

Стехиометрические законы: закон сохранения массы, закон постоянства состава. Их роль в химии и современная трактовка. Газовые законы: закон Авогадро, уравнение Менделеева-Клапейрона.
Стехиометрия — раздел химии, в котором рассматриваются массовые или объемные отношения между реагирующими веществами.
ЗАКОН СОХРАНЕНИЯ МАССЫ (первоначальная формулировка): масса веществ, вступающих в реакцию, равна массе веществ, образующихся в результате реакции (число атомов определенного сорта в исходных веществах и продуктах реакции одинаково).
СОВРЕМЕННАЯ ФОРМУЛИРОВКА: сумма массы вещества системы и массы, эквивалентной энергии, которая получена или отдана этой системой, постоянна.
Закон сохранения массы в современной формулировке следует применять только для ядерных реакций, сопровождающихся выделением огромного количества энергии.
Установление первых двух стехиометрических законов позволило приписать атомам химических элементов строго определенную массу. Закон сохранения массы и закон постоянства состава применяются при проведении количественных расчетов.
ЗАКОН ПОСТОЯНСТВА СОСТАВА: любое химически чистое соединение независимо от способа его получения состоит из одних и тех же химических элементов, причем отношения их масс постоянны, а относительные числа их атомов выражаются целыми числами.
Закон выполняется только для химических соединений, состоящих из молекул. Для сложных в-в, которые состоят из атомов или ионов, его нельзя считать справедливым. Для них определяют границы состава, а не точное число атомов в химическом соединении.
ЕСЛИ ХИМИЧЕСКИЕ РЕАКЦИИ ПРОТЕКАЮТ С УЧАСТИЕМ ГАЗООБРАЗНЫХ В-В, ТО ДЛЯ ОПРЕДЕЛЕНИЯ КОЛИЧЕСТВА ЭТИХ В-В ИСПОЛЬЗУЮТ ЗАКОН АВОГАДРО И УРАВНЕНИЕ КЛАЙПЕРОНА-МЕНДЕЛЕЕВА.
ЗАКОН АВОГАДРО: в равных объемах различных газов, взятых при одинаковых значениях температуры и давления, содержится одно и то же число молекул.
Следствие: один моль любого газа при одинаковых условиях занимает одинаковый объем.
Закон Авогадро справедлив только для идеального газа, в котором единственно возможным видом взаимодействия частиц является их упругое соударение. При нормальных условиях (275K и давлении 1 атм) объем 1 моля идеального газа =22,414 л/моль.
ОБЪЕДИНЕННЫЙ ГАЗОВЫЙ ЗАКОН ИЛИ УРАВНЕНИЕ КЛАЙПЕРОНА-МЕНДЕЛЕЕВА.
p ⋅V = ν ⋅ R ⋅T, где р – давление газа, Па; V – объем, м³; ν – число молей газа; T – абсолютная температура, K; R – универсальная газовая постоянная (R = 8,314 Дж/(K⋅моль))
Если объем или давление выражены в каких-либо иных единицах, следует использовать другие значения R (0,082 (л⋅атм)/(моль⋅K); 62,4 (л⋅мм рт. ст.)/(моль⋅K))
Если в химической реакции участвуют несколько газов, давление газовой постоянной смеси равно сумме давлений, создаваемых каждым ее компонентов. Давление компонента газовой смеси называют парциальным давлением.
Основные понятия и определения термодинамики. Энтальпия. Энтальпия химической реакции. Экспериментальное определение энтальпии реакции (на примере реакции нейтрализации).
Термодинамика – наука, изучающая закономерности превращения теплоты, работы и прочих форм энергии друг в друга. Химическая термодинамика применяет общие законы термодинамики к химическим процессам. С ее помощью определяют, сколько энергии выделяется или поглощается при химической реакции. Ее законы позволяют определить, каким станет состав смеси в-в к тому моменту, когда химические превращения в системе завершатся. Для применения законов химической термодинамики нужно знать только свойства исходных в-в и продуктов реакции.
Термодинамика не говорит ничего ни о скорости, ни о конкретном пути химической реакции; нельзя использовать законы термодинамики для описания отдельных частиц.
Под ТЕРМОДИНАМИЧЕСКОЙ СИСТЕМОЙ понимают часть Вселенной, выделенную при помощи реальных или мысленных границ.
Все, что находится вне системы, называют ВНЕШНЕЙ СРЕДОЙ.
ВСЕЛЕННАЯ — СИСТЕМА + ВНЕШНЯЯ СРЕДА
Изолированная система не обменивается с внешней средой ни энергией, ни в-вом.
Закрытая система – обменивается энергией, но не обменивается в-вом.
Открытая система – обменивается и энергией, и в-вом.
Параметры системы делят на экстенсивные и интенсивные.
Экстенсивным называются параметры, значения которых зависят от общего объема системы. (m, V, n)
Интенсивными называются параметры, значения которых не зависят от общего объема системы. (p, T, C)
КОМПОНЕНТЫ — вещества, образующие систему, количества которых можно менять независимо друг от друга.
Раствор NaCl
Компоненты H2O и NaCl
Состоит из молекул H2O, ионов Na+ и Cl–
Часть системы, в пределах которой химический состав, плотность и другие интенсивные св-ва постоянные или изменяются плавно, без скачков, называют фазой. При переходе из одной фазы в другую, т.е. на границе раздела фаз, эти св-ва изменяются скачком.
Систему, состоящую из одной фазы называют гомогенной, из нескольких фаз – гетерогенной.
ЭНТАЛЬПИЯ
ЗАКОН СОХРАНЕНИЯ ЭНЕРГИИ: если в результате какого-либо процесса в закрытой или открытой системе внутренняя энергия изменяется, то избыток энергии выделяется во внешнюю среду, а недостаток энергии – восполняется из внешней среды.
U – внутренняя энергия системы (сумма потенциальной и кинетической энергий всех составляющих систему частиц.) ∆U ─ изменение внутренней энергии системы в процессе
Q ─ тепловой эффект процесса A ─ работа (A = p⋅ ∆V ) ∆U + Q + A = 0 Q = –(∆U + p⋅ ∆V)
const V A = 0 Qv = –∆U
const р Qp = –∆(U + p⋅ V) = –∆H
Энтальпия химической реакции равна по величине и противоположна по знаку тепловому эффекту этой реакции, измеренному при постоянном давлении и постоянной температуре.
Экзотермический процесс Q > 0, ∆H < 0, ∆U < 0
Эндотермический процесс Q < 0, ∆H > 0, ∆U > 0
Энтальпии реакций, в которых исходные вещества и продукты находятся в стандартных состояниях, называют стандартными энтальпиями реакций ∆rH0
Экспериментальное определение энтальпии реакции (на примере реакции нейтрализации).
Нейтрализация сильной кислоты сильным основанием
Практикум 2 опыт 3.
NaCl + KOH = KCl+H₂O
СHCL = 0,6 M CKOH = 6 M
Ход работы:
Ждем постоянства температуры в системе калориметра
Измеряем температуру( пока она не перестанет изменятся) после добавления щелочи ∆Т1
Дожидаемся нормализации температуры, а затем включаем нагреватель( сопротивление “R” и напряжение “U” нагревателя известны). Ждем когда температура перестанет увеличиваться(засекаем сколько прошло времени с момента нагрева - ∆t) изменение температуры при нагреве ∆Т2
Производим расчеты:
Кол-во
теплоты, затраченное на нагрев калориметра
q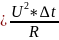
Тепловое
значение калориметра K
=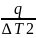
Кол-во теплоты выделившееся в рез-те реакции q(реакции) = K * ∆T1
Тепловой
эффект Q =
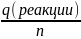
Энтальпия ∆Н = -Q
Энтальпия образования вещества. Стандартное состояние элемента и веще- ства. Расчет энтальпий реакций по стандартным энтальпиям образования веществ (на конкретном примере).
Энтальпия химической реакции равна по величине и противоположна по знаку тепловому эффекту этой реакции, измеренному при постоянном давлении и постоянной температуре.
ЭНТАЛЬПИЯ ОБРАЗОВАНИЯ ВЕЩЕСТВА :
Стандартная энтальпия образования вещества ∆fH0 — энтальпия реакции образования 1 моль этого вещества в стандартном состоянии из соответствующих простых веществ.
Стандартное состояние вещества – состояние данного вещества в чистом виде при заданной температуре и давлении 1 атм.
Стандартное состояние элемента – наиболее устойчивое просто в-во, образуемое данным элементом при стандартной температуре 298,15K и стандартном давлении 1 атм.
Стандартное состояние растворенного вещества – 1М идеальный раствор при раствор при заданной температуре и давлении 1 атм
Энтальпия связи (лучше знать) – энтальпия образования 1 моля в-ва в стандартном состоянии из идеального газа свободных атомов элементов, образующих данное вещество.
Стандартная энтальпия образования простого вещества в стандартном состоянии равна нулю.
Если элемент образует несколько простых веществ равной нулю считается стандартная энтальпия образования только одного из них (наиболее распространенного при 298 К (25°С)).
Пример расчета энтальпии реакции по стандартным энтальпиям образования веществ:
2H2O(г.) + CH4(г.) = CO2(г.)+4H2(г.) ∆rH0=∑ jпрод. ∆fH0 прод. - ∑ iисх. ∆fH0 исх.
H2 – простое в-во, ∆fH0=0
|
СO2(г.) |
H2O(г.) |
CH4(г.) |
∆fH0, кДж/моль |
-393,5 |
-241, 8 |
-74,8 |
ТОГДА: ∆rH0 = -393,5 – 2 (-241,8) + 74,8 = 164,9 кДж/моль > 0 реакция эндотермическая
Закон Гесса. Следствия из закона Гесса. Способы расчета энтальпий реакций с использованием закона Гесса (на конкретных примерах).
ЗАКОН ГЕССА - энтальпия химического процесса не зависит от его пути, а определяется только начальным и конечным состоянием системы, т.е. состоянием исходных веществ и продуктов реакции.
Следствие 1 - энтальпия химического процесса определяется энтальпиями тех реакций, комбинируя которые можно получить этот процесс
Пример 1 (следствие 1): вычислите энтальпию реакции: (3) 3/2О2(г.) = О3(г.); по следующим данным: (1) As2O3(к.) + О2(г.)= As2O5(к.) + 271 кДж, (2) 3As2O3(к.) + 2O3(г.) = 3As2O5(к.) + 1096 кДж.
(3) = 3/2(1) – 1/2(2)
∆rH0(1) = -271 кДж; ∆rH0(2) = -1096 кДж
∆rH0(3) = 3/2 (-271) – 1/2(-1096) = 141,5 кДж
Следствие 2 - Энтальпия реакции определяется как разность энтальпий образования продуктов реакции и энтальпий образования исходных веществ (с учетом стехиометрических коэффициентов)
Пример 2 (следствие 2): ∆rH= Σjпрод.∆fHпрод. – Σ iисх.в-в.∆fHисх.в-в.
2H2O(г.) + CH4(г.) = CO2(г.)+4H2(г.) ∆rH0=∑ jпрод. ∆fH0 прод. - ∑ iисх. ∆fH0 исх.
H2 – простое в-во, ∆fH0=0
|
СO2(г.) |
H2O(г.) |
CH4(г.) |
∆fH0, кДж/моль |
-393,5 |
-241, 8 |
-74,8 |
ТОГДА: ∆rH0 = -393,5 – 2 (-241,8) + 74,8 = 164,9 кДж/моль > 0 реакция эндотермическая
Следствие 3 – Энтальпия обратной реакции равна энтальпии прямой реакции, взятой с обратным знаком.
Например: 2SO2 + O2 = 2SO3 (обратимая реакция)
5. Самопроизвольные и несамопроизвольные процессы (примеры). Макро-и микросостояния системы. Термодинамическая вероятность и энтропия. Возрастание энтропии как движущая сила самопроизвольного процесса.
С термодинамической точки зрения процессы делят на самопроизвольные и несамопроизвольные.
Самопроизвольный процесс протекает без всякой помощи извне, хотя для его начала иногда необходимо внешнее воздействие. Пример: H2+F2 = 2HF, C+O2 = CO2 (необходимо нагреть до температуры около 800 ͦС)
Несамопроизвольные процессы не могут протекать без постоянной затраты работы извне. Пример: электролиз расплава хлорида натрия – 2NaCl = 2Na + Cl2
Макросостоянием называют состояние системы, которое можно охарактеризовать определенными, реально наблюдаемыми параметрами (Т, р, состав).
Способы реализации макросостояний – микросостояния
Микросостояние – состояние системы, для которого известно все, что происходит с каждой частицей системы.
Число способов (микросостояний), которыми может быть реализовано макросостояние называют термодинамической вероятностью (W)
Процессы от состояния с низкой термодинамической вероятностью к состоянию с высокой термодинамической вероятностью являются самопроизвольными.
