

1. Первый закон термохимии (Лавуазье и Лаплас, 1780—1784):
тепловой эффект образования данного соединения в точности равен, но обратен по знаку тепловому эффекту его разложения.
Из закона Лавуазье—Лапласа следует невозможность построить вечный двигатель I рода, использующий энергию химических реакций.
Второй закон термохимии (Г. И. Гесс, 1840):
тепловой эффект химической реакции не зависит от характера и последовательности отдельных ее стадий и определяется только начальными и конечными продуктами реакции и их физическим состоянием (при p=const или при v=const).
Г. И. Гесс первый принял во внимание физическое состояние реагирующих веществ, так как теплоты изменения агрегатных состояний веществ накладываются на тепловой эффект реакции, увеличивая или уменьшая его.
Утверждение закона Гесса о том, что тепловой эффект процесса не зависит от его отдельных стадий и их последовательности, дает возможность рассчитывать тепловые эффекты реакций для случаев, когда их определить экспериментально или очень трудно, или вообще невозможно.
Применение закона Гесса чрезвычайно расширило возможности термохимии, позволяя производить точные расчеты тепловых эффектов образования целого ряда веществ, опытные данные по которым получить было трудно.
Закон Гесса в наши дни применяют главным образом для расчета термодинамических функций—энтальпий, которые сейчас используются для термохимических расчетов. Термохимия, исторически сложившаяся раньше термодинамики, в настоящее время претерпела некоторые изменения и стала разделом химической термодинамики
2. Термохимические уравнения включают в себя кроме химических формул тепловой эффект реакции. Числовое значение в уравнении реакции строго соответствует количествам веществ, участников реакции, т.е. коэффициентам. Благодаря этому соответствию, можно установить пропорциональные отношения между количеством вещества или массой и количеством теплоты в этой реакции.
Тепловой эффект химической реакции или изменение энтальпии системы вследствие протекания химической реакции — отнесенное к изменению химической переменной количество теплоты, полученное системой, в которой прошла химическая реакция и продукты реакции приняли температуру реагентов.
Теплотой реакции (тепловым эффектом реакции) называется количество выделенной или поглощённой теплоты Q. Если в ходе реакции теплота выделяется, такая реакция называется экзотермической, если теплота поглощается, реакция называется эндотермической.Теплота реакции определяется, исходя из первого закона (начала) термодинамики, математическим выражением которого в его наиболее простой форме для химических реакций является уравнение: Q = ΔU + рΔV (2.1) ,где Q - теплота реакции, ΔU - изменение внутренней энергии, р -давление, ΔV - изменение объёма.Термохимический расчёт заключается в определении теплового эффекта реакции. В соответствии с уравнением (2.1) численное значение теплоты реакции зависит от способа её проведения. В изохорном процессе, проводимом при V=const, теплота реакции QV=ΔU, в изобарном процессе при p=const тепловой эффект QP =ΔH. Таким образом, термохимический расчёт заключаетсяв определении величины изменения или внутренней энергии, или энтальпии в ходе реакции. Поскольку подавляющее большинство реакций протекает в изобарных условиях (например, это все реакции в открытых сосудах. протекающие при атмосферном давлении), при приведении термохимических расчётов практическивсегда производится расчёт ΔН. ЕслиΔН<0, то реакция экзотермическая, если жеΔН>0, то реакция эндотермическая.Термохимические расчёты производятся, используя или закон Гесса, согласно которому тепловой эффект процесса не зависит от его пути, а определяется лишь природой и состоянием исходных веществ и продуктов процесса, или,чаще всего, следствие из закона Гесса: тепловой эффект реакции равен сумме теплот (энтальпий ) образования продуктов за вычетом суммы теплот ( энтальпий ) образования реагентов.В расчётах по закону Гесса используются уравнения вспомогательных реакций, тепловые эффекты которых известны. Суть операций при расчётах по закону Гесса заключается в том, что над уравнениями вспомогательных реакций производят такие алгебраические действия, которые приводят к уравнению реакции с неизвестным тепловым эффектом
3. Теплота́ сгора́ния — это количество выделившейся теплоты при полном сгорании массовой (для твердых и жидких веществ) или объёмной (для газообразных) единицы вещества. Измеряется в джоулях или калориях. Теплота сгорания, отнесённая к единице массы или объёма топлива, называетсяудельной теплотой сгорания (дж или кал на 1 кг, м³ или моль).
теплота образования — тепловой эффект реакции образования химических соединений из простых веществ в стандартном состоянии
4. Термодинамическая стабильность химических соединенийопределяется знаком и величиной изменения изобарно-изотермического потенциала при их образовании из простых веществ. Наиболее важным и общим методом расчета изменения изобарно-изотермического потенциала AGr является определение его из данных химического равновесия по уравнению изотермы химическойреакции. С точки зрения термодинамики направление химической реакциимежду металлом и окружающей средой определяется изменением изобарно-изотермического потенциала
5. Энтропи́я (от др.-греч. ἐντροπία — поворот, превращение) — широко используемый в естественных и точных наукахтермин. Впервые введён в рамках термодинамики как функция состояния термодинамической системы, определяющая меру необратимого рассеивания энергии. В статистической физике энтропия является мерой вероятности осуществления какого-либо макроскопического состояния. Кроме физики, термин широко употребляется в математике: теории информации и математической статистике. Энтропия может интерпретироваться как мера неопределённости (неупорядоченности) некоторой системы (например, какого-либо опыта (испытания), который может иметь разные исходы, а значит, и количество информации[1][2]). Другой интерпретацией этого понятия является информационная ёмкость системы. С данной интерпретацией связан тот факт, что создатель понятия энтропии в теории информации Клод Шеннонсначала хотел назвать эту величину информацией. В широком смысле, в каком слово часто употребляется в быту, энтропия означает меру неупорядоченности системы; чем меньше элементы системы подчинены какому-либо порядку, тем выше энтропия.
6. Изучение тепловых эффектов химических процессов показало, чтоэкзотермические реакции, особенно сопровождающиеся значительным выделением теплоты, протекают самопроизвольно и часто весьма бурно. Более спокойно, но также самопроизвольно, т. е. без притока энергии извне, протекают экзотермические реакции с малым тепловым эффектом, многие из которых при повышении температуры обратимы. На основе этих наблюдений был сформулирован обп ий принцип (Бертло, 1867), утверждавший, что мерой химического сродства служит тепловой эффект реакции и что самопроизвольно протекают лишь такие ироцессы, которые сопровождаются выделением теплоты.
