

Квантово-механическая модель атома водорода. Периодический закон. Периодическая система д.И. Менделеева. Электронные формулы элементов
Основные вопросы:
Квантово-механическая модель атома водорода. Волновое уравнение Шредингера.
Квантовые числа, характеризующие поведение электрона в атоме (главное, орбитальное, магнитное, спиновое).
Принципы минимума энергии, запрета Паули, правила Гунда и Клечковского.
Периодический закон Д.И. Менделеева. Периодическая система химических элементов Д.И. Менделеева.
Периодические свойства химических элементов.
Классическая механика Ньютона не пригодна для описания поведения микорообъектов: элементарных частиц, ядер, атомов, молекул, ионов. Эту задачу решает квантовая механика, оперирующая сложным математическим аппаратом. Не приводя математических выкладок, рассмотрим некоторые выводы, которые следуют из квантово-механических моделей атомов и молекул.
В 1924 г. Луи де Бройль высказал гипотезу о проявлении волновых свойств теми микрообъектами, которые считались частицами. Связь между
9
массой микрообъекта и его волновыми характеристиками он выразил уравнением:
![]()
где h 6,625 1034 Дж с – постоянная Планка; v – скорость частицы; m – ее масса; λ – соответствующая этой массе длина волны.
Уже в 1927 г. гипотеза де Бройля получила блестящее экспериментальное подтверждение: были обнаружены волновые свойства электрона, а в дальнейшем и нейтрона.
В 1926 г. Э. Шредингер представил состояние электрона в атоме как трехмерную стоячую волну и описал его дифференциальным уравнением второй степени в частных производных. В уравнение Шредингера входит волновая функция Ψ, которая является математическим описанием волнового поведения электрона. Решение уравнения Шредингера указывает на вероятность нахождения электрона в том или ином участке пространства и не связывает пребывание электрона в атоме с траекторией. Квадрат волновой функции Ψ2 характеризует вероятность нахождения электрона в элементе объема пространства dV.
Электрон в атоме водорода можно представить как бы размазанным в пространстве в виде электронного облака. Электронное облако бесконечно. Выделяют пространство, в котором вероятность пребывания электрона равна определенной, заданной величине, которую называют орбиталью. Наиболее удобно принять 0,9 (90 %), однако эта величина условна и может быть увеличена или уменьшена, что приведет к увеличению или уменьшению размера орбитали, но не скажется на ее форме.
При математическом описании состояния электрона появляются простые целые числа, определяющие дискретность его характеристик. Числа называются квантовыми. Их четыре – столько, сколько у электрона степеней свободы (три, обусловленные движением электрона в пространстве, и еще одна, не связанная с ним).
1.1. Главное квантовое число n. Из решения уравнения Шредингера следует, что энергия электрона в атоме водорода равна
E m e 4 ,
2 2 n 2
где m 9,11 1031 кг – масса электрона; е – заряд электрона, 1,60 1019 Кл; n главное квантовое число, принимающее целочисленные действительные
10
положительные значения: 1, 2, 3, 4 и т.д.; 2h – приведенная постоянная
![]()
Планка.
В атоме водорода n однозначно определяет главную характеристику электрона – энергию, а также характеризует размер орбитали (с увеличением n увеличивается средняя удаленность электрона от ядра).
Совокупность электронных состояний с определенным значением главного квантового числа называют квантовым (или электронным) уровнем (или слоем).
Орбитальное квантовое число l. Движение электрона в поле ядра характеризуется орбитальным моментом количества движения (ОМКД), который связан с формой орбитали: чем меньше ОМКД, тем совершенней симметрия орбитали. Из решения уравнения Шредингера следует, что ОМКД дискретен. Его модуль относительно одной из координат (например, z) определяется уравнением
М
z
![]()
![]() l(l
1)
,
l(l
1)
,
где l – орбитальное квантовое число, которое принимает целочисленные положительные значения от 0 до (n-1). Его принято обозначать не только цифрами, но и латинскими буквами: 0(s), 1(p), 2(d), 3(f), 4(g)…
Совокупность электронных состояний с определенным значением n и l называют квантовым (электронным) подуровнем.