Величину lnW, умноженную на постоянную Больцмана k (R/NA) (1,38 ∙ 10-23 Дж/К), называют энтропией и обозначают буквой S (мера беспорядка системы)
S = k lnW
Самопроизвольные процессы идут в направлении возрастания термодинамической вероятности, а следовательно, и энтропии системы. Поэтому возрастание энтропии в изолированной системе является условием самопроизвольности протекающего в этой системе процесса.
6. Энтропия вещества. Зависимость энтропии вещества от температуры, объема, агрегатного состояния. Энтропия химической реакции. Способы расчета энтропии химической реакции (на конкретных примерах). Энтропия образования вещества.
Энтропия – мера беспорядка системы.
Значение энтропии 1 моля в-ва в его стандратном состоянии называют стандартной энтропией в-ва.
Энтропия – экстенсивная величина.
∆S = Q/T ∆S = - ∆rH/T ∆Sобщ. = ∆rS + ∆Sвнешн. среды.
Энтропия увеличивается при переходе от твердого к жидкому и особенно к газообразному состоянию
∆Sтв.< ∆Sж.< ∆Sг.
Энтропия обычно возрастает при растворении твердого в-ва в воде или каком-нибудь другом растворителе. Энтропия газа уменьшается при его растворении в воде или другом растворителе. Энтропия 1 моля в-ва тем больше, чем сложнее его химический состав. При одинаковой сложности состава в-ва в одном и том же агрегатном состоянии энтропия в-ва тем больше, чем больше молекулярная масса.
Стремление системы к беспорядку проявляется тем больше, чем выше температура. Произведение изменения энтропии системы на температуру T ΔS количественно оценивает эту тенденцию и называется энтропийным фактором. Энтропия РЕАКЦИИ мало зависит от температуры.
Чем больше объем, тем больше энтропия. Зависимость энтропии от объема может быть обусловлена возможностью перемещения частиц, образующих систему, в предоставленном им пространстве. (НЕ УВЕРЕНА)
ЭНТРОПИЯ ХИМИЧЕСКОЙ РЕАКЦИИ – разность суммы энтальпий образования продуктов и суммы энтальпий образования исходных в-в с учетом стехиометрических коэффициентов.
Энтропией образования в-ва называют энтропию реакции в которой 1 моль этого в-ва образуется из простых в-в, являющихся стандартными состояниями элементов.
Способы расчета энтропии:
∆rS = Σjпрод.Sпрод. — Σiисх. в-вSисх. в-в ∆rS = 143 – 64,7 – 1/2∙239,2 – 3/2∙205 = -348, 7 Дж/Моль∙К
K(к.) + 1/2 Cl2(г.)+ 3/2 О2(г.) = KClO3(к.)
S0(K(к.)) = 64,7 Дж/Моль∙К
S0(Cl2(г.)) = 239,2 Дж/Моль∙К
S0(О2(г.)) = 205 Дж/Моль∙К
S0(KClO3(к.))= 143 Дж/Моль∙К
Через энергию Гиббса: ∆fG° = ∆fH° –T∙∆fS°
∆rG = Σjпрод.∆fGпрод. – Σ iисх.в-в.∆fGисх.в-в.
2H2O(г.) + CH4(г.) = CO2(г.)+4H2(г.) ∆rH0=∑ jпрод. ∆fH0 прод. - ∑ iисх. ∆fH0 исх.
H2 – простое в-во, ∆fH0=0, ∆fG0= 0
|
СO2(г.) |
H2O(г.) |
CH4(г.) |
∆fH0, кДж/моль |
-393,5 |
-241, 8 |
-74,8 |
∆fG0, кДж/моль |
-394,4 |
-228,6 |
-50,8 |
∆fS0, Дж/К∙моль |
213,7 |
188,7 |
186,3 |
ТОГДА: ∆rH0 = -393,5 – 2 (-241,8) + 74,8 = 164,9 кДж/моль > 0 реакция эндотермическая
∆rG0= -394,4 – 2(-228,6) +50,8 =113,6 кДж/моль
Вычислите, чему равна энтропия 1 г натрия в форме идеального кристалла при 0 k.
В 1 г натрия содержится (m/M)/NA = 2,61∙1022 атомов.
Поскольку при 0K тепловое движение отсутствует, термодинамическая вероятность = кол-ву атомов.
S = k lnW = 7,12 ∙ 10-22 Дж/K
7. Энергия Гиббса. Энергия Гиббса и самопроизвольность процесса: энталь- пийный и энтропийный факторы. Способы расчета энергии Гиббса химической реакции (на конкретном примере).
∆rG < 0 — можно получить полезную работу ∆rG > 0 — необходимо затратить работу
H2(г.) + 1/2 O2(г.) = H2O(ж.) 6CO2(г.) + 6H2O(ж.) → C6H12O6 (к.) + 6O2(г.)
∆rG° = –237,2 кДж ∆rG° = 2870,4 кДж
Энергия Гиббса ∆rG° = ∆rH° –T∙∆rS°
Для реакций образования вещества ∆fG° = ∆fH° –T∙∆fS°
ЭНЕРГИЯ ГИББСА ХИМИЧЕСКОЙ РЕАКЦИИ – разность суммы энергий Гиббса образования продуктов и суммы энергий Гиббса образования исходных в-в с учетом стехиометрических коэффициентов.
Процессы с ∆rG < 0 являются самопроизвольными
При ∆rH > 0, ∆rS > 0 реакции могут протекать самопроизвольно при высоких температурах
При ∆rH < 0, ∆rS > 0 реакции могут протекать самопроизвольно при любых температурах
При ∆rH > 0, ∆rS < 0 реакции не могут протекать самопроизвольно ни при каких температурах
При ∆rH < 0, ∆rS < 0 реакции могут протекать самопроизвольно при низких температурах
Благоприятные факторы - ∆rH < 0, ∆rS > 0
∆rG =∆fG + RT ln a
Активность в-в стандартном состоянии = 1
Для газообразных в-в активность равна парциальному давлению (в атм). Для растворенных веществ и ионов активность численно близка к концентрации, выраженной молей на литр.
Способы расчета энергии Гиббса химической реакции:
Через стандартные энергии Гиббса образования
∆rG = Σjпрод.∆fGпрод. – Σ iисх.в-в.∆fGисх.в-в.
2H2O(г.) + CH4(г.) = CO2(г.)+4H2(г.) ∆rH0=∑ jпрод. ∆fH0 прод. - ∑ iисх. ∆fH0 исх.
H2 – простое в-во, ∆fH0=0, ∆fG0= 0
|
СO2(г.) |
H2O(г.) |
CH4(г.) |
∆fG0, кДж/моль |
-394,4 |
-228,6 |
-50,8 |
ТОГДА: ∆rG0= -394,4 – 2(-228,6) +50,8 =113,6 кДж/моль, реакция не самопроизвольна
Через энтропию и энтальпию
Вычислить энергию Гиббса CH4 + H2O = CO +3H2 при температурах 750°.
При 750°С (1023 К) вода находится в газообразном состоянии, поэтому уравнение реакции выглядит так: CH4(г.) + H2O(г.) = CO(г.) + 3H2(г.). Для расчета энергии Гиббса этой реакции при 1023 К воспользуемся формулой ΔrG° = ΔrH° – 1023∙ΔrS°. ΔrH°298 = 206,1кДж, ΔrS°298 = 214 Дж/К
ΔrG°1023 = 206100 – 1023∙214 = –12822 Дж = –12,822 кДж. Энергия Гиббса реакции меньше нуля, поэтому при 750°С и стандартных состояниях веществ реакция является термодинамически самопроизвольной.
Через концентрации ионов
Для реакции Ag+ (р.) + Cl– (р.) = AgCl(к.) при 25°С определите энергию Гиббса ΔrG при равных 1,0·10–5 моль/л концентрациях ионов Ag+ и Cl– в растворе.
Сначала определим энергии Гиббса образования ионов Ag+ и Cl– при концентрациях 1,0·10–5 моль/л. Поскольку в разбавленных растворах активность иона принимают равной его концентрации,
ΔfG(Ag+ (р.)) = ΔfG°(Ag+ (р.)) + RТ ln a(Ag+ (р.)) = 77100 + 8,314∙298∙ln (10–5) = = 48576 Дж = 48,6 кДж/моль; ΔfG(Cl– (р.)) = ΔfG°(Cl– (р.)) + RТ ln a(Cl– (р.)) = –159,8 кДж/моль.
ΔrG = ΔfG°(AgCl(к.)) – ΔfG(Ag+ (р.)) – ΔfG(Cl– (р.)) = –109,8 – (48,6 – 159,8) = 81,4 кДж > 0
8. Стандартная энергия Гиббса образования вещества. Термодинамическая активность вещества. Расчет энергии Гиббса реакции с учетом термодина- мической активности ее участников.
Стандартная энергия Гиббса образования ∆fG° — изменение энергии Гиббса при образовании 1 моля вещества в стандартном состоянии из простых веществ в стандартных состояниях элементов.
Энергия Гиббса образования ∆fG° простого вещества в стандартном состоянии элемента при любой температуре равна нулю
Для того, чтобы охарактеризовать отклонение энергии Гиббса образования вещества (или иона), находящегося в газовой смеси или в растворе, от его свойств в стандартном состоянии, используют величину, называемую термодинамической активностью данного вещества или иона. Ее обозначают буквой a и используют для определения энергии Гиббса образования вещества:
∆rG =∆fG + RT ln a
Активность в-в стандартном состоянии = 1
Для газообразных в-в активность равна парциальному давлению (в атм). Для растворенных веществ и ионов активность численно близка к концентрации, выраженной молей на литр
Пример:
Для реакции Ag+ (р.) + Cl– (р.) = AgCl(к.) при 25°С определите энергию Гиббса ΔrG при равных 1,0·10–5 моль/л концентрациях ионов Ag+ и Cl– в растворе.
Сначала определим энергии Гиббса образования ионов Ag+ и Cl– при концентрациях 1,0·10–5 моль/л. Поскольку в разбавленных растворах активность иона принимают равной его концентрации,
ΔfG(Ag+ (р.)) = ΔfG°(Ag+ (р.)) + RТ ln a(Ag+ (р.)) = 77100 + 8,314∙298∙ln (10–5) = = 48576 Дж = 48,6 кДж/моль; ΔfG(Cl– (р.)) = ΔfG°(Cl– (р.)) + RТ ln a(Cl– (р.)) = –159,8 кДж/моль.
ΔrG = ΔfG°(AgCl(к.)) – ΔfG(Ag+ (р.)) – ΔfG(Cl– (р.)) = –109,8 – (48,6 – 159,8) = 81,4 кДж > 0
9. Скорость химической реакции. Зависимость скорости химической реакции от концентраций реагирующих веществ. Кинетическое уравнение и порядок реакции. Методы экспериментального определения порядка реакции (конкретный пример).
Химическая кинетика изучает закономерности протекания химических реакций во времени.
Если в системе протекает несколько реакций, то каждая из них подчиняется основному закону химической кинетики и протекает независимо от других реакций.
В состоянии химического равновесия отношение концентраций продуктов в степенях их стехиометрических коэффициентов, к концентрациям исходных веществ, в степенях их стехиометрических коэффициентов, величина постоянная (K РАВНОВЕСИЯ)
Скорость гомогенной реакции — изменение количества вещества n в единице объема V в единицу времени, τ
τ ∆ = ± 1/V ∙ Δn/Δt = ± ΔC/Δt
Скорость гетерогенной реакции — изменение количества вещества n в единицу времени на единице площади S, τ
τ = ± 1/S ∙ Δn/Δt
Стадии гетерогенного процесса – подвод реагентов к границе раздела,
Зависимость скорости химической реакции от концентрации.
Скорость реакции зависит: 1. Природы реагирующих веществ 2. Концентрации реагирующих веществ 3. Температуры 4. Присутствия катализатора (ингибитора)
τ = k ∙ ∏i Ciai
Сi — концентрация i-го компонента ai — порядок реакции по i-му компоненту n = Σαi — общий порядок реакции k — константа скорости.
основной закон химической кинетики – скорость реакции пропорциональна произведению концентраций реагирующих в-в, в степенях, называемых порядками реакций по соответствующим элементам. Порядок реакции устанавливается экспериментально, для простых реакций совпадает со стехиометрическими коэффициентами.