7. Для обратимых экзотермических реакций с повышением температуры равновесный выход продукта непрерывно уменьшается, а действительный выход увеличивается при низких температурах и уменьшается при высоких, проходя через максимум при оптимальной температуре.Абсолютное значение максимального выхода и соответствующее ему значение оптимальной температуры изменяются в зависимости от активности катализатора, концентрации реагирующих веществ и других условий процесса, но оптимальная температура всегда понижается с увеличением степени превращения.Эти закономерности справедливы для всех простых обратимых экзотермических газовых реакций, в частности для промышленных каталитических процессов: гидрирования, окисления, гидратации и подобного, для реакций синтеза аммиака, каталитического окисления SO2, конверсии оксида углерода, окисления этилена в оксид этилена и др.Сложным может быть влияние температуры на каталитические процессы, в которых повышение температуры до некоторого предела вызывает протекание вредных побочных реакций (например, для синтеза метанола и эталона, окисления аммиака и др.). При этом необходимо анализировать влияние температуры на каждую реакцию в отдельности.По фазовому состоянию реагентов реакции бывают гомогенные (однородные) и гетерогенные(неоднородные). В гомогенных реакциях все взаимодействующие вещества находятся в одной фазе (газовой, жидкой или твердой). Зоной реакции при проведении гомогенных реакций служит весь реакционный объем. В гетерогенных процессах реагенты, принимающие участие в реакции, находятся в разных фазах. В реакционном объеме одновременно находятся две или более фазы, а химическая реакция протекает на границе раздела фаз или в объеме одной из фаз.Гетерогенные двухфазные реакции в зависимости от агрегатного состояния исходных веществ бывают следующих типов: 1. в системе «газ – твердое тело»; 2. между двумя несмешивающимися жидкостями; 3. в системе «газ – жидкость»; 4. в системе «жидкость – твердая фаза».
Реакция сжигания угля служит примером гетерогенной реакции в системе «газ – твердое тело». Твердая фаза – углерод, взаимодействуя с газообразным кислородом, дает газообразный продукт реакции – углекислый газ.Примером реакции между несмешивающимися жидкостями может служить реакция нитрования толуола азотной кислотой, в которой оба исходных вещества являются взаимонерастворимыми жидкостями.Примером реакции в системе «газ – жидкость» является реакция поглощения ангидрида водой с образованием жидкой серной кислоты.Реакция между серной кислотой и окисью меди служит примером гетерогенной реакции «жидкость – твердая фаза».Гетерогенные системы бывают также трехфазными.
8. Скоростью химической реакции называют количество элементарных актов реакции, совершающихся в единицу времени.
Для удобства расчетов скорость химической реакции определяют по изменению концентрации реагирующих веществ за единицу времени. Для определения скорости реакции достаточно знать изменение концентрации во времени только одного из участвующих в реакции веществ, причем безразлично какого вещества - исходного или конечного. Различают среднюю и истинную скорость реакции. Средней скоростью реакции называют отношение изменения концентрации вещества (уменьшения концентрации исходного вещества или увеличение концентрации продукта реакции) к промежутку времени, в течение которого это изменение произошло:
![]()
Скорость реакции всегда считается величиной положительной, но отношение Δc/Δt может быть отрицательным, если υ изучается по изменению концентрации исходных веществ.
Чем ближе сдвигаются между собой t1 и t2, тем меньше становится Δt и Δc. Тогда отношение Δc/Δt все больше приближается к значению истинной скорости. Предельная величина Δc/Δt, когда Δt стремится к нулю как к своему пределу (Δt → 0), называется истинной скоростью химической реакции:
υ = lim (± Δc/Δt) = ± dc/dt,
где dc/dt - производная от концентрации по времени. Истинную скорость еще называют мгновенной скоростью реакции, т.е. скорость в данный момент времени.
9. Одним из основных постулатов химической кинетики является представление, согласно которому химическое взаимодействие является результатом столкновения частиц реагирующих веществ. Увеличение числа частиц в данном объеме (концентрации) приводит к более частым их столкновениям, т.е. к увеличению скорости химической реакции. Таким образом, скорость химической реакции пропорциональна концентрации реагирующих веществ. Эту зависимость скорости реакции от концентрации реагирующих веществ выражает закон действующих масс, открытый норвежскими учеными К. Гульдбергом и П. Вааге в 1867 г. Математическое выражение данной зависимости называют кинетическим уравнением реакции. Например, для реакции
2NO + O2 = 2NO2
зависимость скорости от концентрации исходных веществ может быть выражена уравнением:
? = k [NO] [NO] [O2] = k [NO]2 [O2],
где k — коэффициент пропорциональности, называемый константой скорости данной реакции, [NO] и [O2] — концентрации оксида азота (II) и кислорода соответственно. Константа скорости химической реакции зависит от природы реагирующих веществ и от температуры, но не зависит от концентрации.
В случае гетерогенных реакций в кинетическое уравнение входят концентрации только тех веществ, которые находятся в газовой фазе или в растворе. Например, для реакции горения угля
C(тв.) + O2(газ) = CO2
закон действующих масс будет представлен в следующем виде:
? = k [O2].
Следует отметить, что закон действия масс для газов может быть выражен как зависимость скорости реакции не только от концентрации исходных веществ, но от их парциальных давлений в системе. Например, для скорости реакции горения угля можно записать:
? = k p (O2),
где p(O2) — парциальное давление кислорода. Таким образом, скорость реакции горения угля прямо пропорциональна парциальному давлению кислорода.
Скоростью химической реакции называется количество вещества, вступающего в реакцию или образующегося при реакции за единицу времени в единице объема системы.
Количество вещества выражают в МОЛЯХ, а объем в ЛИТРАХ. В этом случае мы получаем удобную для работы величину - КОНЦЕНТРАЦИЮ вещества в моль/л, которая ИЗМЕНЯЕТСЯ в ходе реакции.
