающих
заводов, при получении моторных масел,
топлива и мазута. Н. и продукты ее
переработки служат сырьем для синтеза
пластмасс, каучука, различных растворителей.
В перспективе они могут быть заменены
альтернативными энергоресурсами. Доля
Н., используемая в нефтехим. производствах
возрастёт скоро до 20%. Происходит
интеграция нефтеперерабатывающих
заводов и промышленности.Комбинированное
нефтеперерабатывающих и нефтехим.
процессов-это
пиролиз, синтез мономеров, пр-во пластмасс;
расширяет возможности выбора оптимальных
схем переработки нефти, повышает индекс
производительности систем для получения
моторных топлив для производства
сырья.Сейчас
исп. процессы рекомбинации, кот.
потенциально могут обеспечивать глубокую
переработку нефти, вплоть до 100%.
Выбор структуры нефтехим. компонентов
зависит от регионального и общего
способа и от ее природоохранных
факторов.Первичная
перегонка нефти-процесс
разделения Н. на фракции перегонки, а
вторичные процессы деструктивная или
химич. переработка нефти и очистка НП.
Перегонка
Н-первичный
технологический процесс НП. При
однократном испарении и последующей
конденсации паров получаем 2 фракции-легкую,
в ней содержится больше низкомолекулярных
компонентов и тяжёлую-меньше
низкомолекулярных компонентов, чем в
исходном сырье. Достичь требуемых
компонентов разделения нефти и получить
конечные продукты нельзя. В связи с этим
пары однократно подвергаются
ректификации-это массообменный процесс
разделения жидкости, различной по
температуре кипения, за счёт противоточного
многократного конденсирования паров
и жидкости. t,
необходимая для проведения процесса
получается в трубчатых печах, оборудованных
горелками. В зависимости от св-в
переработанной Н., ректификацию получают:
атмосф. трубчатых установок; установок,
сочетающих атмосф. вакуумные трубчатые
установки.Н.
подаётся через теплообменник, где она
нагревается до 170-175 теплотой продуктов
перегонки и подаётся в 1. Нагретая
до 350ºС Н подаётся в испарительную часть
2, работающую под атм. р. Здесь происходит
однократное испарение Н. При впуске в
испаритель Н, нагретая в 1 мгновенно
испаряется впоследствии резкого снижения
давления. При этом расходуется часть
теплоты. Пары низкомолекулярных фракций
устремляются вверх навстречу стекающей
вниз жидкости-флегме, при соприкосновении
с которой они охлаждаются и частично
конденсируются. Жидкость при этом
нагревается и из неё испаряются более
летучие фракции, те жидкости обогащается
высококипящими УВ, а пары-легколетучими.С10По
высоте колоны отбираются дистиляты
различного состава в строго определенном
интервале t.
При t=300-350
конденсируется и отбирается соляровое
масло, при 200-300С керосин, t=160-200
лигроиновая фракция. Из верхней части
колоны выводятся пары бензина, которые
охлаждаются и конденсируются в 3
и 4.
Часть жидкого бензина подают на орошение
колоны в 2.
В нижней части собирается мазут, при
дальнейшей переработке получают
смазочные масла во 2 ректификационной
колоне 6,
работающей под вакуумом. Для перегонки
мазута вакуум используется с целью
предотвращения расщепления углеводородов
под воздействием высоких t. Предварительно
мазут отправляют в 5
где он нагревается до 400-500С. Образующиеся
пары поступают в ректификационную
колону 6
которая поддерживает р=5-8 кПа. Стекающая
вниз по колоне жидкость продувается
водяным паром для облегчения испарения
лёгких компонентов и снижение t
в нижней части колоны. Ассортимент
вакуумной перегонки мазута зависит от
варианта переработки-масляный или
топливный. По
1 схеме получаются фракции: легкая,
средняя, тяжелый масленный дистилят.
По
2 схеме 1-ая фракция вакуумная газоида,
которая используется как kat крекинга
или гидрокрекинга.
Дистиляты из 1-ой схемы подвергаются
специальной очистке и затем смешиваются
в различных соотношениях для получения
различных сортов масел. Из нижней части
колоны выводят остаток перегонки нефти.