КОНСТАНТА СКОРОСТИ — коэффициент в кинетическом уравнении, численно равный скорости реакции при концентрациях всех реагентов равных 1 моль/л
τ = k [A]ᵅ[B]ᵝ
Экспериментально установленные кинетические уравнения для некоторых реакций:
2N2O5(p.) = 2N2O4(p.)+O2(г.) τ = k С(N2O5)
H2(г.) + Br2(г.) = 2HBr(г.) τ = k С(H2)(C(Br2))1/2
H2(г.) + I2(г.) = 2HI(г.) τ = k С(H2)C(I2)
Методы экспериментального определения порядка реакции
1.Метод изолирования Оствальда на примере необратимой сложной реакции с участием трех исходных в-в:
A + B + C = D + F
Кинетическое уравнение имеет вид: k∙ C(A)ᵅC(B)ᵝC(C)ᵞ
Проводится несколько опытов с различной концентрацией одного и того же реагента, чтобы изучить зависимость скорости реакции от концентраций каждого из исходных в-в по отдельности. Все остальные реагенты берут в большом избытке, в этом случае их концентрации можно считать постоянными и пренебречь их расходованием. Чтобы определить порядок реакции а, измеряют скорость реакции в зависимости концентрации реагента А. Тогда кинетическое уравнение можно записать в виде: k’ C(A)ᵅ; k’= k∙ C(B)ᵝC(C)ᵞ - величина постоянная и не меняется от опыта к опыту.
a = (lnr2 – lnr1)/(lnC2 – lnC1)
2. Графический способ
10. Скорость химической реакции. Зависимость скорости химической реакции от температуры. Уравнение Аррениуса. Энергетический профиль химической реакции. Экспериментальное определение энергии активации химической реакции (конкретный пример).
Химическая кинетика изучает закономерности протекания химических реакций во времени.
Если в системе протекает несколько реакций, то каждая из них подчиняется основному закону химической кинетики и протекает независимо от других реакций.
В состоянии химического равновесия отношение концентраций продуктов в степенях их стехиометрических коэффициентов, к концентрациям исходных веществ, в степенях их стехиометрических коэффициентов, величина постоянная (K РАВНОВЕСИЯ)
Скорость гомогенной реакции — изменение количества вещества n в единице объема V в единицу времени, τ
τ ∆ = ± 1/V ∙ Δn/Δt = ± ΔC/Δt
Скорость гетерогенной реакции — изменение количества вещества n в единицу времени на единице площади S, τ
τ = ± 1/S ∙ Δn/Δt
Стадии гетерогенного процесса – подвод реагентов к границе раздела,
Зависимость скорости химической реакции от концентрации.
Скорость реакции зависит: 1. Природы реагирующих веществ 2. Концентрации реагирующих веществ 3. Температуры 4. Присутствия катализатора (ингибитора)
Зависимость скорости реакции от температуры:
Скорость подавляющего большинства реакций возрастает при увеличении температуры. Причиной этого является увеличение с ростом температуры доли активных молекул, т.е. молекул, имеющих достаточную энергию для того, чтобы вступить в реакцию.
Уравнение Аррениуса Ea - энергия активации процесса, A – предэкспоненциальный множитель
![]()
Энергия активации - минимальная избыточная энергия, которую необходимо иметь реагирующим молекулам для того, чтобы они могли вступить в химическую реакцию.
Ea = RT1T2/(T2-T1) ln(k2/k1)
С повышением температуры число активных молекул увеличивается и, следовательно, возрастает скорость химической реакции.
Кроме того, скорость реакции зависит от вероятности благоприятной пространственной ориентации молекул в момент их столкновения (стерический фактор). (входит в предэкспоненциальный множитель, мало зависит от T)
Правило Вант-Гоффа – при увеличении температуры на 10 градусов, скорость реакции возрастает в 2-4 раза. Правило пригодно для небольших интервалов температуры.
11. Катализ. Влияние катализатора на скорость химической реакции. Причины влияния. Гомогенный и гетерогенный катализ. Автокатализ. Ферментативный катализ. Примеры практического использования катализаторов для изменения скорости реакции. Ингибирование реакций.
Катализ – явление ускорения химических реакций под действием малых количеств веществ - катализаторов, которые сами в процессе реакции не расходуются и после ее окончания остаются неизменными. Катализаторы – вещества, ускоряющие химическую реакцию. Вступают в промежуточное химическое взаимодействие с участниками реакции, а затем восстанавливают свой химический состав. С помощью катализатора реакция направляется по выгодному пути с меньшей энергией активации. Катализатор может не только ускорять реакцию, но и направлять ее по пути, наиболее благоприятному для образования определенного продукта.
Гомогенный катализ - катализатор и реагирующие в-ва находятся в 1 фазе.
Гетерогенный катализ - катализатор и реагирующие в-ва находятся в разных фазах.
Химическую реакцию называют автокаталитической, если ее продукт служит ее же катализатором.
Практически все химические процессы, протекающие в живых организмах, ускоряются биокатализаторами – ферментами. Большинство известных ферментов имеет белковую природу. Реакция, катализируемая белком протекает не на любом участке белковой молекулы, а только в так называемом активном центре, состоящем из нескольких аминокислотных остатков. Помимо аминокислотных остатков активный центр может содержать также ионы металла, органические и комплексные молекулы.
Каталитическая активность ферментов гораздо выше, чем обычных катализаторов. В реакциях, катализируемых ферментами высокий стерический фактор, благоприятная организация молекул, что приводит к росту предэкспоненциального множителя.
Вещества, понижающие скорость химической реакции называют ингибиторами. Ингибирование реакции не связано с повышением ее энергии активации. Роль ингибитора обычно сводится к разрушению или связыванию активных частиц, являющихся промежуточными веществами в реакции, например свободных радикалов. В гетерогенных процессах ингибиторы обычно адсорбируются на активных участках поверхности.
Примеры практического использования катализаторов для изменения скорости реакции.
2H2O2 = 2H2O + O2 При комнатной температуре реакция идет медленно, катализатор – MnO2
Автокатализ 2KMnO4 + 5H2C2O4+ 3H2SO4 = 2MnSO4 +K2 SO4 + 8 H2O +10CO2
Сначала реакция идет очень медленно, однако после того, как в реакционной смеси появляются ионы Mn2+, механизм реакции изменяется. Ионы MnO4- окисляют ионы Mn2+ до Mn3+
MnO4- + 4Mn2++8H+= 5Mn3++ 4H2O
После чего ионы Mn3+ быстро взаимодействуют с щавелевой кислотой.
2Mn3++H2C2O4 = 2Mn2+ + 2H++ 2CO2
12. Химическое равновесие. Условия химического равновесия. Константа равновесия химической реакции. Термодинамический вывод константы равновесия.
Состояние, не изменяющееся во времени, в котором реакционная смесь содержит как исходные вещества, так и продукты реакции называется химическим равновесием.
ΔrG = - RTlnK
УСЛОВИЯ ХИМИЧЕСКОГО РАВНОВЕСИЯ:
1. Состояние равновесия может быть достигнуто только в закрытой системе как со стороны исходных веществ, так и со стороны продуктов реакции
2. В состоянии равновесия ∆rGпр. = ∆rGобр. = 0
3. В состоянии равновесия rпр. = rобр.
4. При изменении внешних условий (температуры, давления, активностей участников) состояние равновесия изменяется (смещается)
В состоянии равновесия отношение произведения активностей продуктов реакции к произведению активностей исходных веществ в степенях, соответствующих – константа равновесия
Смещение химического равновесия это изменение относительных количеств участников реакции (реагентов и продуктов), вызванное действием внешних условий:
1. Изменение концентраций реагентов или продуктов реакции (изменение давления или объема)
2. Изменение температуры
3. Участие в реакции катализатора
Термодинамический вывод константы равновесия
Определим термодинамические условия химического равновесия на примере обратимой реакции, протекающей в закрытой системе:
2NO2(г.) ↔ N2O4(г.)
Эту реакцию можно представить как совокупность двух одновременно протекающих реакций: 2NO2(г.) → N2O4(г.)
N2O4(г.) → 2NO2(г.)
∆rGпр=∆fG (N2O4) – 2∆fG(NO2)
Для обратной реакции величина ∆rGобр. в каждый момент реакции имеет такое же абсолютное значение, что и ∆rGпр , но противоположный знак
По ходу прямой и обратной реакций кол-во оксидов N2O4 и NO2 изменяется, поэтому ∆fG необходимо пересчитать.
∆fG = ∆fG0 + RTlna
∆fG(N2O4) = ∆fG0(N2O4) + RTlnp(N2O4)
∆fG(NO2) = ∆fG0(NO2) + RTlnp(NO2 )
Подставим эти значения в первоначальную формулу:
∆rGпр= ∆fG0(N2O4) + RTlnp(N2O4) –2 (∆fG0(NO2) + RTlnp(NO2 ))
∆rGпр= (∆fG0(N2O4)– 2∆fG0(NO2)) + RT (lnp(N2O4) -2 lnp(NO2 ))
Первое слагаемое – выражение для ∆rG0 пр
∆rGпр=∆rG0 пр + RTln (p(N2O4))/ (p2(NO2 ))
∆rGобр.=∆rG0обр. +RTln (p2(NO2 )) /(p(N2O4))
Если общее давление в газовой смеси равно p, то очевидно, что парциальные давления оксидов могут принимать значения от 0 до p. Если в начальный момент времени в системе имеется только оксид NO2,а оксид N2O4 отсутствует, то p(NO2 ) = p, p(N2O4) = 0. В этом случае получим ln[p(N2O4)/ p2(NO2)] = - ∞, соответственно и энергия Гиббса прямой реакции тоже равна - ∞. Отрицательное значение энергии Гиббса означает, что в отсутствие в системе оксида N2O4 процесс димеризации NO2 является самопроизвольным.
Если же в начальный момент времени в смеси присутствует только N2O4, то ln[p(N2O4)/ p2(NO2)] = +∞, а значит и энергия Гиббса равна +∞. Тогда самопроизвольной становится обратная реакция.
Система находится в состоянии равновесия, когда ∆rG = 0. Это означает, что прямой и обратный процессы полностью уравновешивают друг друга и реагирующая смесь находится в равновесном состоянии.
∆rG= ∆rG0 + RTln[p(N2O4)/ p2(NO2)]
∆rG0 = - RTln[p(N2O4)/ p2(NO2)]
Kравновесия
=
[p(N2O4)/
p2(NO2)]
=
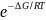
13. Смещение химического равновесия при изменении внешних условий. Принцип Ле Шателье и его термодинамическое обоснование.
УСЛОВИЯ ХИМИЧЕСКОГО РАВНОВЕСИЯ:
1. Состояние равновесия может быть достигнуто только в закрытой системе как со стороны исходных веществ, так и со стороны продуктов реакции
2. В состоянии равновесия ∆rGпр. = ∆rGобр. = 0
3. В состоянии равновесия rпр. = rобр.
4. При изменении внешних условий (температуры, давления, активностей участников) состояние равновесия изменяется (смещается)
В состоянии равновесия отношение произведения активностей продуктов реакции к произведению активностей исходных веществ в степенях, соответствующих – константа равновесия
Смещение химического равновесия это изменение относительных количеств участников реакции (реагентов и продуктов), вызванное действием внешних условий:
1. Изменение концентраций реагентов или продуктов реакции (изменение давления или объема)
2. Изменение температуры
3. Участие в реакции катализатора
Принцип Ле Шателье: если на систему, находящуюся в равновесии, оказывается внешнее воздействие, смещающее это равновесие, то данное равновесие смещается в сторону, ослабляющее это воздействие, до тех пор, пока нарастающее в системе противодействие не станет равным оказываемому действию.
K
= ∏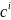 равн.
прод./
∏
равн.
прод./
∏ равн.
исх.в-в.
∆fG° = ∆fH°
–T∙∆fS°
равн.
исх.в-в.
∆fG° = ∆fH°
–T∙∆fS°
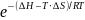 =
=
 +
+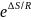
Видно, что температурная зависимость константы равновесия определяется знаком энтальпии реакции. Если энтальпия отрицательная, т.е. реакция экзотермическая, константа равновесия при повышении температуры уменьшается, а при снижении возрастает. В случае, если энтальпия реакции положительна, константа равновесия увеличивается при нагревании и уменьшается при охлаждении.
Изменение исходных активностей (концентраций) реагентов и продуктов реакции не изменяет константу равновесия реакции K. (∆rG° – не зависит от исходных а(С) реагентов и прод. реакции, поэтому от а(С) не зависит константа равновесия K).
Изменение температуры реакции Т изменяет константу равновесия реакции K.
Участие в реакции катализатора не изменяет константу равновесия реакции K. ∆rG° – не зависит от механизма процесса, поэтому от механизма реакции не зависит и константа равновесия K.
14. Кинетическое описание химического равновесия (связь константы равновесия реакции с константами скоростей прямого и обратного процессов). Смещение химического равновесия при изменении внешних условий. Принцип Ле Шателье и его кинетическое обоснование.