Таким образом, скоростью реакции называют изменение концентрации какого-нибудь вещества, участвующего в реакции, за единицу времени (например, за секунду или за минуту). Отсюда другое определение скорости реакции:
Скоростью химической реакции называется ИЗМЕНЕНИЕ КОНЦЕНТРАЦИИ реагента или продукта в единицу времени.
Разницу между тем, что было и тем, что стало, часто обозначают буквой греческого алфавита Δ (дельта) Следовательно, только что приведенное определение математически можно выразить так:
![]()
10. Молекулярность реакции – это минимальное число молекул, участвующих в элементарном химическом процессе. По молекулярности элементарные химические реакции делятся на молекулярные (А →) и бимолекулярные (А + В →); тримолекулярные реакции встречаются чрезвычайно редко.
Если реакция протекает последовательно через несколько гомогенных или гетерогенных элементарных стадий, то суммарная скорость всего процесса определяется самой медленной его частью, а молекулярность заменяется порядком реакции – формальным показателем при концентрации реагирующих веществ. Поэтому весь процесс в целом лучше характеризует порядок реакции.
Кинетическое уравнение реакции только для элементарных стадий совпадает с выражением ЗДМ. В этих случаях молекулярность и порядок реакции совпадают, хотя и не всегда. Так, при избытке одного из компонентов элементарной реакции А + В (А >> В) скорость реакции будет практически зависеть от изменения концентрации вещества В (А = const), поэтому порядок бимолекулярной реакции понижается до первого. Аналогично тому, что скорость реакции может характеризоваться по любому веществу, участвующему в реакции, для реакции aА + bВ → кинетические уравнения по веществу А и веществу В выглядят соответственно
|
|
|
а общее кинетическое уравнение –
|
|
(5.2) |
Здесь z = x + y – общий порядок реакции. Запишем кинетическое уравнение в дифференциальной форме для разных исходных реагентов:
|
|
|
Разделение переменных и интегрирование в пределах от нуля до τ дает приведенные в таб. 5.1 уравнения для реакций первого, второго и третьего порядков.
|
||||||||||||
Таблица 5.1 Кинетические уравнения и период полупревращения реакций первого, второго и третьего порядков |
 [с–1]
[с–1]
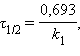 [с–1]
[с–1]
 [л∙моль∙с–1]
[л∙моль∙с–1]
 [л∙моль–1∙с–1]
[л∙моль–1∙с–1]
 [л2∙моль–2∙с–1]
[л2∙моль–2∙с–1]
 [л2∙моль–2∙с–1]
[л2∙моль–2∙с–1]
Решения кинетических уравнений 2-го и 3-го порядка, приведенные в таблице 5.1, справедливы только при равных начальных концентрациях веществ
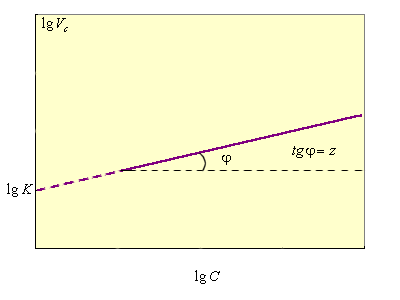
В каждом случае кинетическое уравнение линейно в соответствующих координатах С(τ), что позволяет графически определить порядок реакции (рис. 5.2).
|
Рисунок 5.2 Зависисмости C = f (τ) для реакций первого, второго и третьего порядков |

Прологарифмировав уравнение (5.2), получим lg V = lg K + z lg C; из графической зависимости (рис. 5.2) получаем lg K и z = tg φ.
Порядок реакции, особенно гетерогенной, может быть не только целочисленным (в том числе и нулевым), но и дробным. Нулевой порядок реакции указывает на постоянство скорости во времени.
Для
гетерогенной реакции C(тв)
+ О2(г)
= СО2(г)
можно создать концентрационнные условия,
при которых порядок реакции будет
меняться в пределах от нуля до единицы.
Действительно, при больших парциальных
давлениях кислорода
![]() в
приповерхностном слое твердого углерода
большой концентрационный градиент
способствует практически мгновенному
восполнению прореагировавшего кислорода.
Следствием этого оказывается постоянство
скорости реакции по кислороду, поскольку
в
приповерхностном слое твердого углерода
большой концентрационный градиент
способствует практически мгновенному
восполнению прореагировавшего кислорода.
Следствием этого оказывается постоянство
скорости реакции по кислороду, поскольку
![]() ~
const. Поэтому реакция горения углерода
будет в этих условиях подчиняться
кинетическому уравнению нулевого
порядка. При уменьшении парциального
давления кислорода, начиная с некоторого
скорость
реакции будет соответствовать
кинетическому уравнению первого порядка
~
const. Поэтому реакция горения углерода
будет в этих условиях подчиняться
кинетическому уравнению нулевого
порядка. При уменьшении парциального
давления кислорода, начиная с некоторого
скорость
реакции будет соответствовать
кинетическому уравнению первого порядка
 При
промежуточных давлениях кислорода
порядок реакции изменяется в интервале
от 0 до 1.
При
промежуточных давлениях кислорода
порядок реакции изменяется в интервале
от 0 до 1.
|
|
11. Гетерогенные реакции, хим. реакции с участием веществ, находящихся в различных фазах и составляющих в совокупности гетерогенную систему. Типичные гетерогенные реакции: термическое разложение солей с образованием газообразных и твердых продуктов (например, СаСО3 -> СаО + СО2), восстановление оксидов металлов водородом или углеродом (например, РbО + С -> Рb + СО), растворение металлов в кислотах (например, Zn + H2SO4 -> ZnSO4 + Н2), взаимодействие твердых реагентов (Аl2О3 + NiO -> NiAl2O4). В особый класс выделяют гетерогенно-каталитические реакции, протекающие на поверхности катализатора, при этом реагенты и продукты могут и не находиться в разных фазах. Например, при реакции N2 + + ЗН2 -> 2NH3, протекающей на поверхности железного катализатора, реагенты и продукт реакции находятся в газовой фазе и образуют гомогенную систему.