Для первого квантового уровня n = 1 значение орбитального квантового числа единственное: l = 0. Такая орбиталь обозначается 1s и имеет форму шара. Знак Ψ-функции в любой точке пространства одинаков.
Во втором квантовом уровне n = 2 орбитальное квантовое число l может принимать два значения: l = 0(2s) l = 1(2p). ОМКД 2р-орбитали отличен от нуля. Она напоминает объемную восьмерку. Функция Ψ2р имеет разные знаки в разных ее частях.
В третьем квантовом уровне n = 3 орбитальное квантовое число l может принимать три значения: l = 0(3s) l = 1(3p) l = 2(3d). Форма орбитали более сложная. Увеличение l приводит к дальнейшему усложнению формы орбитали.
Орбитальное квантовое число определяет в атоме величину ОМКД и, как следствие этого, форму орбитали (рисунок 1).
11
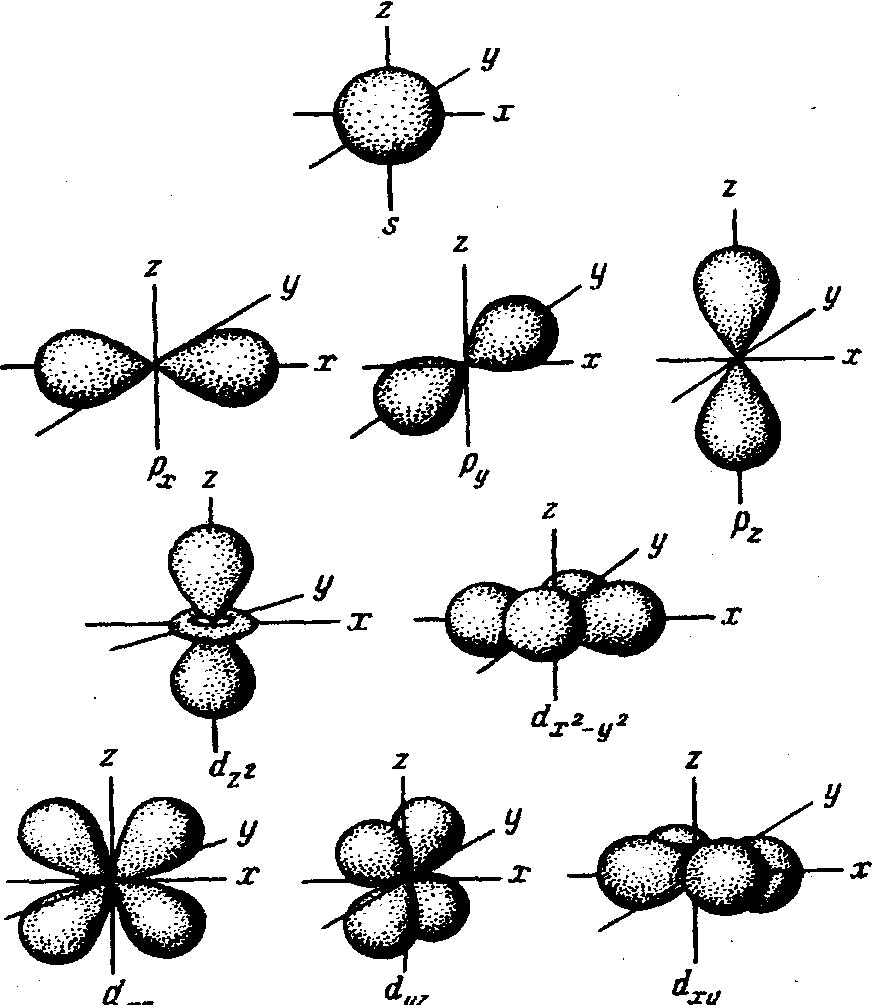
Рис. 1. Формы электронной орбитали
Магнитное квантовое число m(ml). Из решения уравнения Шредингера проекция ОМКД на направление поля (ось z) равна
M z m ,
где m – магнитное квантовое число, принимающее целочисленные значения от –l до +l, включая нуль.
Магнитное квантовое число определяет величину проекции определяет величину проекции ОМКД на направление магнитного поля, т.е. ориентацию орбитали в пространстве.