Смещение химического равновесия это изменение относительных количеств участников реакции (реагентов и продуктов), вызванное действием внешних условий:
1. Изменение концентраций реагентов или продуктов реакции (изменение давления или объема)
2. Изменение температуры
3. Участие в реакции катализатора
Химическое равновесие с точки зрения кинетики
В начальный момент протекания обратимой химической реакции скорость прямой реакции имеет ненулевое значение, зависящее от исходных концентраций реагентов и константы скорости прямой реакции. Скорость обратной реакции равна 0, поскольку являющиеся для нее исходными веществами продукты прямой реакции в системе пока отсутствуют. В ходе процесса скорость прямой реакции медленно убывает из-за расходования исходных в-в, в то время, как скорость обратной возрастает. Когда эти скорости становятся равными, концентрации участников перестают меняться, т.е. устанавливается химическое равновесие. Кинетическим условием равновесия является равенство прямой и обратной скоростей.
РАССМОТРИМ ОБРАТИМУЮ РЕАКЦИЮ:
СО2(р.) + OH-(р.) = HCO3- (p.) τпр.= kпр.С(СО2)С(OH-) τпр.= kобр.С(HCO3- ) τпр.= τпр.
kобр.С(HCO3- ) = kпр.С(СО2)С(OH-)
С(HCO3- ) / (С(СО2)С(OH-)) = kпр./ kобр.= K
Принцип Ле Шателье: если на систему, находящуюся в равновесии, оказывается внешнее воздействие, смещающее это равновесие, то данное равновесие смещается в сторону, ослабляющее это воздействие, до тех пор, пока нарастающее в системе противодействие не станет равным оказываемому действию.
K = ∏ равн. прод./ ∏ равн. исх.в-в.
K = kпр./kобр.
ln K = ln A – Ea/RT
Изменение исходных концентраций реагентов и продуктов реакции не изменяет константу равновесия реакции K. kпр. и kобр. реакций не зависят от исходных С реагентов и прод. реакции, поэтому от С не зависит константа равновесия K.
Изменение температуры реакции Т изменяет константу равновесия реакции K. kпр. и kобр. реакций увеличиваются с ростом температуры и уменьшаются с понижением температуры неодинаково.
15. Растворы. Образование растворов: термодинамические закономерности. Растворы неэлектролитов. Коллигативные свойства растворов. Осмос, причины его возникновения, осмотическое давление. Биологическая роль осмоса.
Раствор – гомогенная система, содержащая несколько компонентов. Компонентами растворов могут быть вещества различной природы: твердые, жидкие и газообразные, органические и неорганические, полярные и неполярные, поэтому свойства растворов чрезвычайно разнообразны.
В насыщенном растворе между раствором и избытком растворяемого в-ва устанавливается равновесие.
Иногда в равновесии с раствором находится не чистое растворяемое в-во, а его соединение с растворителем. Если растворителем является вода, то такое соединение называют кристаллогидратом.
В пересыщенном растворе концентрация в-ва превышает растворимость данного в-ва.
Среди растворителей и растворяемых в-в принято выделять полярные и неполярные. Наиболее типичным полярным растворителем является вода. К полярным растворяемым в-вам относятся кислоты, основания и соли, а также органические соединения, молекулы которых могут принимать участие в образовании водородных связей (глюкоза, мочевина). Неполярными растворяемыми в-вами являются: простые в-ва молекулярного строения и соединения элементов, мало отличающихся по электроотрицательности; среди органических соединений – углеводороды и другие в-ва, в молекулах которых имеются длинные углеводородные цепи.
Полярные в-ва лучше растворяются в полярных растворителях, а неполярные – в неполярных.
Электролитическая диссоциация – распад молекул или кристаллов на ионы. Благодаря подвижности ионов такие растворы хорошо проводят электрический ток, т.е. являются электролитами. В химии электролитами чаще называют сами в-ва, дающие электропроводящие водные растворы. Вещества, в растворах которых диссоциация не происходит, называют неэлектролитами.
Термодинамические закономерности образования растворов:
Мысленно разделим процесс образования раствора на 2 части (только для разбавленных растворов): 1) разрушение структуры растворяемого в-ва с образованием свободных частиц. 2) образование раствора при взаимодействии этих частиц (молекул или ионов) с растворителем. Соединения, которые частицы растворенного вещества образуют с молекулами растворителя – сольваты.
В соответствии с законом Гесса энтальпию, энтропию и энергию Гиббса растворения можно считать суммой соответствующих величин для первой и второй стадий.
Очевидно, что первая часть требует затраты энергии, поэтому ее энтальпия положительная. Энтальпия процесса сольватации, напротив, имеет отрицательное значение. Их сумма может быть как больше, так и меньше 0.
При растворении газов первая стадия отсутствует, так что энтальпия всегда отрицательна. Растворы, в которых энтальпия растворения равна 0 называют идеальными. В идеальных растворах термодинамические активности частиц растворенного в-ва равны их мольным концентрациям, а термодинамическая активность раствора – его мольной доле.
Энтропия растворения газа в жидкости всегда отрицательная из-за уменьшения объема системы, при растворении твердых и жидких в-в в жидкости – чаще всего энтропия положительна.
Если известны стандартные энтальпия и энтропия растворения, можно вычислить стандартную энергию Гиббса растворения: ∆раст.G° = ∆раст.H° –T∙∆раст.S°
Образование растворов неэлектролитов.
Энтальпии растворения неэлектролитов имеют как правило, небольшие положительные или отрицательные значения. Это связано прежде всего с тем, что разрушение структуры неэлектролитов не требует больших энергетических затрат, также неэлектролиты не образуют прочных сольватов.
Намного более сильная сольватация наблюдается при растворении полярных молекулярных в-в в воде. Соединение частиц растворенного вещества с молекулами воды здесь происходит за счет образования водородных связей.
Энтропия системы возрастает при образовании раствора твердого в-ва и уменьшается при растворении в воде газа.
(на всякий случай лучше знать) Закон Генри: равновесная концентрация малорастворимого газа в растворе пропорциональна его парциальному давлению в газовой фазе над раствором.
Коллигативные св-ва растворов. (у электролитов проявляются сильнее)
Некоторые св-ва растворов зависят от природы растворителя и числа частиц растворенного в-ва, но практически не зависят от природы этих частиц, т.е. их размера, массы, химического состава и т.д. Такие св-ва называют коллигативные. Коллигативные св-ва: понижение давления пара растворителя над раствором (закон Рауля); понижение температуры замерзания раствора; повышение температуры кипения раствора; возникновение осмотического давления.
Закон Рауля: давление насыщенного пара растворителя над жидким раствором меньше, чем над растворителем. При этом относительное понижение давления пара растворителя над идеальным раствором равно мольной доле растворенного в-ва Xp.в:
(p0 – p)/ p0 = Xp.в где p0 – давление пара растворителя на растворителем, р – над раствором
Для реальных растворов закон Рауля справедлив лишь при невысоких концентрациях растворенного в-ва, так как в концентрированных растворах активности нельзя считать равными их концентрациям.
Понижение температуры замерзания и повышение температуры кипения раствора.
Как растворитель, так и раствор закипают при такой температуре, когда давление пара становится равным внешнему давлению. Если растворенное в-во нелетучее, т.е. практически не образует паров, общее давление пара над раствором равно давлению пара растворителя.
Поскольку понижение давления пара растворителя на раствором пропорциональьно мольной доле растворенного в-ва, повышение температуры кипения также пропорционально концентрации раствора:
∆Tкипения = ECm E – коэффицент пропорциональности, который называют эбулиоскопической константой растворителя; Cm – моляльная концентрация раствора, моль/кг
Эбулиоскопическая константа растворителя численно равна раности температур кипения одномоляльного идеального раствора и растворителя. Понижение температуры начала кристаллизации раствора также пропорционально концентрации растворенного в-ва:
∆Tзам = KCm K – криоскопическая константа растворителя. Криоскопическая константа растворителя численно равна разности температурзамерзания одномоляльного идеального раствора и растворителя.
Из формул следует, что понижение температуры замерзания и повышение температуры кипения раствора не зависят от природы частиц растворенного в-ва, а зависят от только от концентрации раствора и природы растворителя. Как и закон Рауля эти формулы справедливы лишь для разбавленных растворов.
Возникновение осмотического давления.
Явление осмоса возникает тогда, когда раствор и растворитель или два раствора различной концентрации разделяют перегородкой – полунепроницаемой мембраной, которая пропускает молекулы растворителя, но не пропускает молекулы растворенного в-ва. В результате растворитель самопроизвольно переходит через перегородку из разбавленного раствора или индивидуального растворителя в более концентрированный раствор.
Осмотическое давление, в случае, когда мембрана отделяет раствор от чистого растворителя равно: ∏ = CRT
Два раствора, имеющие одинаковое осмотическое давление, называют изотоническими. Если взаимодейтсвуют два раствора с различными осмотическими давлениями, то раствор с большим осомтическим давлением называется гипертоническим, с меньшим – гипотоническим.
Биологическая роль осмотического давления. Транспорт в-в по проводящим тканям растений. Благодаря осмосу регулируется поступление воды в клетки и межклеточные структуры, упругость клеток (тургор). Животные и растительные клетки имеют оболочки или поверхностный слой протоплазмы, обладающие свойствами полупроницаемых мембран. При помещении этих клеток в растворы с различной концентрацией наблюдается осмос.
16. Сильные и слабые электролиты. Равновесие диссоциации в растворах слабых электролитов. Степень диссоциации и константа диссоциации слабого электролита. Влияние концентрации и температуры на степень диссоциации слабого электролита.
Электролиты – в-ва (кислоты, основания и соли), растворы которых проводят электрический ток за счет движения ионов. Сильные электролиты в растворах полностью распадаются на ионы, т.е. их диссоциация носит необратимый характер. В растворах слабых электролитов между ионами и молекулами растворенного в-ва устанавливается химическое равновесие. Равновесия в растворах слабых электролитов, как и любые другие, характеризуются константами равновесия.
К сильным электролитам принадлежат гидроксиды щелочных металлов, некоторые кислоты (соляная, азотная, хлорная и несколько других) и большая часть солей.
Активность i-того иона ai в растворе равна произведению его концентрации Сi на коэффициент активности fi ai = fi∙Ci
Ионная сила раствора: I = ½ ∑i Сi Zi2 Z – заряд ионов
Равновесие, устанавливающееся при диссоциации слабого электролита, описывают константой диссоциации K. Для характеристики процесса диссоциации слабого электролита используют также степень диссоциации ꭤ - отношение количества в-ва, распавшегося на ионы, к общему количеству растворенного в-ва. Kд = (ꭤ2 C)/(1 - ꭤ)
При малых значениях степени диссоциации (ꭤ<0,1(10%)) пользуются упрощенными формулами:
Kд =
ꭤ2 C, ꭤ =
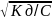 (Закон разбавлений Оствальда) Из этого
следует, что степень диссоциации слабого
электролита уменьшается с увеличением
его концентрации и, напротив, увеличивается
с ее уменьшением.
(Закон разбавлений Оствальда) Из этого
следует, что степень диссоциации слабого
электролита уменьшается с увеличением
его концентрации и, напротив, увеличивается
с ее уменьшением.
Слабые многоосновные кислоты и слабые многокислотные гидроксиды металлов диссоциируют ступенчато. Каждая ступень диссоциации характеризуется собственной ступенчатой константой диссоциации, при этом вторая константа обычно значительно меньше первой, третья меньше второй и т.д.
Уменьшение или увеличение концентраций продуктов диссоциации или исходных в-в смещает равновесие, но не изменяет константу диссоциации. Влияние температуры на равновесие диссоциации слабого электролита определяется энтальпией этого процесса. Энтальпии диссоциации большинства слабых электролитов не превышают нескольких килоджоулей, поэтому константа диссоциации и степень диссоциации от температуры зависят слабо. Исключением является вода, диссоциация которой заметно усиливается при повышении температуры. Влияние давления на диссоциацию проявляется только при очень больших его значениях.
17. Кислоты и основания. Теория кислот и оснований Бренстеда и Лоури. Протолитические равновесия (на примере процессов диссоциации и нейтрализации кислот и оснований). Понятие о теории кислот и оснований Льюиса.
Теория Аррениуса (НА ВСЯКИЙ СЛУЧАЙ) Согласно теории Аррениуса кислота – водородосодержащее соединение, при диссоциации которого в водном растворе образуются ионы H+ и анионы; основание – это гидроксидосодержащее соединение при диссоциации которого в водном растворе образуются гидроксид-ионы OH- и катионы.
В теории Аррениуса при нейтрализации кислота и основание исчезают.