Особенности гетерогенных реакций обусловлены участием в них конденсированных фаз. Это затрудняет перемешивание и транспорт реагентов и продуктов; возможна активация молекул реагентов на поверхности раздела фаз. Кинетика любой гетерогенные реакции определяется как скоростью самого химического превращения, так и процессами переноса (диффузией), необходимыми для восполнения расхода реагирующих веществ и удаления из реакционной зоны продуктов реакции. В отсутствие диффузионных затруднений скорость гетерогенной реакции пропорциональна размерам реакционной зоны; так называемая удельная скорость реакции, рассчитанная на единицу поверхности (или объема) реакционной зоны, не изменяется во времени; для простых (одностадийных) реакций она может быть определена на основе действующих масс закона. Этот закон не выполняется, если диффузия веществ протекает медленнее, чем химическая реакция; в этом случае наблюдаемая скорость гетерогенные реакции описывается уравнениями диффузионной кинетики.
При гетерогенных реакциях с участием одного или нескольких твердых реагентов часто образуются твердофазные продукты. Такие реакции, как правило, локализованы на поверхности раздела фаз или в поверхностном слое и обычно протекают нестационарно. Они характеризуются периодом индукции. в течение которого возникают зародыши (ядра) новой фазы. Их образование связано с перестройкой атомной структуры твердого реагента и требует затраты энергии. Поэтому такие гетерогенные реакции чувствительны ко всем нарушениям структуры, облегчающим образование зародышей, и могут быть активированы термическими, радиационными, механическими и другими воздействиями, увеличивающими концентрацию дефектов, в первую очередь плотность дислокаций (см. Дефектыв кристаллах). Кинетическое уравнение реакции в этом случае отражает изменение во времени не только концентраций реагирующих веществ, но и поверхности раздела твердых фаз реагента и продукта: по мере роста зародышей поверхность раздела увеличивается и скорость реакции сначала возрастает, затем проходит через максимум и снижается вследствие соприкосновения растущих зародышей и образования сплошного слоя твердого продукта.
В природе гетерогенные реакции входят в комплекс процессов, приводящих к образованию осадочных пород и выветриванию. В химической технологии гетерогенные реакции газа с жидкостью (окисление воздухом, кислородом, озоном, хлорирование и др.) обычно проводят при интенсивном перемешивании специальными механическими устройствами или самим газом (в так называемом барботажном реакторе). Реакции термического разложения составляют основу фотографического процесса, реакции между газами или жидкостями и твердыми веществами - основу обжига, восстановления и окисления металлов, горения, производства твердых катализаторов, выщелачивания, экстракции и др. Часто сочетаются гетерогенные реакции в трех- и многофазных системах, например хлорирование твердых оксидов металлов газообразным хлором в присутствии твердого углеродсодержащего восстановителя. Важная область использования гетерогенные реакции - получение тонких поверхностных слоев и покрытий при взаимодействии твердого тела с жидкостью. При низких температурах диффузия в глубь твердого материала протекает медленно, что позволяет получать стабильные тонкие поверхностные слои, а в отдельных случаях - двухмерные фазы, толщина которых по порядку величины близка к параметру кристаллической решетки. Иногда стабильные поверхностные слои образуются самопроизвольно; таковы защитные оксидные пленки на металлах, препятствующие дальнейшему окислению.
12. Зависимость скорости протекания химической реакции от температуры определяется правилом Вант-Гоффа.
Голландский химик Вант-Гофф Якоб Хендрик, основатель стереохимии, в 1901 г. стал первым лауреатом Нобелевской премии по химии. Она была присуждена ему за открытие законов химической динамики и осмотического давления. Вант-Гофф ввёл представления о пространственном строении химических веществ. Он был уверен, что прогресса в фундаментальных и прикладных исследованиях по химии можно достичь, применяя физические и математические методы. Разработав учение о скорости реакций, он создал химическую кинетику.
Итак, кинетикой химических реакций называют учение о скорости протекания, о том, какое химической взаимодействие происходит в процессе реакций, и о зависимости реакций от различных факторов. У различных реакций скорость протекания различна.
Скорость химической реакции напрямую зависит от природы химических веществ, вступающих в реакцию. Некоторые вещества, такие как NаОН и НCl, способны реагировать за доли секунды. А некоторые химические реакции длятся годами. Пример такой реакции – ржавление железа.
Скорость реакции зависит также и от концентрации реагирующих веществ. Чем выше концентрация реагентов, тем выше и скорость реакции. В ходе реакции концентрация реагентов уменьшается, следовательно, замедляется и скорость реакции. То есть, в начальный момент скорость всегда выше, чем в любой последующий.
Скоростью химической реакции принято считать изменение концентрации реагирующих веществ в единицу времени.
V = (Cкон – Снач)/( tкон – tнач)
Концентрации реагентов определяют через определённые промежутки времени.
Важным фактором, от которого зависит скорость протекания реакций, является температура.
Все молекулы сталкиваются с другими. Число соударений в секунду очень велико. Но, тем не менее, химические реакции не протекают с огромной скоростью. Так происходит, потому что в ходе реакции молекулы должны собраться в активированный комплекс. А образовать его могут только активные молекулы, кинетической энергии которых достаточно для этого. При малом количестве активных молекул реакция протекает медленно. При повышении температуры увеличивается число активных молекул. Следовательно, и скорость реакции будет выше.
Вант-Гофф считал, что скорость химической реакции – это закономерное изменение концентрации реагирующих веществ в единицу времени. Но оно не всегда является равномерным.
Правило Вант-Гоффа гласит, что при повышении температуры на каждые 10о скорость химической реакции увеличивается в 2-4 раза.
Математически правило Вант-Гоффа выглядит так:
![]()
где V2 – скорость протекания реакции при температуре t2, а V1 – скорость протекания реакции при температуре t1;
ɣ - температурный коэффициент скорости реакции. Этот коэффициент есть отношение констант скоростей при температуре t+10 и t.
Так, если ɣ = 3, а при 0оС реакция длится 10 минут, то при 100оС она будет продолжаться всего 0,01 сек. Резкое увеличение скорости протекания химической реакции объясняется увеличением количества активных молекул при повышении температуры.
Правило Вант-Гоффа применимо только в температурном диапазоне 10-400оС. Не подчиняются правилу Вант-Гоффа и реакции, в которых участвуют большие молекулы.
13.
Уравне́ние
Арре́ниуса
устанавливает зависимость константы
скорости
k
{\displaystyle k}
 химической
реакции
от температуры
T
{\displaystyle T}
.
химической
реакции
от температуры
T
{\displaystyle T}
.