При l = 0 возможно единственное значение m = 0. Действительно, на шарообразной s-орбитали возможен единственный способ ее ориентации в пространстве.
Для р-орбитали (l = 1) трем значениям магнитного квантового числа
(m = 1; 0; -1) отвечают три варианта ориентации вектора ОМКД и, соответственно, три способа расположения орбитали в пространстве:
Спиновое квантовое число s (или ms). Электрон кроме ОМКД имеет собственный момент количества движения (СМКД), аналогичный тому, который в макросистемах возникает при вращении тела вокруг своей оси.
12
Однако объяснить наличие СМКД электрона его механическим вращением нельзя. Эта четвертая степень свободы электрона приводит к появлению четвертого квантового числа, которое называется спиновым, или спином. Проекция СМКД на направление магнитного поля:
M s s ,
где s – спин электрона. Спин принимает лишь два значения (±1/2). Знак указывает на ориентацию вектора СМКД относительно магнитного поля.
Орбиталь является совокупностью электронных состояний с определенными значениями n, l, m. Часто ее называют квантовой ячейкой. Стрелками обозначают электроны. Направление стрелки символизирует знак спина.
1.2. Принципы построения электронной оболочки в многоэлектронных атомах. Одним из основных положений квантовой механики является принцип (запрет) Паули (1925 г): в атоме не может быть двух электронов, имеющих одинаковый набор всех четырех квантовых чисел, или в атоме любой электрон отличается от любого другого, по крайней мере, одним квантовым числом.
Состояние электрона в атоме должно удовлетворять термодинамическому условию устойчивости системы, что отражается принципом наименьшей энергии: электрон в атоме самопроизвольно занимает состояние с наименьшей энергией. Оно называется нормальным или стационарным. Для атома водорода это 1s. При ультрафиолетовом облучении атом может поглощать квант света. Это приводит к переходу электрона в возбужденное состояние. Время жизни электрона порядка 10-10–10-8 с, после чего он самопроизвольно переходит в стационарное состояние, излучая количество энергии, равное поглощенному, в виде одного или несколько квантов.
В отличие от атома водорода, в многоэлектронных атомах энергия электрона возрастает с ростом главного и орбитального квантовых чисел. В.М. Клечковским (1951 г.) показано, что между суммой (n+l) и энергией электрона в атоме имеется прямая зависимость. На основании принципа наименьшей энергии им сформулировано правило последовательности заполнения орбиталей электронами:
Заполнение атомных орбиталей идет от орбиталей с меньшим значением , n+l суммы квантовых чисел к орбиталям с большим значением n+l (первое правило Клечковского).
13
При равных величинах n+l порядок заполнения электронами атомных орбиталей происходит последовательно в направлении от меньшего значения n к большему (второе правило Клечковского).
Последовательное квантованное возрастание возрастание энергии подуровней происходит в следующем порядке:
1s–2s –2p–3s –3p –4s –3d–4 p –5s –…
Такой порядок увеличения энергии подуровней отражен в электронных формулах атомов элементов.
Принципу наименьшей энергии также подчиняется заселение электронами орбиталей в пределах квантового подуровня. Электроны занимают равные орбитали, поскольку средняя удаленность их друг от друга в этом случае наибольшая, электростатическое отталкивание минимальное, следовательно, энергия системы меньше. Это отражается правилом Хунда (1927 г.): в пределах квантового подуровня наименьшую энергию имеет состояние с максимальным значением суммарного спина.
Распределение электронов по квантовым уровням и подуровням представляют электронными формулами. Форма записи для каждого подуровня атома (Э) с зарядом ядра (Z) имеет вид: Z Э...nl x где n –главное квантовое число; l – буквенное обозначение орбитального квантового числа; х – число электронов на подуровне. Наряду с электронными формулами используют графическое изображение распределения электронов по орбиталям (квантовым ячейкам), которое дает дополнительную информацию
магнитном и спиновом квантовом числах.