Теория Бренстеда-Лоури. КИСЛОТЫ – частицы, которые отдают протон (доноры протона)
ОСНОВАНИЯ – частицы, которые принимают протон (акцепторы протона)
АМФОЛИТЫ – частицы, которые и принимают и отдают протон
В рамках теории Бренстеда-Лоури каждой кислоте отвечает сопряженное с ней основание, которое образуется при потере кислотой протона, и соответственно каждому основанию – сопряженная с ним кислота, которая возникает при присоединении основанием протона
Кислота = Основание + H+
Например, кислоте HCL отвечает сопряженное основание – ион Cl- ; с основанием NH3 сопряжена кислота NH4+
По Бренстеду-Лоури кислотами и основаниями могут быть не только нейтральные молекулы, но и заряженные частицы – ионы.
Кислота является сильной, если легко отдает протон; основание – напротив, если прочно его удерживает. Поэтому чем сильнее кислота, тем слабее сопряженное с ней основание.
Согласно теории Бренстеда-Лоури в кислотно-основных взаимодействиях обязательно участвуют две пары сопряженных кислот и оснований: кислота отдает протон, превращаясь в сопряженное с ней основание, а основание, присоединив протон, становится сопряженной с ним кислотой:
Кислота 1 + Основание 2 = Основание 1 + Кислота 2
Реакции переноса протона от кислоты к основанию называют протолитическими, а сам процесс переноса протона – протолизом. В результате образуются новое основание и новая кислота, сопряженные с исходными.
Протолитические равновесия (на примере процессов диссоциации и нейтрализации кислот и оснований)
Реакции диссоциации Реакции нейтрализации
HСl + H2O = Cl− + H3O+ H3O+ + OH– = H2O + H2O
СH3COOH + H2O = CH3COO– + H3O+ СH3COOH + OH– = CH3COO– + H2O
H2O + NH3 = OH– + NH4+ H3O+ + NH3 = H2O + NH4+
Теория кислот и оснований Льюиса
Кислотный характер веществ не всегда обусловлен их способностью отдавать протоны. Например, кислая среда водного раствора борной кислоты обусловлена не отщеплением протона от молекулы B(OH)3 , а тем, что эта молекула отрывает гидроксид-ион от молекулы воды: B(OH)3 + H2O = B(OH)4- + H+
Кислоты – это молекулы или ионы, которые при образовании ковалентной связи принимают пару электронов, т.е. являются акцепторами электронной пары, а основания – это частицы, которые при образовании ковалентной связи отдают пару электронов, т.е. являются ее донорами.
18. Вода. Автопротолиз воды. Ионное произведение воды. Влияние температуры на ионное произведение воды. Водородный показатель рН.
Вода – слабый электролит, в наибольшей степени диссоциирует на ионы H+ и OH-. Н2О = H+ + OH-
Ион H+ в свободном виде не существует, но для простоты пишем так
Константа равновесия этой реакции равна произведению активностей ионов H+ и OH- и называется ионным произведением воды.
Kв = a(H+) a(OH-)
Поскольку из значения константы видно, что равновесие диссоциации воды сильно смещено влево, активности ионов считают равными их равновесным концентрациям и чаще записывают ионное произведение воды так:
Kв = [H+]∙[OH-]
Ионное произведение воды довольно заметно зависит от температуры. При температурах, близких к комнатным, используют Kв= 10-14. Постоянство произведения концентраций ионов H+ и OH- соблюдаются во всех водных системах.
В теории Бренстеда-Лоури процесс диссоциации воды представляет собой реакцию автопротолиза – переноса протона между двумя одинаковыми молекулами – H2O + H2O = H3O+ + OH-
Ионное произведение воды зависит от температуры – чем выше температура, тем выше больше константа воды, но не зависит от концентраций ионов H+ и OH-в растворе. Энтальпия диссоциации воды равна +56,8 кДж/моль, т.е. это сильно эндотермический процесс. В соответствии с принципом Ле Шателье повышение температуры смещает равновесие диссоциации воды вправо, то есть увеличивает константу воды.
Концентрацию ионов H+ в водных растворах характеризуют водородным показателем pH, который представляет собой отрицательный десятичный логарифм от активности ионов водорода в растворе. В приближенных расчетах активность заменяют на концентрацию. pH = - lg a(H+) = - lg[H+]
pH + pOH = = -lgKв=pKв В воде и нейтральных растворах pH=pOH.
pH + pOH = - lg 10-14 = 14
Водные растворы, в которых концентрация ионов водорода больше, чем концентрация гидроксид-ионов называют кислыми. pH меньше 7.
Водные растворы, в которых концентрация ионов водорода меньше, чем концентрация гидроксид-ионов называют щелочными. pH больше 7.
Приближенно значение pH можно определить с помощью индикаторов.
|
Кислая среда |
Нейтральная среда |
Щелочная среда |
Метил-оранж |
Красно-розовый |
Оранжевый |
Желтый |
Фенолфталеин |
Бесцветный |
Бесцветный |
Малиновый |
Лакмус |
Красный |
Фиолетовый |
Синий |
Универсальный индикатор |
Красный |
Желтый |
Синий |
19. Гидролиз как пример протолитического равновесия (гидролиз 19. Гидролиз как пример протолитического равновесия (гидролиз катиона и аниона). Константа и степень гидролиза. Зависимость степени гидролиза от концентрации гидролизующегося иона и температуры. Необратимый гидролиз.
1. Гидролизу подвергаются ионы, а не молекулы. 2. Равновесие гидролиза (обычно, но не всегда) смещено в сторону исходных веществ. 3. Чем слабее кислота или основание, образующие соль, тем больше значение константы гидролиза и тем сильнее рН раствора отличается от 7. 4. Гидролиз катиона слабого основания и гидролиз аниона многоосновной кислоты идет преимущественно по первой ступени. 5. рН растворов солей, образованных катионом слабого основания и анионом слабой кислоты, определяется соотношением констант диссоциации этой кислоты и этого основания. 6. Нельзя получить растворы солей, образованных очень слабыми, нерастворимыми или летучими кислотами и основаниями. 7. Гидролиз усиливается при разбавлении и нагревании.
Гидролиз – реакция ионов соли с водой.
АНИОНЫ СИЛЬНЫХ КИСЛОТ не конкурируют с водой за протон
АНИОНЫ СЛАБЫХ КИСЛОТ конкурируют с водой за протон
рН среды растворов солей > 7 CO32–, PO43–, CN–, S2–
CO32– + H2O = HCO3– + OH– 1 ступень гидролиза
Kг1
=
 ∙
=
∙
=
 =
2∙10-4
=
2∙10-4
При взаимодействии аниона слабой кислоты с водой образуется избыток гидроксид-ионов OH-
Константа
гидролиза аниона рассчитывается по
уравнению: Kг =
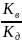 ,
где Кв – ионное произведение
воды, а Kд – константа
диссоциации кислоты, образующей соль.
Если кислота двух- или трехосновная, то
при расчете константы гидролиза по
первой ступени в формулу подставляют
константу диссоциации кислоты по
последней ступени.
,
где Кв – ионное произведение
воды, а Kд – константа
диссоциации кислоты, образующей соль.
Если кислота двух- или трехосновная, то
при расчете константы гидролиза по
первой ступени в формулу подставляют
константу диссоциации кислоты по
последней ступени.
Расчет концентраций ионов OH- в растворе соли, образованной слабой кислотой и сильным основанием, при малых значениях констант гидролиза (т.е. при константе диссоциации больше константы воды), сводится к решению уравнения:
[ОН-]
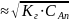
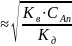
При взаимодействии катиона слабого основания с водой образуется избыток ионов Н+
Константа гидролиза рассчитывается по уравнению: Kг = , где Кв – ионное произведение воды, а Kд – константа диссоциации основания, образующего соль. При расчете константы гидролиза по первой ступени используют константу диссоциации последней ступени.
Расчет концентрации ионов H+ при малых значениях констант гидролиза можно выполнить по уравнению:
[Н+]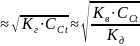
Степень
гидролиза h – отношение
кол-ва гидролизованных ионов к общему
кол-ву ионов в растворе заданной
концентрации С. Чаще всего рассчитывают
по упрощенной формуле: h
=

Однако, если предварительная оценка показывает, что степень гидролиза достаточно велика, упрощенная формула неприменима, т.к. нельзя пренебрегать h в знаменателе соотношения, связывающего константу и степень гидролиза:
Kг
=
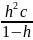
Гидролиз – процесс, в результате которого происходит образование ионов H+ или ОН- , изменяющее pH среды растворов солей. Соли сильных кислот и сильных оснований гидролизу в растворах не подвергаются.
Разбавление раствора увеличивает степень гидролиза. Гидролиз – эндотермический процесс, поэтому в соответствии с принципом Ле Шателье возрастание температуры увеличивает константу гидролиза, а понижение температуры – уменьшает.
Гидролиз соли, образованной слабой кислотой и сильным основанием (гидролиз по аниону). Гидролиз соли, образованной слабым основанием и сильной кислотой (гидролиз по катиону). Гидролиз соли, образованной слабой кислотой и слабым основанием.
В теории Бренстеда-Лоури гидролиз рассматривают как протолитическую реакцию между ионом соли и водой. При этом изменение концентрации ионов Н+ в растворе происходит тогда, когда гидратированный катион-кислота или анион-основание способны конкурировать за протон с водой. Например, анион CH3COO- (анион слабой кислоты) в рамках теории Бренстеда-Лоури довольно сильное основание, способное отщеплять протон от молекул воды: CH3COO- + H2O = CH3COOН + ОН-
В отсутствии такой конкуренции гидролиз не происходит и концентрации ионов гидроксония и гидроксид-ионов в растворе определяются только диссоциацией воды.
Что касается катионов, то в растворах они гидратированы, т.е. находятся в окружении молекул воды. Между положительно заряженным катионом и протонами молекул гидратной оболочки возникают силы отталкивания, в результате чего облегчается отрыв иона Н+ от молекул гидратной оболочки и перенос его к молекулам воды-растворителя. Гидратированный катион при этом проявляет свойства кислоты: Al(H2O)63+ + H2O = AlOH(H2O)52+ + H3O+
Чем меньше катион и чем больше его положительных заряд, тем легче отщепляется протон от гидратной оболочки катиона и тем в большей степени идет гидролиз.
С точки зрения теории Бренстеда-Лоури гидролиз катиона слабого основания рассматривается как протолитическое равновесие между двумя парами сопряженных кислот и оснований:
NH4+ + H2O = NH3 + H3O+
Al(H2O)63+ + H2O = AlOH(H2O)52+ + H3O+
Кислота 1 - NH4+, Al(H2O)63+
Основание 2 - H2O
Основание 1 - NH3, AlOH(H2O)52+
Кислота 2 - H3O+
Необратимый гидролиз солей – соли, образованные чрезвычайно слабыми, нерастворимыми или летучими кислотами и основаниями, при контакте с водой необратимо гидролизуются.
20. Буферные системы. Расчет рН буферной системы (на примере ацетатного буфера). Механизм действия буфера. Биологические буферные системы.
Растворы, водородный показатель которых незначительно изменяется при разбавлении или при добавлении к ним сильной кислоты или сильного основания называют буферными. Буферные растворы, или буферные системы могут быть: 1) смесью растворов слабой кислоты и ее соли; например, СН3СООН и NaCH3СОО 2) смесью растворов слабого основания и его соли; например, NH3 и NH4Cl 3) смесью растворов двух солей многоосновной кислоты, анионы которых содержат различное число атомов водорода; например NaH2PO4 и Na2HPO4
Водородный показатель буферного раствора можно рассчитать, зная концентрации составляющих его в-в. На примере ацетатного буфера: концентрация уксусной кислоты – Сk, концентрация ее соли, например ацетата натрия – Сс.
NaCH3СОО = СН3СОО- +Na+ Уксусная кислота – слабый электролит, процесс ее диссоциации обратим: СН3СООН = СН3СОО- + Н+, Kд = [СН3СОО-][ Н+]/[СН3СООН] [ Н+] = Kд[СН3СООН]/[СН3СОО-] Концентрация ионов водорода в ацетатном буферном растворе определяется соотношением равновесных концентраций уксусной кислоты и ацетат-ионов. Равновесная концентрация недиссоциированной уксусной кислоты в растворе равна: [СН3СООН] = Сk - [ Н+]
Равновесная концентрация ацетат-ионов складывается из концентрации ионов СН3СОО-, образовавшихся при диссоциации кислоты, и концентрации ионов, образовавшихся при диссоциации соли. [СН3СОО-] = Сс + [ Н+]
Уксусная кислота является слабым электролитом, так что степень ее диссоциации обычно невелика. В присутствии ионов СН3СОО- равновесие смещается в сторону недиссоциированных молекул СН3СООН, в результате чего концентрация ионов Н+ в смеси уксусной кислоты и ее соли становится меньше, чем в растворе кислоты той же концентрации. Поэтому величиной [ Н+] можно пренебречь и принять равновесные концентрации приближенно равными исходным. [СН3СОО-] = Сс [СН3СООН] = Сk , подставим равновесные концентрации в формулу
[ Н+] = Kд [СН3СООН]/[СН3СОО-]
[ Н+] = Kд∙Сk /Сс pH= -lgKд -lg (Сk /Сс )=pKд -lg (Сk /Сс ) Формулу используют для рассчета pH всех буферных растворов, приготовленных из слабой одноосновной кислоты и ее соли.