Гудрон используется как сырье для
термического крекинга, коксования,
производства бутилена, вывод масел.
CO2+
«С»,
2Ag+2C=Ag2C2. Жидкий
метанол поступает в кипятильник 2
внутри которого теплообменники обогрев.
горяч. водой. Снизу через слой метанола
пропускается очищенная азотоводородная
смесь, воздух насыщается парами метанола
и поступает в теплообменник 5.
Обогрев до t=800С. Метанол-азотоводородная
смесь нагревается до 750С и поступает. в
контактный аппарат 6,
внутри которого серебряная сетка или
слой пемзы, пропитанный серебром.
Требования:
отсутсвие следов железа (при их наличии:
CH3OH=CO2+H2,
CO=CO2+C(сажа),2Ag+
2C=Ag2C2).
При
этом cat отравляется необрат. Поэтому
ахотоводородная смесь и метанол очищают,
а контакт. аппарат изготавливают из
алюминия. При 750С метанол окисл. на 90%.
Газовая смесь проходит теплообменник
1
и отдаёт часть тепла на объём воды и газ
идёт в котёл 8
и охлаждается до 20С и поступает в общий
распределитель 9,
отчасти идёт в аппарат 10.
Здесь получается 30-40% формальдегида, в
котором 10% метанола, который явл.
ингибитором ре-ции полимеризации
формальдегида. Часть формальдегида
поступ. в рассольн. холодильник 11
и при 21С превращается в жидкость.Пр-во
этанола.В
настоящее время в России млрд тонн.
Основное кол-во-пр-во дивинила, эфиров,
растворителей, пищевых продуктов
топлива. Сущ.
4 метола получения:1.биохимический
способ;2.Серно-кислая
гидратациях этилена;3.Парообразная
гидратация этилена; 4.
Гидролиз древесины.1-исп.
картофель, фрукты, зерно.
Исх.
сырье разматывают, обрабатывают горяч.
водой до Клейстера, выдерж. несколько
часов до охлаждения 60С и добавл. солод
(проросшее зерно, в котором есть фермент
амилаза и под его действием происходит
гидролиз целлюлозы до мальтозы. Ра-р
охлаждают и добавляют дрожжи. Глюкоза
под действием дрожжей переходит в 2 моля
СО2 и 2 моля С6Н5ОН. Получ. бражка, которую
фильтрую и ректифицироваться по 2
ступеням. Сначала получ. спирт-81%, при
вторич. ректификации получают
спирт-ректификат (96,5-97,5%). 2-C3.
98% ацетилен сжим. компрессором до 15 атм
и пропускается через гидрататор 2
по всей высоте которого есть ситуативно
тарелки и водяной теплообменник для
поддержания 80-90С. Сверху аппарат орошает
98% H2SO4
и отрабатывают алкил-сульфатом. Содержащая
примеси смесь, промыв. в реакторе 3
водой, а в 4
NaOH,
и сжигают на факеле. Ра-р алкилсульфата
поступает в гидролизер 5,
где образуется спирт и диэтиловый эфир.
Образовавшуюся кислоту (50 %) подвергают
бараб. концентрирование в 6
и далее 98% H2SO4
собирают в приемнике 7
и далее может совращаться в пр-во. Пары
содержат спирт, эфир, воду и H2SO4.
Они охлаждаются в 8
и поступает в
9,
заполненный 29% Ca(OH)2.
Смесь пересушивают и H2SO4
удаляется. Гибс отделяют в центрифуге
10,
а водяной растворов спирта, эфира
поступают на 2-ух ступенчатый ректификатор
11.
Здесь отделяется эфир, затем спирт и с
конц. 83% его отправляют на дополнительную
ректификацию в аппарат 12.
Пары спирта концентрируют а холодильнике
13,
а жидкий спирт собирают в 14.
Часть спирта исп. на орошение башни 12.
Исходный ацетилен получают пиролизом
2
при 800С, при этом образуется пирогаз,
который содержит этилены и бутонных.
Пирогаз охлажд. и разделяют, пропускают
через колонку, орошают H2SO4
разной концентрации.