Согласно простой модели столкновений, химическая реакция между двумя исходными веществами может происходить только в результате столкновения молекул этих веществ. Но не каждое столкновение ведёт к химической реакции. Необходимо преодолеть определённый энергетический барьер, чтобы молекулы начали друг с другом реагировать. То есть молекулы должны обладать некой минимальной энергией (энергия активации E A {\displaystyle E_{A}} ), чтобы этот барьер преодолеть. Из распределения Больцмана для кинетической энергии молекул известно, что число молекул, обладающих энергией E > E A {\displaystyle E>E_{A}} , пропорционально exp ( − E A R T ) {\displaystyle \exp {\left(-{\frac {E_{A}}{RT}}\right)}} . В результате скорость химической реакции представляется уравнением, которое было получено шведским химиком Сванте Аррениусом из термодинамических соображений:
k = A e − E a / R T {\displaystyle k=Ae^{{-E_{a}}/{RT}}}
Здесь A {\displaystyle A} характеризует частоту столкновений реагирующих молекул, R {\displaystyle R} — универсальная газовая постоянная.
В рамках теории активных соударений A {\displaystyle A} зависит от температуры, но эта зависимость достаточно медленная:
A = a ⋅ T {\displaystyle A=a\cdot {\sqrt {T}}}
Оценки этого параметра показывают, что изменение температуры в диапазоне от 200 °C до 300 °C приводит к изменению частоты столкновений A {\displaystyle A} на 10 %.
В рамках теории активированного комплекса получаются другие зависимости A {\displaystyle A} от температуры, но во всех случаях более слабые, чем экспонента.
Уравнение Аррениуса стало одним из основных уравнений химической кинетики, а энергия активации — важной количественной характеристикой реакционной способности веществ.
14. Влияние катализатора Одно из наиболее эффективных средств воздействия на скорость химических реакций - использование катализаторов. Катализаторы - это вещества, которые изменяют скорость реакции, а сами к концу процесса остаются неизменными по составу и по массе. Иначе говоря, в момент самой реакции катализатор активно участвует в химическом процессе, но к концу реакции реагенты изменяют свой химический состав, превращаясь в продукты, а катализатор выделяется в первоначальном виде. Обычно роль катализатора заключается в увеличении скорости реакции, хотя некоторые катализаторы не ускоряют, а замедляют процесс. Явление ускорения химических реакций благодаря присутствию катализаторов носит название катализа, а замедления - ингибирования. Некоторые вещества не обладают каталитическим действием, но их добавки резко увеличивают каталитическую способность катализаторов. Такие вещества называются промоторами. Другие вещества (каталитические яды) уменьшают или даже полностью блокируют действие катализаторов, этот процесс называется отравлением катализатора. Существуют два вида катализа: гомогенный и гетерогенный. При гомогенном катализе реагенты, продукты и катализатор составляют одну фазу (газовую или жидкую). В этом случае отсутствует поверхность раздела между катализатором и реагентами. Особенность гетерогенного катализа состоит в том, что катализаторы (обычно твердые вещества) находятся в ином фазовом состоянии, чем реагенты и продукты реакции. Реакция развивается обычно на поверхности твердого тела. При гомогенном катализе происходит образование промежуточных продуктов между катализатором и реагирующим веществом в результате реакции с меньшим значением энергии активации. При гетерогенномый катализ объясняется адсорбцией реагирующих веществ на поверхности катализатора. В результате этого их концентрация увеличивается и скорость реакции растет. Особым случаем катализа является аутокатализ. Смысл его заключается в том, что химический процесс ускоряется одним из продуктов реакции.
15. 2. Константа равновесия
Равновесная концентрация веществ
Равновесная концентрация веществ – это концентрации веществ в реакционной смеси, находящихся в состоянии химического равновесия. Равновесная концентрация обозначается химической формулой вещества, заключенной в квадратные скобки.
Например,
следующая запись![]() обозначает,
что равновесная концентрация
водорода в равновесной
системе составляет 1
моль/л.
обозначает,
что равновесная концентрация
водорода в равновесной
системе составляет 1
моль/л.

Рис. 2
Химическое равновесие (Рис. 2) отличается от привычного для нас понятия «равновесие». Химическое равновесие – динамическое. В системе, находящейся в состоянии химического равновесия, происходят и прямая, и обратная реакции, но их скорости равны, и поэтому концентрации участвующих веществ не меняются. Химическое равновесие характеризуется константой равновесия, равной отношению констант скоростей прямой и обратной реакций.
![]()
Константы скорости прямой и обратной реакции – это скорости данной реакции при концентрациях исходных для каждой из них веществ в равных единицах. Также константа равновесия равна отношению равновесных концентраций продуктов прямой реакции в степенях стехиометрических коэффициентов к произведению равновесных концентраций реагентов.
![]()
Если
![]() ,
то в системе больше исходных
веществ. Если
,
то в системе больше исходных
веществ. Если ![]() ,
то в системе больше продуктов
реакции.
,
то в системе больше продуктов
реакции.
3. Обратимые и необратимые химические реакции
Если константа равновесия значительно больше 1, такую реакцию называют необратимой.
Необратимыми называются химические реакции, которые происходят только в одном направлении до полного расходования одного из реагентов.
Например, это реакция:
4Р+5О2 =2Р2О5 (2)
Обратимыми называются химические реакции, которые осуществляются во взаимно противоположных направлениях при одних и тех же условиях.
4. Факторы, влияющие на смещение равновесия
Если изменить внешние условия, то состояние химического равновесия нарушится. Смещение равновесия в зависимости от изменения внешних условий в общем виде определяется
· Принципом Ле Шателье: если на систему, находящуюся в равновесии, оказывают воздействие извне путем изменения какого-либо из условий, определяющих положение равновесия, то оно смещается в направлении того процесса, протекание которого ослабляет эффект произведённого воздействия.
Так, повышение температуры вызывает смещение равновесия в направлении того из процессов, течение которого сопровождается поглощением тепла, а понижение температуры действует в противоположном направлении.
Равновесие смещается вправо, если повысились равновесные концентрации продуктов прямой реакции. Если повышаются равновесные концентрации исходных веществ прямой реакции, то равновесие смещается влево. Какие факторы можно изменять, чтобы сместить равновесие? Это
· Температура
· Давление
· Концентрации веществ
· Добавление катализатора
· Изменение площади реакционной поверхности гетерогенных реакций
Добавление катализатора и изменение площади реакционной поверхности гетерогенных реакций не оказывают влияние на смещение химического равновесия.