Для расчета pH буферных систем, содержащих слабое основание и его соль, например аммиачного буферного раствора, можно получить аналогичные выражения, где [ ОН-] = Kд∙Со /Сс pH =14 - pKд + lg (Сk /Сс )
При равных концентрациях в буферных растворах кислоты и ее соли или основания его соли выполняются соотношения: pH = pKд(к.), pH = 14 – pKд(о)
Значение рН буферного раствора не должно изменяться при разбавлении раствора, поскольку отношение концентраций кислоты и соли или основания и соли при этом остается постоянным.
Механизм действия буферного раствора. Вычислим, как изменится рН ацетатного буферного раствора, в 1 л которого содержится 1 моль СН3СООН и 1 моль NaCH3СОО, в результате добавления к нему 0,1 моля сильного основания NaOH или 0,1 моля сильной кислоты HCl. При добавлении сильного основания гидроксид-ионы свяжут часть ионов водорода, образующихся при диссоциации кислоты, в слабый электролит – воду, и равновесие сместится вправо СН3СООН = СН3СОО- + Н+. При этом равновесная концентрация кислоты в растворе уменьшится на 0,01 моль/л.
[СН3СООН] = Сk – 0,01 [СН3СОО-] = Сс + 0,01
В результате pH будет равен pH = pKд -lg ([СН3СООН] /[СН3СОО-]) = 4,75 – lg((1-0,01)/(1+0,01)) = 4, 84
Найденное значение отличается от исходного значения только на 0,09 единицы pH.
Число молей кислоты или щелочи, вызывающее изменение рН 1 л буферного раствора на единицу, называют буферной емкостью раствора.
Природные буферные системы обеспечивают постоянные концентрации ионов Н+ и ОН- в природных водах, почвах, а также внутри- и внеклеточных жидкостях живых организмов. Например, значение рН плазмы крови, равное 7,4, поддерживается благодаря гидрокарбонатной и фосфатной буферным системам, а также буферному действию находящихся в крови белков.
21. Буферные системы. Расчет рН буферной системы (на примере аммиачного буфера). Механизм действия буфера. Биологические буферные системы.
Растворы, водородный показатель которых незначительно изменяется при разбавлении или при добавлении к ним сильной кислоты или сильного основания называют буферными. Буферные растворы, или буферные системы могут быть: 1) смесью растворов слабой кислоты и ее соли; например, СН3СООН и NaCH3СОО 2) смесью растворов слабого основания и его соли; например, NH3 и NH4Cl 3) смесью растворов двух солей многоосновной кислоты, анионы которых содержат различное число атомов водорода; например NaH2PO4 и Na2HPO4
Водородный показатель буферного раствора можно рассчитать, зная концентрации составляющих его в-в. На примере аммиачного буфера: концентрация аммиака кислоты – Со, концентрация его соли, например хлорида аммония – Сс.
NH3 + H2O = NH4+ + OH-
Аммиак – слабый электролит, процесс его диссоциации обратим: Kд = [NH4+][ ОН-]/[NH3] [ОН-] = Kд[NH4+]/[NH3] Концентрация гидроксид-ионов в аммиачном буферном растворе определяется соотношением равновесных концентраций аммиака и ионов аммония. Равновесная концентрация недиссоциированного аммиака в растворе равна: [NH3] = Со - [ОН-]
Равновесная концентрация ионов аммония складывается из концентрации ионов NH4+, образовавшихся при диссоциации основания, и концентрации ионов, образовавшихся при диссоциации соли. [NH4+] = Сс + [ОН-]
Аммиак является слабым электролитом, так что степень его диссоциации обычно невелика. В присутствии ионов NH4+ равновесие смещается в сторону недиссоциированных молекул NH3, в результате чего концентрация ионов OH- в смеси аммиака и его соли становится меньше, чем в растворе аммиака той же концентрации. Поэтому величиной [ОН-] можно пренебречь и принять равновесные концентрации приближенно равными исходным.
[NH4+] = Сс [NH3] = Со , подставим равновесные концентрации в формулу
[OH-] = Kд [NH3]/[ NH4+]
[OH-] = Kд∙Сo /Сс pOH= -lgKд -lg (Сo /Сс )=pKд -lg (Сo /Сс )
Для расчета pH буферных систем, содержащих слабое основание и его соль, pH =14 - pKд + lg (Сk /Сс ), т.к. pH + pOH = 14.
При равных концентрациях в буферных растворах кислоты и ее соли или основания его соли выполняются соотношения: pH = pKд(к.), pH = 14 – pKд(о)
Значение рН буферного раствора не должно изменяться при разбавлении раствора, поскольку отношение концентраций кислоты и соли или основания и соли при этом остается постоянным.
Механизм действия буферного раствора. Вычислим, как изменится рН ацетатного буферного раствора, в 1 л которого содержится 1 моль СН3СООН и 1 моль NaCH3СОО, в результате добавления к нему 0,1 моля сильного основания NaOH или 0,1 моля сильной кислоты HCl. При добавлении сильного основания гидроксид-ионы свяжут часть ионов водорода, образующихся при диссоциации кислоты, в слабый электролит – воду, и равновесие сместится вправо СН3СООН = СН3СОО- + Н+. При этом равновесная концентрация кислоты в растворе уменьшится на 0,01 моль/л.
[СН3СООН] = Сk – 0,01 [СН3СОО-] = Сс + 0,01
В результате pH будет равен pH = pKд -lg ([СН3СООН] /[СН3СОО-]) = 4,75 – lg((1-0,01)/(1+0,01)) = 4, 84
Найденное значение отличается от исходного значения только на 0,09 единицы pH.
Число молей кислоты или щелочи, вызывающее изменение рН 1 л буферного раствора на единицу, называют буферной емкостью раствора.
Природные буферные системы обеспечивают постоянные концентрации ионов Н+ и ОН- в природных водах, почвах, а также внутри- и внеклеточных жидкостях живых организмов. Например, значение рН плазмы крови, равное 7,4, поддерживается благодаря гидрокарбонатной и фосфатной буферным системам, а также буферному действию находящихся в крови белков.
22. Равновесие осадок-раствор. Произведение растворимости. Условия выпадения и растворения осадка. Образование коллоидных систем при выделении малорастворимых веществ.
В насыщенных растворах сильных электролитов между твердым веществом (осадком) и ионами в растворе устанавливается равновесие. AnBm (к.) = nAp+(p.) + mBq-(p.) K = anAp+ + am Bq- . Активность твердой фазы AnBm равна единице и не входит в выражение.
Для малорастворимых электролитов активности ионов в растворе можно заменить на концентрации и получить константу, которую называют произведением растворимости соединения AnBm: ПР = [Ap+]n [Bq-]m Произведением растворимости малорастворимого электролита называют произведение равновесных концентраций ионов, образующих данный электролит, в степенях, равных их стехиометрическим коэффициентам. Произведение растворимости есть постоянная величина для данных значений температуры, давления и данного растворителя.
Зная произведение растворимости, можно также определить, будет ли при заданных концентрациях ионов в растворе происходить образование или растворение осадка. Для этого нужно вычислить произведение фактических концентраций этих ионов в степенях, равных их стехиометрическим коэффициентам в уравнении реакции образования осадка, и сравнить полученный результат с произведением растворимости. Если произведение фактических концентраций больше ПР, то для достижения равновесия осадок должен выделяться, если меньше, то присутствующий в системе осадок должен растворяться.
Из определения ПР как константы равновесия процесса следует, что при увеличении концентрации одного из ионов в растворе происходит уменьшение равновесной концентрации другого, т.е. происходит смещение равновесия в сторону образования осадка. Растворимость малорастворимого электролита понижается при введении в его раствор солей с одноименными ионами.
Образование коллоидных систем при выделении малорастворимых веществ.
При определенных условиях осадок малорастворимого в-ва выделяется из раствора в виде чрезвычайно тонкой взвеси (дисперсной фазы), состоящей из частиц микроскопического размера. В результате этого образуется коллоидная система. Коллоидная система является гетерогенной. (НЕ КОЛЛОИДНЫМ РАСТВОРОМ). Устойчивость коллоидной системы, как правило, обусловлена тем, что частицы дисперсной фазы имеют одноименные электрические заряды и поэтому отталкиваются друг от друга.
Пример возникновения заряда на коллоидных частицах. Допустим, что в сосуде смешали сильноразбавленные растворы нитрата серебра и иодида калия:
AgNO3 + KI = AgI + KNO3 и получили осадок AgI в виде очень мелких (коллоидных) частиц. Коллоидная частица формируется следующим образом. Сначала образуется микрокристалл AgI, кристаллическая решетка которого способна достраиваться ионами, входящими в ее состав. Если при смешивании растворов в избытке был взят KI, то это – ионы I-. Микрокристалл AgI с присоединившимися к нему дополнительными иодид-ионами образует отрицательно заряженное ядро коллоидной частицы. Это ядро притягивает к себе некоторое кол-во противоионов – катионов K+ и вместе с ними образует коллоидную частицу. Эта частица тоже заряжена отрицательно, но ее заряд меньше, чем заряд ядра. Силы отталкивания между одинаково заряженными частицами препятствуют их соединению друг с другом. Заряженную частицу, вместе с противоионами, компенсирующими ее заряд, называют мицеллой. Если в избытке будет нитрат серебра, частица будет положительно заряжена.
Добавление к коллоидной системе концентрированного раствора электролита приведет к исчезновению заряда дисперсной частицы вледствие присоединения к ней противоположно заряженных ионов. После этого начнется объединение частиц друг с другом, т.е. разрушение коллоидной системы (коагуляция).
23. Окислительно-восстановительные реакции (примеры). Важнейшие окислители и восстановители. Продукты восстановления перманганата калия и бихромата калия в зависимости от рН среды растворов.
В настоящее время окислительно-восстановительными называют все реакции, в которых происходит перенос электронов от одних атомов, молекул или ионов к другим. Частицу, отдающую электроны, называют восстановителем, а соответствующий процесс – окислением. Частицу, принимающую электроны называют окислителем, а соответствующий процесс – восстановлением. Оксиление невозможно без одновременно протекающего восстановления и наоборот.
Простейший пример окислительно-восстановительного процесса – образование ионного соедитения, например хлорида натрия, из простых в-в – натрия и хлора: 2Na + Cl2 = 2 NaCl. В этой реакции атомы натрия теряют электроны и превращаются в положительно заряженные ионы, то есть окисляются. Молекулы хлора приобретают электроны и превращаются в отрицательно заряженные ионы Сl- , т.е. восстанавливаются.
В большинстве ОВР от одних частиц к другим переходят не только электроны, но и атомы, например: Сa(OCl)2 + 2H2O2 = CaCl2 + 2O2 + 2H2O в таких случаях не всегда понятно, какие частицы отдают, а какие принимают электроны. Поэтому каждому элементу приписывают степень окисления.
Степень окисления – формальный заряд атома элемента, вычисленный при условии, что все полярные химические связи в молекуле этого соединения являются ионными.
Окислителем в реакции является то в-во, в составе которого есть атом, понижающий свою степень окисления. В составе восстановителя присутствует атом, повышающий свою степень окисления.
Примеры ОВР: 3MnCO3 + KClO3 = 3MnO2 + KCl + 3CO2
Fe2O3 + 3CO = 2Fe +3CO2
2(NH4)2CrO4 = Cr2O3 + N2 +5H2O + 2NH3
K2Cr2O7 + 4H2SO4 + 3H2S = Cr2(SO4)3 + 7H2O + 3S(т) + K2SO4
Важнейшие восстановители |
Важнейшие окислители |
Металлы, Водород, Уголь, Окись углерода (II) (CO) Сероводород (H2S), Оксид серы (IV) (SO2), Cернистая кислота H2SO3 и ее соли, Галогеноводородные кислоты и их соли, Катионы металлов в низших степенях окисления: SnCl2, FeCl2, MnSO4, Cr2(SO4)3, Азотистая кислота HNO2, Аммиак NH3, Гидразин NH2NH2, Оксид азота(II) (NO), Катод при электролизе.
|
Галогены, Перманганат калия(KMnO4) манганат калия (K2MnO4) оксид марганца (IV) (MnO2) Дихромат калия (K2Cr2O7) хромат калия (K2CrO4) Азотная кислота (HNO3) Серная кислота (H2SO4) концентрированная Оксид меди(II) (CuO) оксид свинца(IV) (PbO2) оксид серебра (Ag2O) пероксид водорода (H2O2) Хлорид железа(III) (FeCl3), Бертоллетова соль (KClO3) Анод при электролизе.
|
Продукты восстановления важнейших окислителей:
Вещество |
Среда |
Продукт |
KMnO4 |
Кислая |
Mn2+ |
Нейтральная или слабощелочная |
MnO2 |
|
Сильнощелочная |
MnO42 |
|
MnO2
|
|
Mn2+ |
K2Cr2O7 |
кислая или нейтральная |
Cr3+ |
щелочная |
CrO2- |
|
K2CrO4 Na2CrO4 |
кислая или нейтральная |
Cr3+ |
щелочная |
CrO2- (СrO3 аналогично) |
|
H2O2 |
кислая или нейт. |
H2O |
щелочная |
2OH- |
http://tehtab.ru/Guide/GuideChemistry/burningandexolisions/OxidationAndDeoxidation/
24. Электродный потенциал. Его возникновение и измерение в гальваническом элементе. Электроды сравнения: водородный электрод, хлорсеребряный электрод. Стандартный электродный потенциал.