Раствора
газов обрабатывают в ситчатых колонках
оставшимся паром и газы отдув., затем
промывают водой и сушат.3-Использование
98% ацетилена. C2H4+H2O(c
H3PO4,
300ºC,
90атм)=C2H5OH+Q.
98%
ацетилен, пары воды и немного H3PO4
необходимо для восполнения её портрет
с поверхности kat. 1
сжигет до 70-89 атм., затем проходит
теплообменник 2
(t=110-150), за счёт постоянных газов из 4
в камеры печи 3
перегревают до 280С. С этой температурой
и давлением исходная смесь поступает
в полочной контакт
4,
внутр. часть которого из меди. За один
проход через катализатор образуется
до 5% этанола и немного эфира.C4.
Контактные
газы вначале проходят теплообменник 2
и водяной холодильник 5
и охлаждаются до 20-30С. При этом, спирты,
эфиры и вода конденсируются и отделяется
в аппарате 6,
а ацетилен приходит в 7
и промывается водой и возвращается в
процесс. Жидкие продукты из 6
и 7
поступают на мойку, которая проводит
Са(ОН)2:H3PO4+Ca(OH)2=
CaHPO4+2H2OВ
результате получается удобрение. Раствор
15% спирта и эфира выступают на
двух-ступенчатый ректификатор. На 1-ой
ступени в аппарате 8
отделяют эфиры, а растворы спирта
перегоняются в 12.
Жидкий спирт в 13.
Часть спирта-флегматик при орошении
11.
Общая степень конверсии 95%.4-При
переработке древесины образуется до
30% отходов/опилок.
В
составе древесины хвойных пород:
целлюлоза, гемицеллюлозы, пектоза,
лигнин, продукты не способные к гидролизу,
т.е. данное сырье является ценным для
производства глюкозы и фурфурола.
Процесс гидролиза при 15-20 атм, 2 часа, в
результате образуется глюкоза. Раствор
её нейтрализует оксидом кальция, далее
процесс брожения.C5.Древесные
отходы загружают в гидролизер, заливают
H2SO4
и гидролизирует, обогревают при 150°
выдерживают 2
часа. Протекает полном гидролиз с
образованием раствора глюкозы и
фурфурола. Через 2 часа жидкость продолжает
поступать в конденсатор 2
за счёт понижение давления от 20 до 1 атм.
Образуется раствор глюкозы и пары
фурфурола. Пары конденсируют в холодильник
3
и сливается в приёмник 4.
Из него получается пластмасса и смолы.
Водный раствор глюкоза поступает в
реактор 5,
заполненный гидроксидом кальция. Здесь
нейтрализуется кислота и образуется
гипс в реакторе 6.
Водный раствор глюкозы упаривается в
вакууме или подвергается брожению в
реакторе 7.
Через 12-14 суток брожение заканчивается,
всё фильтруют и ректифицируют. Пары
спирта кантеры в холодильнике 10.
Часть спирта идёт на орошение колонны,
а лигнин отмывают водой, сушиться и
используют для пиролиза для получения
фенола. При этом получается ароматич.
карбоновые кислоты.Пр-во
уксусной кислоты.
Она используется как сырье и среда для
проведения реакции окисления Ar УВ.
Сейчас укс.кисл получается kat окислением
ацетальдегида и мягкой бензиновой
фракцией или н-бутана. Если kat явл.
ацетальдегид Cu и добавляется этилметил
кетон то образуется только ангидрид
укс.кисл, тк кетоны образуют азиотропы
с водой, которая уходит из колоны и
реакция гидростанции не протекает.С6
Укс.альдегид и очищенная N-H смесь насосами
1
и 2
подаётся в окислительную колону 3 по
всей высоте катализатора-Стичатые
тарелки и по ним сверху стекает раствор
ацетата Mn в укс.кисл; есть в колоне водный
теплообменник и по нему циркулирует
горячая вода, тк промежуточный продукт
надуксусная кислота, которая взрываюоопасна.
Пр-во
уксусной кислоты окислением С4Н10.