Остальные факторы рассматриваем более детально.
Температура
Реакция синтеза аммиака (Рис. 3)
относится
к экзотермическим
реакциям.
При прохождении прямой
реакции теплота выделяется,
а при прохождении обратной
– поглощается. Если
увеличить температуру,
то, согласно правилу Ле
Шателье, равновесие
сместится в таком направлении,
чтобы уменьшить это воздействие.
В данном случае влево,
так как теплота поглощается.
Реакция синтеза аммиака
проводится при температуре
около 500![]()
Если реакция эндотермическая, то повышение температуры приведет к смещению равновесия вправо.
Изменение концентрации веществ
При увеличении концентрации какого-либо из веществ, участвующих в равновесной реакции, равновесие реакции сместится в сторону его расходования, а соответственно, при уменьшении концентрации какого-либо из веществ – в сторону реакции его образования. Например, при увеличении концентрации азота в реакции синтеза аммиака, равновесие сместится вправо, т. е. в сторону расходования азота. Если же в этой реакции удалять из реакционной смеси аммиак, то равновесие сместится в сторону его образования. Сделать это можно, например, при растворении аммиака в воде.
Изменение давления
Изменение давления может оказывать влияние только на реакции с участием газообразных веществ. Если в реакции синтеза аммиака увеличить давление, равновесие сместится в сторону уменьшения числа моль газа. Если слева число моль газа больше, чем справа, равновесие сместится в сторону образования аммиака.
Если число моль газа одинаково и слева и справа, например, в реакции получения оксида азота (II),
N2
+O2![]() (3)
(3)
то изменение давления не будет оказывать влияние на положение химического равновесия в таких реакциях. Изучение химического равновесия имеет большое значение, как для теоретических исследований, так и для решения практических задач. Определяя положение равновесия для различных температур и давлений, можно выбрать наиболее благоприятные условия проведения химического процесса. Окончательный выбор условий требует учета влияния их и на скорость процесса.
16. Принцип Ле Шателье
При известных ΔH реакции или при Δn ≠ 0 на химическое равновесие можно воздействовать изменением температуры или давления. Химическое равновесие может быть смещено изменением концентраций реагентов. Другими словами, равновесие можно сместить внешним воздействием, руководствуясь принципом Ле Шателье: если на равновесную систему оказывать внешнее воздействие, то равновесие смещается в сторону, противодействующую этому воздействию.
Влияние температуры. Для реакций, идущих с уменьшением энтальпии (экзотермических), повышение температуры будет препятствовать протеканию прямого процесса, то есть смещать реакцию в сторону исходных веществ. Эндотермические реакции при этом будут смещаться в сторону конечных продуктов. Например, при обычных условиях реакция N2 + O2 не идет (ΔH > 0), но повышение температуры может сделать эти реакцию осуществимой. Реакция CO + 1/2O2 = CO2, ΔH < 0 с повышением температуры будут смещаться в сторону исходных веществ.
Влияние давления. Если реагируют газообразные вещества, то при неизменном числе молей начальных и конечных реагентов повышение общего давления не приведет к смещению равновесия. Если число молей при реакции меняется, то изменение общего давления приведет к смещению равновесия. В частности, реакция 2CO + O2 = 2CO2, протекающая с уменьшением Δn, при повышении общего давления сместится в сторону образования СO2.
Влияние концентраций. В тех реакциях, в которых лучше оперировать концентрациями (реакции в растворах), увеличение концентраций исходных веществ приводит к смещению равновесия в сторону конечных продуктов и наоборот. Так, в реакции этерификации (образование сложного эфира)
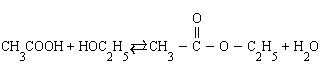
увеличение концентрации уксусной кислоты или этанола увеличивает выход этилацетата, а добавление в систему воды приводит к омылению, т. е. образованию исходных продуктов.
17. Концентрация раствора может выражаться как в безразмерных единицах (долях, процентах), так и в размерных величинах (массовых долях, молярности, титрах, мольных долях).
Концентрация – это количественный состав растворенного вещества (в конкретных единицах) в единице объема или массы. Обозначили растворенное вещество - Х, а растворитель - S. Чаще всего использую понятие молярности (молярная концентрация) и мольной доли.
Способы выражения концентрации растворов.
1. Массовая доля (или процентная концентрация вещества) – это отношение массы растворенного вещества m к общей массе раствора. Для бинарного раствора, состоящего из растворённого вещества и растворителя:
 ,
,
где:
ω – массовая доля растворенного вещества;
mв-ва – масса растворённого вещества;
mр-ра – масса растворителя.
Массовую долю выражают в долях от единицы или в процентах.
2. Молярная концентрация или молярность – это количество молей растворённого вещества в одном литре раствора V:
 ,
,
где:
C – молярная концентрация растворённого вещества, моль/л (возможно также обозначение М, например, 0,2 М HCl);
n – количество растворенного вещества, моль;
V – объём раствора, л.
Раствор называют молярным или одномолярным, если в 1 литре раствора растворено 1 моль вещества, децимолярным – растворено 0,1 моля вещества, сантимолярным – растворено 0,01 моля вещества, миллимолярным – растворено 0,001 моля вещества.
3. Моляльная концентрация (моляльность) раствора С(x) показывает количество молей n растворенного вещества в 1 кг растворителя m:
 ,
,
где:
С (x) – моляльность, моль/кг;
n – количество растворенного вещества, моль;
mр-ля – масса растворителя, кг.
4. Титр – содержание вещества в граммах в 1 мл раствора:
 ,
,
где:
T – титр растворённого вещества, г/мл;
mв-ва – масса растворенного вещества, г;
Vр-ра – объём раствора, мл.
5. Мольная доля растворённого вещества – безразмерная величина, равная отношению количества растворенного вещества n к общему количеству веществ в растворе:
 ,
,
где:
N – мольная доля растворённого вещества;
n – количество растворённого вещества, моль;
nр-ля – количество вещества растворителя, моль.
Сумма мольных долей должна равняться 1:
N(X) + N(S) = 1.