Гальванический элемент. Реакция проводится так, чтобы процессы окисления и восстановления протекали в разных сосудах, а перенос электронов от восстановителя к окислителю осуществлялся по внешнему проводнику. Гальванический элемент состоит из двух сосудов, в одном из которых находится восстановитель, а в другом окислитель; соединяющего эти два сосуда проводника и трубки с насыщенным раствором хлорида калия (эту трубку называют солевым мостиком). В сосуде 1 проходит полуреакция окисления, в результате которой высвобождаются электроны. По внешнему проводнику эти электроны переходят в сосуд 2, где с их участием протекает полуреакция восстановления. Заряд электронов, переходящих по проводнику от восстановителя к окислителю, компенсируется зарядом движущихся по солевому мостику ионов. В гальваническом элементе совершается работа А = nF∆Е, где n – число электронов, переходящих от восстановителя к окислителю, F – число Фарадея (96485 Кл/моль), равное по модулю заряду 1 моля электронов; ∆Е – разность электрических потенциалов, возникающая между металлическими пластинками 1 и 2 гальванического элемента. A = - ∆rG ∆rG= - nF∆Е
При контакте металла и раствора через поверхность их раздела могут проходить ионы металла. Если они переходят из металла в раствор, о поверхность металла, на которой остался избыток электронов, заряжается отрицательно, а раствор в слое около пластинки – положительно. Если, наоборот, ионы металла из раствора захватываются поверхностью пластинки, металл заряжается положительно, а раствор вокруг пластинки за счет накопления анионов – отрицательно. И в том, и в другом случае накопление электрического заряда на пластинке металла затрудняет дальнейший переход ионов в раствор или из него. Поэтому через некоторое время на границе раздела устанавливается равновесие: Mn+ +ne = M. Абсолютное значение потенциала, возникающего на границе металл-раствор, измерить невозможно, поэтому его определяют относительно какого-либо электрода, который называют электродом сравнения. Для получения сопоставимых значений электродные потенциалы различных электродов измеряют относительно одного и того же электрода сравнения, в качестве которого выбран стандартный водородный электрод.

Платиновая пластинка, покрытая слоем мелких частиц платины, погружена в сосуд с раствором серной или соляной кислоты, активность ионов водорода в котором равна 1. Через раствор кислоты пропускают водород под давлением 1 атм, который адсорбируется на поверхности платины и находится в равновесии с ионами Н+: 2Н+ + 2е = Н2 Потенциал этой реакции, т.е. потенциал стандартного водородного электрода, принимают равным нулю при любой температуре.
Электродный потенциал металлического электрода – потенциал гальванического элемента, составленного из исследуемого электрода и стандартного водородного электрода. Если измерения проводят при стандартной температуре 298K и при этом обеспечивают условия, при которых активность металла в растворе равна 1, электродный потенциал называют стандартным электродным потенциалом. Чем больше значение стандартного электродного потенциала металла, тем более сильные окислительные св-ва проявляют его ионы в р-рах. Отрицательное значение означает, что в паре с водородным электродом на нем самопроизвольно происходит окисление, т.е. металл превращается в ионы.
Строение хлорсеребряного электрода
Представляет собой серебряную проволоку, покрытую хлоридом серебра и помещенную в раствор калия. Устойчив к высоким температурам. Электродный потенциал = 0,201В

Стандартный электродный потенциал полуреакции Е° — это разность электродных потенциалов, возникающая в гальваническом элементе, составленном из стандартного водородного электрода и электрода, в котором при 298К и при активностях всех участвующих в исследуемой реакции частиц равных единице протекает исследуемая полуреакция.
Электродные потенциалы характеризуют окислительную и восстановительную способность в-в: чем больше электродный потенциал, тем выше окислительная способность окисленной формы и ни же восстановительная способность восстановленной. Электродный потенциал полуреакции называют стандартным, если активности всех участников полуреакции равны 1.
25. Зависимость электродного потенциала от условий проведения реакции. Уравнение Нернста. Направление протекания окислительно-восстановительной реакции. Константа равновесия окислительно-восстановительной реакции.
Зависимость электродного потенциала от условий проведения реакции Стандартные электродные потенциалы, которые приводят в справочниках, соответствуют активностям всех участников равным единице. При изменении активностей электродные потенциалы тоже меняются. Выражение, определяющее зависимость электродного потенциала от активностей компонентов окисленной и восстановленной форм полуреакции, называют уравнением Нернста:
На практике активности считают равными концентрациям.

При 298K:
![]()
Электродный потенциал может быть pH зависим. Зависит от концентраций компонентов системы, температуры.
Значения электродных потенциалов используют для расчетов разность потенциалов ∆Е окислительно-восстановительной реакции и определения направления ее протекания. При положительном значении самопроизвольно протекает реакция в прямом направлении, при отрицательном – в обратном.
Расчет константы равновесия окислительно-восстановительной реакции.
lnK = -∆rG/RT
lnK = nF∆E/RT При стандартной температуре уравнение принимает вид: lnK = n∆E/0,059 Электролиз – окислительно-восстановительные процессы, происходящие при прохождении через электролит электрического тока.
26. Квантовое описание строения атома. Атомные орбитали и квантовые числа. Графическое представление атомных орбиталей. Порядок заполнения атомных орбиталей в многоэлектронных атомах.
Порядок заполнения атомных орбиталей в многоэлектронных атомах.
1s < 2s < 2p < 3s < 3p < 4s < 3d < 4p < 5s < 4d < 5p < 6s < 5d > 4f < 6p < 7s
Принцип Паули: в атоме и любой другой многоэлектронной системе не может быть более двух электронов, состояние которых описывается одной и той же волновой функцией, или, что то же самое, тремя одинаковыми квантовыми числами n, l и m. При этом два электрона, занимающие одну атомную орбиталь, должны отличаться друг от друга направлением спина.
Правило Хунда: электроны на атомных орбиталях располагаются так, чтобы их суммарный спин был максимальным.
Строение атома.
Планетарная модель: вокруг положительно заряженного ядра вращаются электроны, компенсирующие ядерный заряд. (Резерфорд)
Первая количественная теория строения атома принадлежит Н. Бору. Опираясь на идею М. Планка о квантовании энергии, Н. Бор постулировал устойчивость некоторых орбит атома Резерфорда.
Квантовая теория:
На сегодняшний день не вызывает сомнения, что атом состоит из электронов и ядра, в состав которого входят протоны и нейтроны. Между атомным ядром и электронами действуют электростатические силы. Они намного больше сил гравитационного и магнитного взаимодействия ядра и электронов, поэтому при описании поведения электронов в атомах достаточно учитывать только силы электростатического взаимодействия.
В атомном ядре протоны и нейтроны притягиваются друг к другу за счет так называемых ядерных сил. На малых расстояниях эти силы в сотки раз превосходят электростатические, но с увеличением расстояния они очень быстро убывают и в химических процессах не играют практически никакой роли.
Поскольку размеры ядра и электронов пренебрежимо малы по сравнению с расстоянием между ними, эти частицы можно рассматривать как материальные точки. Масса электрона намного меньше массы атомных ядер. Поэтому считают, что массивное ядро в атоме неподвижно, а электроны движутся вокруг него.
Для описания поведения электронов в атомах используют понятия квантовой механики. Электрон в атоме должен находиться в одном из стационарных состояний, каждое из которых характеризуется строго определенной энергией Е. Стационарное состояние с наименьшей возможной энергией называют основным, все остальные – возбужденными. При переходе электрона из одного стационарного состояния в другое его энергия изменяется скачкообразно, т.е. определенными порциями, которые называют квантами.
Электрон не может иметь точно определенные координаты, так как в этом случае полностью неопределенным станет его импульс. (принцип неопределенности Гейзенберга)
Переход электрона из одной области пространства в другую происходит так, как будто он движется не по одному, а одновременно по всем возможным путям.
Квантовая механика позволяет определить в какой области пространства вероятность обнаружения больше, а в какой – меньше. Эту вероятность можно рассчитать по уравнению Шредингера:
Атомные орбитали. Волновые функции, получаемые при решении уравнения Шредингера для атома водорода, называют атомными орбиталями.
Атомные орбитали характеризуются при помощи 3-х атомных чисел.
Главное квантовое чило принимает значение от 1 до +∞, все атомные орбитали водорода с одинаковыми значениями n и различными другими квантовыми числами имеют одинаковую энергию. Совокупность атомных орбиталей, имеющих одинаковое значение n, называют энергетическим уровнем.
Орбитальное квантовое число l определяет тип пространственного распределения волновой функции. На каждом энергетическом уровне оно может принимать значения от 0 до n-1.
Орбитальное квантовое число |
0 |
1 |
2 |
3 |
4 |
5 |
Обозначение |
s |
p |
d |
f |
g |
h |
Магнитное квантовое число ml принимает значения от –l до +l.
Графическое представление атомных орбиталей.
При графическом представлении атомных орбиталей в виде элекронных облаков в пространстве вокруг ядра выделяются области, внутри которых преимущественно находится электрон . Границы областей соответствуют определенным значениям угловой части волновой функции или ее квадрата. Поскольку характер симметрии электронных облаков при этом сохраняется, такое различие в большинстве случаев несущественно.
Иногда графики волновых функций представляют в виде изолиний, т.е. линий, соединяющий точки с одинаковым значением волновой функции. Изолинии, советующие положительным значениям

27. Периодический закон и Периодическая система Д. И. Менделеева. Свойства атомов (радиус атома, энергия ионизации, сродство к электрону, электроотрицательность) и закономерности их изменения. Связь строения многоэлектронного атома с его положением в Периодической системе.
Принцип Паули: в атоме и любой другой многоэлектронной системе не может быть более двух электронов, состояние которых описывается одной и той же волновой функцией, или, что то же самое, тремя одинаковыми квантовыми числами n, l и m. При этом два электрона, занимающие одну атомную орбиталь, должны отличаться друг от друга направлением спина.
Правило Хунда: электроны на атомных орбиталях располагаются так, чтобы их суммарный спин был максимальным.
Периодический закон: свойства химических элементов (т.е. свойства и форма образуемых ими соединений) находятся в периодической зависимости от заряда ядра атомов химических элементов.
Ковалентный радиус атома представляет собой половину длины одинарной ковалентной связи между одинаковыми атомами в молекулах или кристаллах простых в-в. При движении по периодам слева направо радиусы атомов уменьшаются, потому что заряд ядра возрастает, а число энергетических уровней остается неизменным. В группах s- и р- элементов радиусы атомов при движении по группам сверху вниз возрастают. В случае d- элементов при переходе от четвертого периода к пятому размеры значительно возрастают, тогда как при переходе от пятого к шестому радиус возрастает очень мало, а нередко даже уменьшается. (происходит лантаноидное сжатие)
Энергия ионизации – энергия, которую необходимо затратить для отрыва электрона от атома. Если удаляют наиболее слабо удерживаемый электрон, то соответствующую энергию называют первую энергию ионизации. I1. При ионизации атомов d-элементов первыми удаляются s-элементы внешнего уровня, а лишь затем d-электроны предвнешнего уровня.
Энергия ионизации в группах s- и р-элементов при движении сверху вниз за редким исключением убывает, а у d-элементов как правило, возрастает, хоть и немонотонно. При движении по периодам слева направо наблюдается тенденция к увеличению энергии ионизации, однако ее рост происходит немонотонно.