К середине XIX в. число известных химических элементов возросло настолько (оно достигло 60), что возникла потребность их упорядочения. В 1869 г Д. И. Менделеев открыл Периодический закон: «…Свойства элементов, а поэтому и свойства образуемых ими простых и сложных тел состоят в периодической зависимости от их атомного веса». Периодический закон был представлен как классификация элементов. На его основе элементы были сгруппированы в нескольких горизонтальных рядах так, что вертикальные столбцы включали элементы, сходные по совокупности химических свойств.
Однако причины периодичности стали ясны только в начале ХХ в. благодаря успехам физики. От элемента к элементу по периодической системе изменяется заряд ядра атома элемента, который определяется числом протонов. В периодической системе это число совпадает с порядковым номером элемента. Атом электронейтрален, поэтому заряд ядра равен числу электронов в атоме. Так как свойства элементов, особенно химические,
определяются, в основном, электронами внешнего квантового слоя,
14
причиной периодичности свойств является периодичность заполнения электронами квантовых подуровней. Фактором, определяющим строение электронных оболочек атомов, а следовательно и свойства элементов, является заряд ядра атома. Современная формулировка периодического закона: свойства элементов и их соединений находятся в периодической зависимости от заряда ядра атома элемента.
Периодический закон представляется в виде периодической таблицы, которая является структорограммой строения атома любого элемента. Положение элемента в таблице однозначно отражает электронное строение его атомов.
Элементы в периодической системе располагаются по периодам и группам. Периодом называется ряд элементов, расположенных в порядке возрастания заряда ядра, начинающийся щелочным металлом с заселения нового квантового уровня и заканчивающийся ближайшим благородным газом с устойчивой октетной структурой ns2np6. Номер периода совпадает со значением главного квантового числа внешнего квантового уровня.
Группа представляет собой вертикальный ряд элементов. Принадлежность к группе и деление ее на подгруппы определяется аналогией структур внешнего (главные, или А-подгруппы) или внешнего и предвнешнего (побочные, или В-подгруппы) квантовых уровней.
В зависимости от того, какой подуровень в атоме заселяет последний электрон, элементы подразделяют на электронные s-, p-, d- и f-семейства.
Семейства s- и p-элементов составляют главные А-подгруппы периодической системы. Валентными, т.е. образующими химические связи, электронами атомов этих элементов являются s- и p-электроны внешнего квантового уровня. Число их равно номеру группы.
Из структуры периодической системы следует:
Номер периода равен максимальному значению главного квантового
числа.
Каждый период начинается с заполнения s-подуровня соответствующего нового квантового уровня и заканчивается заполнением р-подуровня этого же уровня.
В четвертом и последующих больших периодах, начиная с третьей группы, идет заполнение d-подуровня предвнешенего уровня.
Суммарное число s- и р-электронов на внешнем квантовом уровне s-
р-элементов равно номеру группы, в которой находится данный элемент. Эти электроны называются валентными.
Каждый период завершает инертный газ, имеющий внешний уровень
ns2np6.
15
Элементы А-подгрупп являются s- и р-элементами.
Элементы побочных В-подгрупп – d-элементы. Валентными являются s-электроны внешнего и d-электроны предвнешенего подуровней.
В шестом и седьмом периодах у f-элементов – лантоноидов и актиноидов – идет заполнение соответственно 4f- и 5f-подуровней.
Валентными являются, как правило, (n-1)d1ns2-электроны.
Периодическая система элементов Д.И. Менделеева относится к важнейшим инструментам науки и химического образования. Взаимосвязи между Периодической системой, строением атома и свойствами элементов весьма способствует систематике и упорядочению химии как естественной науки.
3. Периодически изменяются следующие свойства атомов: радиус атомов, энергия ионизации, сродство к электрону, электроотрицательность.
За радиус атома принимают половину расстояния между ядрами соседних атомов в кристалле простого вещества. В пределах одного периода с увеличением заряда ядра проявляется тенденция к уменьшению размера атомов. С ростом заряда ядра притяжение между ним и электроном внешнего квантового уровня усиливается, что и является причиной уменьшения радиуса атома. Электроны последующего квантового слоя более удалены от ядра, чем предыдущего, отсюда при переходе к следующему периоды радиусы атомов возрастают. Поэтому в пределах подгруппы с возрастанием заряда ядра размеры атомов увеличиваются.