где N(X) - мольная доля растворенного вещества Х;
N(S) - мольная доля растворенного вещества S.
Иногда при решении задач необходимо переходить от одних единиц выражения к другим:

ω(X) - массовая доля растворенного вещества, в %;
М(Х) – молярная масса растворенного вещества;
ρ= m/(1000V) – плотность раствора.6. Нормальная концентрация растворов (нормальность или молярная концентрация эквивалента) – число грамм-эквивалентов данного вещества в одном литре раствора.
Грамм-эквивалент вещества – количество граммов вещества, численно равное его эквиваленту.
Эквивалент – это условная единица, равноценная одному иону водорода в кислотоно-основных реакциях или одному электрону в окислительно – восстановительных реакциях.
Для записи концентрации таких растворов используют сокращения н или N. Например, раствор, содержащий 0,1 моль-экв/л, называют децинормальным и записывают как 0,1 н.
 ,
,
где:
СН – нормальная концентрация, моль-экв/л;
z – число эквивалентности;
Vр-ра – объём раствора, л.
Растворимость вещества S - максимальная масса вещества, которая может раствориться в 100 г растворителя:

Коэффициент растворимости – отношение массы вещества, образующего насыщенный раствор при конкретной температуре, к массе растворителя:

18. Электролитическая диссоциация — процесс распада электролита на ионы при его растворении или плавлении.
Диссоциация в растворах
Диссоциация на ионы в растворах происходит вследствие взаимодействия растворённого вещества с растворителем; по данным спектроскопических методов, это взаимодействие носит в значительной мере химический характер. Наряду с сольватирующей способностью молекул растворителя определённую роль в электролитической диссоциации играет также макроскопическое свойство растворителя — его диэлектрическая проницаемость (Схема электролитической диссоциации).
Диссоциация при плавлении
Под действием высоких температур ионы кристаллической решётки начинают совершать колебания, кинетическая энергия повышается, и наступит такой момент (при температуре плавления вещества), когда она превысит энергию взаимодействия ионов. Результатом этого является распад вещества на ионы.
Классическая теория электролитической диссоциации
Классическая теория электролитической диссоциации была создана С. Аррениусом и В. Оствальдом в 1887 году. Аррениус придерживался физической теории растворов, не учитывал взаимодействие электролита с водой и считал, что в растворах находятся свободные ионы. Русские химики И. А. Каблуков и В. А. Кистяковский применили для объяснения электролитической диссоциации химическую теорию растворов Д. И. Менделеева и доказали, что при растворении электролита происходит его химическое взаимодействие с водой, в результате которого электролит диссоциирует на ионы.
Классическая теория электролитической диссоциации основана на предположении о неполной диссоциации растворённого вещества, характеризуемой степенью диссоциации α, то есть долей распавшихся молекул электролита. Динамическое равновесие между недиссоциированными молекулами и ионами описывается законом действующих масс . Например, электролитическая диссоциация бинарного электролита KA выражается уравнением типа:
KA ⇄ K + + A − {\displaystyle {\mbox{KA}}\rightleftarrows {\mbox{K}}^{+}+{\mbox{A}}^{-}}
Константа диссоциации K d {\displaystyle K_{d}} определяется активностями катионов a K + {\displaystyle a_{K^{+}}} , анионов a A − {\displaystyle a_{A^{-}}} и недиссоциированных молекул a K A {\displaystyle a_{KA}} следующим образом:
K d = a K + ⋅ a A − a K A {\displaystyle K_{d}={\frac {a_{K^{+}}\cdot a_{A^{-}}}{a_{KA}}}}
Значение K d {\displaystyle K_{d}} зависит от природы растворённого вещества и растворителя, а также от температуры и может быть определено несколькими экспериментальными методами. Степень диссоциации (α) может быть рассчитана при любой концентрации электролита с помощью соотношения:
K d = α 2 1 − α f ± {\displaystyle K_{d}={\frac {\alpha ^{2}}{1-\alpha }}f^{\pm }} ,
где f ± {\displaystyle f^{\pm }} — средний коэффициент активности электролита.
Слабые электролиты
Слабые электролиты — химические соединения, молекулы которых даже в сильно разбавленных растворах незначительно диссоциированны на ионы, которые находятся в динамическом равновесии с недиссоциированными молекулами. К слабым электролитам относится большинство органических кислот и многие органические основания в водных и неводных растворах.
Слабыми электролитами являются:
почти все органические кислоты и вода;
некоторые неорганические кислоты: HF, HClO, HClO2, HNO2, HCN, H2S, HBrO, H2CO3, H2SiO3, H2SO3 и др.;
некоторые малорастворимые гидроксиды металлов: Fe(OH)3, Zn(OH)2 и др.
Сильные электролиты
Сильные электролиты — химические соединения, молекулы которых в разбавленных растворах практически полностью диссоциированны на ионы. Степень диссоциации таких электролитов близка к 1. К сильным электролитам относятся многие неорганические соли, некоторые неорганические кислоты и основания в водных растворах, а также в растворителях, обладающих высокой диссоциирующей способностью (спирты, амиды и др.).
Классическая теория электролитической диссоциации применима лишь к разбавленным растворам слабых электролитов. Сильные электролиты в разбавленных растворах диссоциированы практически полностью, поэтому представления о равновесии между ионами и недиссоциированными молекулами лишено смысла. Согласно представлениям, выдвинутым в 20—30-х гг. 20 в. В. К. Семенченко (СССР), Н. Бьеррумом (Дания), Р. М. Фуоссом (США) и др., в растворах сильных электролитов при средних и высоких концентрациях образуются ионные пары и более сложные агрегаты. Современные спектроскопические данные показывают, что ионная пара состоит из двух ионов противоположного знака, находящихся в контакте («контактная ионная пара») или разделённых одной или несколькими молекулами растворителя («разделённая ионная пара»). Ионные пары электрически нейтральны и не принимают участия в переносе электричества. В сравнительно разбавленных растворах сильных электролитов равновесие между отдельными сольватированными ионами и ионными парами может быть приближённо охарактеризовано, аналогично классической теории электролитической диссоциации, константой диссоциации (или обратной величиной — константой ассоциации). Это позволяет использовать вышеприведённое уравнение для расчёта соответствующей степени диссоциации, исходя из экспериментальных данных.