Сродство атома к электрону. A. Энергия, которая выделяется или поглощается при присоединении к атому электрона, называется сродством к электрону. Если энергия выделяется, то сродство к электрону считают положительным, если поглощается – отрицательным. Сродство к электрону s-элементов в пределах группы изменяется довольно слабо. В группах р-элементов сродство к электрону при переходе от 2р- к 3р- элементу возрастает, а при дальнейшем движении постепенно падает. Атомы d- и f- элементов, как правило, тенденции к присоединению электронов не проявляют
Электроотрицательность. Способность атома сохранять свои электроны и присоединять электроны других атомов. Самый электроотрицательный элемент – фтор.
Электроотрицательность = (I1+A)/2
Электрооттрицательность s-, p-элементов убывает при движении по группам сверху вниз, а по периодам – возрастает слева направо.
28. Образование химической связи, ее характеристики: энергия, длина, поляр- ность. Перекрывание АО. Связи σ- и π-типа. Описание ковалентной хими- ческой связи методом молекулярных орбиталей на примере молекулы H2. Энергетические диаграммы МО ионов H2 + , H2 – и молекулы HeH. Кратность связи.
Хим. св. обуславливается взаимным притяжением электр-в и ат. ядер; им. электростатическую природу.
Виды х\с:
-ковалентная – обобществление валентных электр-в разных атомов; если они в равной степени принадлежат данным атомам, то связь неполярна; если происходит смещение к одному из атомов – полярная;
- ионная – как правило образ-ся между атомами элементов, сильно отличающихся по э.о., при чем валентные эл-ны практически полностью переходят от одного атома к другому (принимающий выступает в роли акцептора, отдающий – донора)=>образуется К+ и А-;
-металлическая – электроны принадлежат не конкретным атомам, а всему кристаллу в целом, и имеют подвижность, придающую электропроводящие св-ва; МЕТАЛЛИЧСКАЯ СВЯЗЬ ХАР-НА ДЛЯ ВЕЩЕСТВ, А НЕ КОНКРЕТНЫХ АТОМОВ;
Энергия х\с – энергия, выделяющиеся при обр-ии молекулы из отдельных атомов\энергия, затрачиваемая при разделении мол-ы на атомы (выражается в электрон-вольтах\кДж на 1 моль в-ва);
Длина х\с – расстояние между ядрами соединившихся атомов, соответствующее наименьшей энергии мол-ы (выражается в пикометрах);
Полярность для двухатомных мол-л определяется дипольным моментом (выражается в дебаях): чем он больше, тем выше полярность ковалентной связи; как правило, дипольный момент тем больше, чем выше разность электроотрицательностей атомов (НЕ ВСЕГДА РАБОТАЕТ)
В физике дипольный момент системы, состоящей из одинаковых по абсолютному значению положительного и отрицательного точечных зарядов, находящихся на определенном расстоянии друг от друга, определяется как произведение заряда на расстояние.
При обр-ии х\с межу атомными ядрами возникает область повышенной электронной плотности – область перекрывания а\о-лей. Если их перекрывание лежит на линии св, то его относят к сигма-типу; если при вз-вии а\о-лей возник две области перекрывания и они наход по разные стороны от линии св – это перекрывание пи-типа. !К обр-ию х\св приводит только перекрывание, при котором знаки волновых ф-ций в области перекрывания совпадают, в ином случае – электронная плотность в межъядерном пространстве не увеличивается и св не возникает (вз-вие s-орбиталей одного а-а с Ру- и Дху- орбиталями другого.
Неэффективное перекрывание – положительное перекрывание полностью компенсируется равным ему по модулю значению отриц перекр-м.
Кратность связи – понятие, характеризующие устойчивость двухатомных м-л в ММО:
n = (Nсвяз – Nразр)\2
Nсвяз- число эл-в на связавающих молекулярных о-лях;
Nразр – число эл-в на разрыхляющих мол.о-лей;

На связывающей мол о-ли иона Н2+ присутствует только один эл-ня, поэтому вполне закономерно, что энергия связи в этом ионе почти в два раза меньше, чем в Н2.
В ионе Н2- на связ мол. о-ли находятся два электрона, а еще один в соответствии с принципом Паули занимает разрыхляющую мол о-ль.
Билет №29. Предсказание геометрического строения молекул методом отталкивания электронных пар (метод Гиллеспи). Геометрия молекул BeCl2, BF3, CH4, NH3 и H2O.
М-д отталкивания электронных пар позволяет предсказать геометрию молекулы, которая явл важной хар-тикой многоатомных м-л.
Суть: области повышенной электронной плотности в валентном окружении центрального атома должны располагаться в пространстве так, чтобы их взаимное отталкивание было наименьшим; такие области соответствуют либо неподеленным электронным парам, либо ковалентным св-м, поэтому св-и и неподеленные пары центрального атома для ослабления межэлектронного отталкивания должны располагаться на максимально возможном удалении, что обеспечивают такие конфигурации как: линейная (2), равносторонний треугольник (3), тетраэдр(4), тригональная бипирамида (5), октаэдр (6)*
*- число областей повышенной электр-ой плотности (n)
Алгоритм м-а Гиллеспи:
1) определяют число областей повышенной электронной плотности в окружении центр а-а, для этого подсчитывают число, образ центр а-ом св-ей, а также число имеющихся у него неподеленных эл-ных пар;
2) выбирают соответствующую числу n фигуру\ многогранник, в центре – центр а-м;
3)удаляют те вершины, соответствующие неподеленным электронным парам – пол форму молекулы!;
В м-ле гидрида бора (BH3) наименьшее отталкивание трех областей повышенной электронной плотности, соответствующих связям В-Н достигается при условии, что эти св-и направлены к вершинам правильного треугольника.
В м-ле СН4 наименьшее отталкивание 4-х областей повышенной эл-ной плотности (С-Н) достигается при их тетраэдрическом расположении вокруг центр а-а.
Атом N в NH3 окружают 4 области повышенной плотности (одна –неподеленная эл-ая пара), из-за чего м-ла аммиака им. форму тригональной пирамиды с а-м азота в ее вершине и 3-мя а-ми водорода в основании.
Соответственно Н2О и


Билет №30. Вещества с молекулярной структурой (примеры). Межмолекулярные взаи- модействия. Силы Ван-дер-Ваальса (три составляющих). Водородная связь. Особенности фтороводорода, воды и аммиака, обусловленные водородны- ми связями.
Молекулярные вещества — это вещества, мельчайшими структурными частицами которых являются молекулы
Например: Вода H2O — жидкость, tпл=0°С; tкип=100°С; Кислород O2 — газ, tпл=-219°С; tкип=-183°С; Оксидазота (V) N2O5 — твердое вещество, tпл=30,3°С; tкип=45°С;
К молекулярным веществам относятся:
большинство простых веществ неметаллов: O2, S8, P4, H2, N2, Cl2;
соединения неметаллов друг с другом (бинарные и многоэлементные): NH3, CO2, H2SO4.
В молекул-х в-вах х\св-и образ между а-ми м-л, а между м-ми образ межмолекулярные св-и. Силы межмол-ного отталкивания появл в результате сил притяж-ия между атомными ядрами одной и эл-нами другой и взаимного отталкивания одноименно заряженных ч-ц.
Мех-мы возникн-ия межмол-ного вз-вия (силы Вандер-Ваальса):
- ориентационное – между полярными м-ми; диполи с противоположно заряженными концам =>возник сила притяжения;
- поляризационное – к примеру, при сближении полярной м-лы с неполярной – при этом эл-ны в неполярной м-ле стремятся удалиться от отриц полюса пол-ной м-лы и приблизится к полож. => сдвиг эл-ной плотности приводит к возник дипольного момента («наведенный»); вз-вие полярных м-л приводит к увеличению их дипольных м-в;
- дисперсионное – располож-ие эл-в в разное время по разные стороны относительно ядра а-а приводит к появл-ию у м-лы постоянно изменяющего свою величину и направление мгновенного д\м; возникающие мгновенные диполи притягиваются=> притягиваются неполярные м-лы;вероятность возникновения таких диполей растет в зависимости от числа эл-нов;
- взаимное отталкивание эл-в разных м-л (!НЕ силы В-В) – если м-лы сближены так, что их электронные оболочки перекрываются взаимное отталкивание эл-нов возраст и препятствует < расс-ия между м-лами;
Водородные св-и – межмолекулярные вз-вия, причиной которых является взаимное притяжение м-л из-за притягивания +-заряженного атома Н одной м-лы к - -заряженному а-у другой. Обр-ие водородных св-ей обусловлено ориентационным вз-вием м-л, НО отлич от обычных В-В сил => малого размера а-а Н м-лы могут сильнее вз-вовать.
Наиболее предрасположены к обр-ию в\с соед-ия Н с эл-тами 2 периода.
А-ы Н преимущественно притягиваются именно! к неподеленной эл-ной паре.
Например: зигзагообраз-ое строение цепочек (HF)n в кристаллическом сост – а-м Н притягивается к одной из 3-х неподеленных пар, которые вместе с одной связывающей эл-ной парой образ его тетраэдрическое окружение.
 Для
аммиака вз-вие им чисто диполь-дипольный
хар-р=> цепочка линейна.
Для
аммиака вз-вие им чисто диполь-дипольный
хар-р=> цепочка линейна.
Две поделенные электронные пары участвуют в образовании двух полярных ковалентных связей, а оставшиеся две неподеленные пары электронов тоже играют важную роль в свойствах воды. Все заместители у атома кислорода, включая неподеленные пары, стремятся расположиться как можно дальше друг от друга. Это приводит к тому, что молекула приобретает форму искаженного тетраэдра с атомом кислорода в центре. В четырех вершинах этого "тетраэдра" находятся два атома водорода и две неподеленные пары электронов. Но если смотреть только по центрам атомов, то получается, что молекула воды имеет угловое строение, причем угол Н–О–Н составляет примерно 105 градусов.
Билет №31. Понятие о зонном строении твердого тела. Металлы, полупроводники и диэлектрики (на примере простых веществ, образованных элементами 14 группы). Общие физические свойства металлов (электропроводность и те- плопроводность).
Все вещества в зависимости от их электрических свойств делятся на диэлектрики, проводники или полупроводники. Различие между ними наиболее наглядно можно показать с помощью энергетических диаграмм зонной теории твердых тел.
Спектральный анализ отдельных атомов показывает, что для атома каждого вещества характерны вполне определенные спектральные линии Это говорит о наличии определенных энергетических состояний (уровней) для разных атомов.
Часть этих уровней заполнена электронами в нормальном , невозбужденном состоянии атома. На других уровнях электроны могут находиться только после того, как атом подвергается внешнему энергетическому воздействию и становится возбужденным. Стремясь снова вернуться к устойчивому состоянию, атом излучает избыток энергии и электроны возвращаются на свои прежние уровни, при которых энергия атома минимальна.
Когда из отдельных атомов образуются молекулы, а из молекул образуется вещество, все имеющиеся у данного типа атома электронные уровни (как заполненные электронами, так и незаполненные) несколько смещаются вследствие действия соседних атомов друг на друга. Таким образом, из отдельных энергетических уровней уединенных атомов в твердом теле образуются целые полосы – зоны энергетических уровней . Нормальные энергетические уровни 1 образуют заполненную электронами зону 2. Уровни возбужденного состояния атома 3 образуют свободную зону 4. Между заполненной зоной и свободной зоной располагается запрещенная зона 5.
Диэлектриками являются такие материалы, у которых запрещенная зона (а следовательно и необходимая для ее преодоления энергия) настолько велика, что в обычных условиях электроны не могут переходить в свободную зону и электронной электропроводности не наблюдается. Таким образом, диэлектрики не проводят электрический ток, они являются изоляторами. Однако такими свойствами диэлектрик обладает до определенного предела. При воздействии очень высоких температур или сильных электрических полей связанные электроны могут переходить в свободную зону. В этом случае диэлектрик теряет свои изоляционные свойства, он перестает быть изолятором и становится проводником.
Полупроводники имеют более узкую запрещенную зону, которая может быть преодолена за счет небольших внешних энергетических воздействий, например температуры, света или других источников энергии. Если подведенная извне энергия будет достаточна для переброса электронов через запрещенную зону, то, став свободным, электроны могут перемещаться и под действием электрического поля, создавать электронную электропроводность полупроводника. При низких температурах полупроводники имеют мало свободных электронов , они плохо проводят электрический ток и практически являются изоляторами. С повышением температуры число носителей заряда растет и сопротивление полупроводников сильно уменьшается.
Проводниками являются материалы, у которых заполненная электронами зона вплотную прилегает к зоне свободных энергетических уровней или даже перекрывается ею. Вследствие этого электроны в металле могут переходить из заполненной зоны в свободную зону даже при слабых напряженностях электрического поля.
Билет №32. Комплексные соединения (примеры). Основные понятия: комплексообра- зователь, лиганд, координационное число. Образование комплексных час- тиц в растворах. Ступенчатые константы образования комплексных частиц и константы их устойчивости.