Радиус катиона всегда меньше, а радиус аниона заметно больше радиуса нейтрального атома. В подгруппе размеры ионов одинакового заряда, как и размеры атомов, возрастают.
Энергия, которую необходимо затратить для отрыва электрона от атома и удаления его на бесконечное расстояние, называется энергией ионизации (I). Это энергетический эффект реакции Э = Э+ + е. Процесс отдачи электронов называется окислением. Атомы, которые окисляются, отдавая электроны, называются восстановителями. Восстановительная способность является мерой активности металла, мерой металличности. С уменьшением энергии ионизации возрастает восстановительная способность, усиливается металличность.
Энергия, которая выделяется при присоединении электрона к атому с образованием аниона, называется сродством к электрону (E). Это энергетический эффект реакции Э +е = Э-. Процесс принятия электронов называется восстановлением. Атомы, которые восстанавливаются, принимая электроны, называются окислителями. Наибольшим сродством к электрону
16
обладают элементы VIIA-подгруппы с конфигурацией ns2np5. Близкие к нулю или отрицательное сродство к электрону имеют металлы и благородные газы. Отчетливо проявляется тенденция возрастания сродства к электрону атомов р-элементов в периоде слева направо и в группе снизу вверх.
Для характеристики способности атомов в соединениях притягивать к себе электроны введено понятие электроотрицательности (ЭО). Электроотрицательность элемента возрастает по периоду и несколько убывает в группах с возрастанием номера периода у элементов I, II, V, VI VII главных подгрупп, III, IV, V – побочных подгрупп имеет сложную зависимость у элементов III главной подгруппы, возрастает с увеличением номера периода у элементов IV, V – побочных подгрупп.
Вопросы для контроля
В чем сущность квантово-механической модели атома водорода?
Что показывают квантовые числа?
Назовите принципы распределения электронов по энергетическим уровня и подуровням.
Для чего записываются электронные формулы элементов?
Сформулируйте Периодический закон Д.И. Менделеева. В чем физический смысл Периодического закона?
Какие элементы называются s-, p-, d-, f-элементами?
Перечислите периодические свойства химических элементов.
Литература
1. Коровин Н.В. Общая химия: Учебник для тех. направл. и спец. Вузов - 6-е
изд., испр. – М.: Высш.шк., 2005. – с. 17-31.
Лекция 3
Строения вещества. Химическая связь
Основные вопросы:
Химическая связь, ее характеристики.
Метод валентных связей. Метод молекулярных орбиталей.
Строение вещества и химическая связь.
17
Химической связью называют взаимодействие двух или несколько атомов, в результате которого образуется химически устойчивая многоатомная система (молекула, кристалл, комплекс и др.)
Учение о связи занимает центральное место в современной химии. В 1915 г. физик Коссель (Германия) дал объяснение химической связи в солях,
1916 г. физико-химик Льюис (США) предложил трактовку химической связи в молекулах. Коссель и Льис исходили из представления о том, что атомы элементов обладают тенденцией к достижению электронной конфигурации благородных газов. Атомы благородных газов, кроме элемента первого периода – гелия, имеют во внешней электронной оболочке устойчивый октет (восемь) электронов. При таком строении способность атомов к вступлению в химические реакции минимальна. Представления Косселя и Льюиса получили в истории химии название октетной теории, или электронной теории валентности.
Различают пять видов связи: ковалентная, ионная, металлическая, водородная и связь межмолекулярного взаимодействия. Связь едина по своей природе и различия между ее видами относительно.
Согласно принципу наименьшей энергии, внутренняя энергия молекулы по сравнению с суммой внутренней энергии образующих ее атомов должна понижаться. Внутренняя энергия молекулы включает сумму энергий взаимодействий каждого электрона с каждым ядром, каждого электрона с каждым другим электроном, каждого ядра с каждым другим ядром. Притяжение должно превалировать над отталкиванием.
Важнейшей характеристикой связи является энергия, определяющая ее прочность. Мерой прочности связи служит как количество энергии, затрачиваемой на ее разрыв (энергия диссоциации связи), так и величина, которая при суммировании по всем связям дает энергию образования молекулы из элементарных атомов.