С4Н10
получают термическим и kat. крекингом
нефти и высокотемпературного мазута,
гудрона и полугудрона. Окислен. С4Н10
приводит к образов. сложной смеси
продуктов. А CH3COOH
получ. в результате разрушения цепи. В
качестве примесей могут быть получены
сложные эфиры. Т.к. целевым яв пр-во
СН3СООН, то окисл. ведут таким обр., чтобы
степень превращения С4Н10-через слой
р-ра kat
не превышала 30-40%. Затем С4Н10 отделяют и
возвращают в пр-во.С7.С4Н10
сжим. в компрессоре 1 до 60атм. при сжигании
газы сильно разогрев. и поэтому после
помпрессора проходят Н2Охолодильник 3
и поступают в окислительную колонну 4.
Сюда же поступ. АВ смесь. Сверху по
ситчатым тарелкам стекает р-р (CH3COO)2Mn
в СН3СООН и дополн. продув. инертным
газом, тк при окислении образуется
пероксидные соединения, которые
взрывоопасны. Продукты окисления сначала
проходят в холодильник 5, t=20,
затем подверг. дрессимилированию 6до
р=1атм. В результате падения р происходит
конденсация всех компонентов за исключ.
С4Н10 и газов. В 7 собир. жидкость, а газовая
смесь проходит через 8 промыв. холодной
Н2О и газы идут в процесс (предварительно
мет. глубоко охлажд. С4Н10 отделяют от Н2
и N2).
Жидкость продолжает продуваться через
башню 7 и 8. подвергается ступенчатой
ректификации, отдел. пары и подаются в
10 и собир. в приемнике 11. Метод очень
дешевый, тк исходн. С4Н10 есть в больших
кол-вах, низкая цена.
Пр-во
высших карбоновых кислот(ВКК).
ВКК
получают каталитическим окислением
н-алканов, при этом в зависимости от
условий производится окисление с
разрушением цепи, так и без него. Можно
регулировать состав продуктов. Окисление
протекает под действием молекул О2 и
лимитируемая t, которая связана с длиной
цепи.
Высшие
парафины. Содержат
С15-С30. Kat-применение MnO2 или CrO3, либо
(CH3COO)2Mn или Со. Инициатором яв. НВr или
NO, NO2. Широко применяется каталит.
окисление-это фракция с t=375-425, соед.
С25-С35. При получении кислот обязательным
является отсутствие в них соединений
с разветвлённой цепью и ароматич. УВ,
тк они яв. сильными канцерогенам.
Биологическая среда водной артерии не
способна потреблять из алкана- Многие
вещества яв. не разлагаемыми(из ВКК).
Учитывая это из исходной фракции нефти,
выделяются изоалканы и ароматич. УВ и
затем обрабвтыв. парафинами или мочевиной.
Мочевина образует адукты с н-алканами,
а ароматич. УВ остаются в растворе.
Выпавшие в осадок адукты отделяются и
затем обрабатываются тёплой водой,
мочевина растворяется и всплывает
парафин с н-алканами, затем ещё раз
отмывается тёплой водой и только после
этого он используется как сырье в методе
Башкирова:C8.Чистый
парафин и KMnO4 загружают в ёмкость 1
снабжённую
водным теплообменником и мешалкой,
парафин расплавляется и KMnO4 в нем
равномерно распределяется, полученая
смесь подаётся в окислительную колонну
3,
продувается N-H смесью разбавленной в 5
раз N2. Данная смесь 24 часа перемешивается
сверху вниз в колоне и при t=105С алкан
окисляется. При этом образуются кислоты,
спирты, кретоны, эфиры, лактоны. Для
уменьшения побочных продуктов степень
окисления парафина за один переход
35-40%. Продукты окисления (оксиданты)
вместе с kat поступают в 4,
где он перемешивается и обрабатывается
конц. HCl: 2KMnO4+16HCl
=2KCl+2MnCl2+5Cl2+8H2O.
Таким образом kat разлагается и переходит
в водную реакционную форму а Cl2 уходит.
Верхний орган. слой идёт в 5,
орошается холодной водой, при этом
отмываются водорастворимые ВКК. Их
отделяют в 6 и подвергают ректификации.