В простейших случаях (большие одноатомные однозарядные ионы) приближённые значения константы диссоциации в разбавленных растворах сильных электролитов можно вычислить теоретически, исходя из представлений о чисто электростатическом взаимодействии между ионами в непрерывной среде — растворителе.
Примеры сильных электролитов: некоторые кислоты (HClO4, HMnO4, H2SO4, HCl, HBr; HI), гидроксиды щелочных и щёлочноземельных металлов (NaOH, KOH, Ba(OH)2); большинство солей.
19. С помощью теории электролитической диссоциации дают определения и описывают свойства кислот, оснований и солей.
Кислотами называются электролиты, при диссоциации которых в качестве катионов образуются только катионы водорода
Н3РО4
![]() Н+
+ Н2РО—4(первая ступень)
Н+
+ Н2РО—4(первая ступень)
Н2РО—4 Н+ + НРO2-4 (вторая ступень)
НРО2-4 Н+ PОЗ—4 (третья ступень)
Диссоциация многоосновной кислоты протекает главным образом по первой ступени, в меньшей степени по второй и лишь в незначительной степени — по третьей. Поэтому в водном растворе, например, фосфорной кислоты наряду с молекулами Н3РО4 имеются ионы (в последовательно уменьшающихся количествах) Н2РО2-4, НРО2-4 и РО3-4.
Основаниями называются электролиты, при диссоциации которых в качестве анионов образуются только гидроксид-ионы.
Например:
KOH K+ + OH—; NH4OH NH+4 + OH—
Основания, растворимые в воде называются щелочами. Их немного. Это основания щелочных и щелочноземельных металлов: LiOH, NaОН, КОН, RbОН, СsОН, FrОН и Са(ОН)2, Sr(ОН)2, Ва(ОН)2, Rа(ОН)2, а также NН4ОН. Большинство оснований в воде малорастворимо.
Кислотность основания определяется числом его гидроксильных групп (гидроксогрупп). Например, NН4ОН — однокислотное основание, Са(ОН)2 — двухкислотное, Fе(ОН)3 — трехкислотное и т.д. Двух— и многокислотные основания диссоциируют ступенчато
Ca(ОН)2 Са(ОН)+ + OH— (первая ступень)
Ca(OH)+ Ca2++OH— (вторая ступень)
Однако имеются электролиты, которые при диссоциации одновременно образуют катионы водорода, и гидроксид—ионы. Эти электролиты называются амфотерными или амфолитами. К ним относятся вода, гидроксиды цинка, алюминия, хрома и ряд других веществ. Вода, например, диссоциирует на ионы Н+ и ОН— (в незначительных количествах):
Н2O Н+ + ОН—
Следовательно, у нее в равной мере выражены и кислотные свойства, обусловленные наличием катионов водорода Н+, и щелочные свойства, обусловленные наличием ионов ОН—.
Диссоциацию амфотерного гидроксида цинка Zn(ОН)2 можно выразить уравнением
2ОН— + Zn2+ + 2Н2О Zn(ОН)2 + 2Н2О [Zn(ОН)4]2-+ 2Н+
Солями называются электролиты, при диссоциации которых образуются катионы металлов а также катион аммония ( NH+4) и анионы кислотных остатков
Например:
(NH4)2SO4 2NH+4 + SO2-4; Na3PO4 3Na+ + PO3-4
Так диссоциируют средние соли. Кислые же и основные соли диссоциируют ступенчато. У кислых солей вначале отщепляются ионы металлов, а затем катионы водорода. Например:
KHSO4 K+ + HSO—4
и далее
HSO—4 H++SO2-4
У основных солей вначале отщепляются кислотные остатки, а затем гидроксид—ионы.
Mg(OH)Cl Mg(OH)++Cl—
и далее
Mg(OH)+ Mg2++OH—
Например:
Например:
НCl Н++ Сl—; СН3СООН Н+ + СН3СОО—
Основностъ кислоты определяется числом катионов водорода, вторые образуются при диссоциации. Так, НCl, HNO3 — одноосновные кислоты — образуется один катион водорода; Н2S, Н2СО3, Н2SO4 — двухосновные, а Н3РО4, Н3АsО4 — трехосновные, так как образуются соответственно два и три катиона водорода. Из четырех атомов водорода, содержащихся в молекуле уксусной кислоты СН3СООН, только один, входящий в карбоксильную группу — СООН, способен отщепляться в виде катиона Н+, — уксусная кислота одноосновная.
Двух— и многоосновные кислоты диссоциируют ступенчато (постепенно).
20. Реакции, протекающие в растворах между электролитами, называются реакциями ионного обмена. (реакции ионного обмена – это реакции между ионами, образовавшимися в результате диссоциации электролитов).
Реакции обмена в растворах электролитов протекают в направлении связывания ионов.
При взаимодействии гидроксида натрия с соляной кислотой образуются хлорид натрия и малодиссоциирующее вещество вода:
NaOH + HCl = NaCl + H2O
Na+ + OH- + H+ + Cl- = Na+ + Cl- + H2O
OH- + H+ = H2O
При взаимодействии гидроксида натрия с раствором сульфата меди(II) образуются сульфат натрия и нерастворимое основание – гидроксид меди(II):
2NaOH + CuSO4 = Na2SO4 + Cu(OH)2?
2Na+ + 2OH- + Cu2+ + SO42- = 2Na+ + SO42- + Cu(OH)2?
2OH- + Cu2+ = Cu(OH)2?
При взаимодействии азотной кислоты с карбонатом калия образуются нитрат калия, вода и углекислый газ:
2HNO3 + K2CO3 = 2KNO3 + H2O + CO2#
2H+ + 2NO3- + 2K+ + CO32- = 2K+ + 2NO3- + H2O + CO2#
2H+ + CO32- = H2O + CO2#
Итак, реакции ионного обмена протекают до конца, если: 1) образуется осадок; 2) выделяется газ; 3) образуется малодиссоциирующее вещество – вода.
Если в растворах нет таких ионов, которые могут связываться между собой с образованием осадка, газа или воды, то реакция является обратимой, например, при взаимодействии растворов хлорида калия и нитрата натрия не происходит связывания ионов:
KCl + NaNO3 D KNO3 + NaCl
K+ + Cl- + Na+ + NO3- D K+ + NO3- + Na+ + Cl-