В органическом слое соединеяются
карбоновые ки-ты, спирты, лактоны, эфиры
и оставшиеся парафин. Для отделения
продуктов окисления из необходимо
перевести в водорастворимую форму-
мыла. Для этого оксидант идёт в 7,
где обрабатывается 20% Na2СO3:2RCOOH
(Na2CO3)->2 RCOONa+CO2+H2O.
Для разрешения лактонов оксиданты идут
в 8 и обрабатываются 30% NaOH и все переходит
в мыла. Образуется
2 слоя: водные
растворимые мыла, твёрдый парафин.
Для
разделения компонентов идут в 9,
верхний слой парафина отбирается и
промывается водой, обезвоживается и
возвращается в процесс, а водный раствор
мыла содержит мало парафина - его сжим.
до 22 атм в 10
и нагревают до 400С в 11.
Это вызывает полное расслоение остатка
парафина, который отделяется в 12.
Для перевода мыла в кислоты его в 13
обрабатывают 10% H2SO4, образуется слой
карболовой кислоты который разделяется
в 15.
Затем вакуумную ректификации в 15
при p 20ммртст получают продукты:
С5-С7-шампунь, С7-С9-шампунь взрослый,
С10-С17- детское мыло, С18-С20-хоз.мыло,
С20-пластифинат. Пр-во
высших спиртов (ВС).
ВС
расходуются на синтез ПАВ, СМС, фарматив.
Исходное сырье-это керосиновая фракция
температурой плавления 270-320С, которая
перед окисление подвергается карбомиды
очистки с целью удаления ароматических
УВ. Окисление проводят при атм р,
t=130-140, 3 часа. Для остановленный реакции
на спиртах добавляют 4-5% H2B2O7, котор. С
образованными спиртами образуют эфиры,
а они уступ. к окислению:3ROH+B(OH)3=(RO)3B+
3H2O.
Образование кислот, сложных эфиров
лактон не значительно. Образованная
смесь подвергается ректификации.
C9Очищенная
парафин. фракция и тв. H3BO4 (5% от m парафина)
поступает в
1,
имеющий мешалку и водонагреватель.
Парафин расплавляется перемешиванием
с H3BO3 и смесь поступает в окислительную
колону 3,
продувается снизу N-H смесью с соединениями
О2 примерно 3,5-4%. Смесь при температуре
130С обрабатывается три часа и за это
время до 40% парафина окисляется в спирты
в небольшом количестве ки-т лактонов.
Продукт окисления из 3
поступает в центрифугу 5,
где отделяется H3BO3, затем вакуумная
ректификация осущ. при давлении 10 ммртс.
Не прореагировавший парафин конденсируют
в холодильник
7 и
поступает в 8
и обрабатывается 30% NaOH. Дальше отделяются
карбоновые кислоты, лактоны, эфиры в
виде мыла. Парафин и мыло разделяется
в 9,
затем парафин промывают водой, обезвреж.
и идёт в процесс, а борные эфиры поступают
на гидролиз в 10,
где обрабвтыв. H3BO3 продувной. Ее отделяют,
снизу поступают спирты, а мыла отдуваются
в башне 12.
Промышленный
органический синтез. Химическая
переработка нефти (Н.). Современные
пр-ва орган. ве-в базируется на ископаемом
органич. сырье: Н. и природный газ.
Доминирующим источником сырья является
Н. Н. различается месторождением, заметно
отличается по своему фракционному
составу, а именно содержание лёгких,
средних и тяжёлых фракций, которые
выкипают до 180С. Основные химич. элементы
нефти: С(82-87%) Н2(11-14%) S(0,1-7%) N2(до 3%) О2(1%).
Общее количество алкенов нефти достигает
30-50%, циклоалканов 25-75%, арены содержаться
в меньших количествах. Соотношение
между группами углеводородов придают
Н. различные свойства и влияют на
номенклатуру полученного продукта.Н.-основной
источник сырья для нефтеперерабатывающих
заводов, при получении моторных масел,
топлива и мазута. Н. и продукты ее
переработки служат сырьем для синтеза
пластмасс, каучука, различных растворителей.
В перспективе они могут быть заменены
альтернативными энергоресурсами. Доля
Н., используемая в нефтехим. производствах
возрастёт скоро до 20%. Происходит
интеграция нефтеперерабатывающих
заводов и промышленности.Комбинированное
нефтеперерабатывающих и нефтехим.
процессов-это
пиролиз, синтез мономеров, пр-во пластмасс;
расширяет возможности выбора оптимальных
схем переработки нефти, повышает индекс
производительности систем для получения
моторных топлив для производства
сырья.Сейчас
исп. процессы рекомбинации, кот.
потенциально могут обеспечивать глубокую
переработку нефти, вплоть до 100%.
Выбор структуры нефтехим. компонентов
зависит от регионального и общего
способа и от ее природоохранных
факторов.Первичная
перегонка нефти-процесс
разделения Н. на фракции перегонки, а
вторичные процессы деструктивная или
химич. переработка нефти и очистка НП.
Перегонка
Н-первичный
технологический процесс НП. При
однократном испарении и последующей
конденсации паров получаем 2 фракции-легкую,
в ней содержится больше низкомолекулярных
компонентов и тяжёлую-меньше
низкомолекулярных компонентов, чем в
исходном сырье. Достичь требуемых
компонентов разделения нефти и получить
конечные продукты нельзя. В связи с этим
пары однократно подвергаются
ректификации-это массообменный процесс
разделения жидкости, различной по
температуре кипения, за счёт противоточного
многократного конденсирования паров
и жидкости. t,
необходимая для проведения процесса
получается в трубчатых печах, оборудованных
горелками. В зависимости от св-в
переработанной Н., ректификацию получают:
атмосф. трубчатых установок; установок,
сочетающих атмосф. вакуумные трубчатые
установки.Н.
подаётся через теплообменник, где она
нагревается до 170-175 теплотой продуктов
перегонки и подаётся в 1. Нагретая
до 350ºС Н подаётся в испарительную часть
2, работающую под атм. р. Здесь происходит
однократное испарение Н. При впуске в
испаритель Н, нагретая в 1 мгновенно
испаряется впоследствии резкого снижения
давления. При этом расходуется часть
теплоты. Пары низкомолекулярных фракций
устремляются вверх навстречу стекающей
вниз жидкости-флегме, при соприкосновении
с которой они охлаждаются и частично
конденсируются. Жидкость при этом
нагревается и из неё испаряются более
летучие фракции, те жидкости обогащается
высококипящими УВ, а пары-легколетучими.С10По
высоте колоны отбираются дистиляты
различного состава в строго определенном
интервале t.
При t=300-350
конденсируется и отбирается соляровое
масло, при 200-300С керосин, t=160-200
лигроиновая фракция. Из верхней части
колоны выводятся пары бензина, которые
охлаждаются и конденсируются в 3
и 4.
Часть жидкого бензина подают на орошение
колоны в 2.
В нижней части собирается мазут, при
дальнейшей переработке получают
смазочные масла во 2 ректификационной
колоне 6,
работающей под вакуумом. Для перегонки
мазута вакуум используется с целью
предотвращения расщепления углеводородов
под воздействием высоких t. Предварительно
мазут отправляют в 5
где он нагревается до 400-500С. Образующиеся
пары поступают в ректификационную
колону 6
которая поддерживает р=5-8 кПа. Стекающая
вниз по колоне жидкость продувается
водяным паром для облегчения испарения
лёгких компонентов и снижение t
в нижней части колоны. Ассортимент
вакуумной перегонки мазута зависит от
варианта переработки-масляный или
топливный. По
1 схеме получаются фракции: легкая,
средняя, тяжелый масленный дистилят.
По
2 схеме 1-ая фракция вакуумная газоида,
которая используется как kat крекинга
или гидрокрекинга.
Дистиляты из 1-ой схемы подвергаются
специальной очистке и затем смешиваются
в различных соотношениях для получения
различных сортов масел. Из нижней части
колоны выводят остаток перегонки нефти.
Гудрон используется как сырье для
термического крекинга, коксования,
производства бутилена, вывод масел.