Развитие квантово-механических представлений о строение атома и создание орбитальной модели атома привели к выработке двух современных научных подходов для объяснения химической связи – метода валентных связей (ВС) и метода молекулярных орбиталей (МО). В 1927 г. уравнение Шредингера было решено для молекулы водорода немецкими физиками Ф. Лондоном и В. Гейтлером. Это была первая удачная попытка применения квантовой механики к решению проблем связи. Из квантово-механического расчета следует, что к образованию связи приводит взаимодействие пары
электронов с противоположными спинами, но с волновыми функциями одинакового знака, которые суммируются. Результатом является увеличение
18
электронной плотности в области перекрывания электронных облаков и стягивания ядер.
Основные положения метода ВС:
связь образуется электронной парой с антипараллельными спинами;
причиной связи является электростатическое взаимодействие ядер и электронов;
прочность связи пропорциональна перекрыванию электронных облаков;
насыщаемость связи обусловлена образованием электронных пар;
направленность связи обусловлена перекрыванием электронных облаков в области их максимальной плотности.
Валентность в методе ВС определяется числом ковалентных связей, которые образует атом.
Механизм образования связи парой электронов с антипараллельными спинами, принадлежавших до образования связи разным атомам, называется обменным. Если учитывать обменный механизм образования связи, то валентность атома определяется числом его неспаренных электронов.
Насыщаемость как свойство ковалентной связи предполагает, что если атом имеет несколько неспаренных электронов, то все они участвуют в образовании ковалентных связей. Это свойство объясняется принципом наименьшей энергии. При образовании каждой дополнительной связи выделяется дополнительная энергия. Поэтому все валентные возможности реализуются полностью. Число ковалентных связей не может превышать числа образующих связи электронных пар.
Химическая связь может быть образована и по донорно-акцепторному
механизму: один атом (донор) представляет на образование связи не поделенную пару электронов, а другой (акцептор) – вакантную квантовую ячейку. Образование химической связи по донорно-акцепторному механизму расширяет валентные возможности атомов. Валентность определяется не только числом неспаренных электронов, но и не поделенных пар электронов и вакантных квантовых ячеек, участвующих в образовании химических связей.
гомоядерных молекулах (H2, F2 и др.) электронная пара, образующая связь, в равной степени принадлежит каждому из атомов, поэтому центры положительного и отрицательного зарядов в молекуле совпадают. Такие молекулы неполярны.
гетероядерных молекулах вклад в связь волновых функций различных атомов неодинаков. В этом случае говорят о смещении электронной пары вблизи одного атома к другому. Для определения
19
направления такого смещения введено понятие электроотрицательности. Имеется несколько шкал электроотрицательности.
Электротрицательность по Малликену есть полусумма энергий ионизации и сродства к электрону. Валентная электронная пара смещается к более электроотрицательному атому. Удобнее пользоваться с относительными значениями электроотрицательности. За единицу принята электроотрицательность лития. Относительная электроотрицательность какого-либо элемента А равна
Х А ЭОА . ЭОLi
Наименьшую электроотрицательность имеют тяжелые щелочные металлы ( X Fr = 0,7). Самый электроотрицательный элемент фтор ( X F = 4,0). В периодах Периодической системы элементов наблюдается общая тенденция роста электроотрицательности, а по подгруппам – ее уменьшение.
Полярность связи определяется смещением валентной электронной пары в двухатомных молекулах и количественно характеризуется дипольным моментом молекулы. Он равен произведению расстояния между ядрами в
![]()
молекуле r и эффективностью заряда δ, соответствующего этому расстоянию:
![]()
![]()
μ r δ .
Так как r считают вектором, направленным от положительного к отрицательного заряду, дипольный момент также является вектором и имеет то же направление. Единицей измерения дипольного момента является дебай
D (1D = 3,33·10-30 Кл·м).
Дипольный момент сложной молекулы определяется как векторная сумма дипольных моментов всех связей. Если молекула АВn симметрична относительно линии каждой связи, суммарный дипольный момент такой
n
м![]() олекулы
равен нулю: μ
μi
0
. Примерами могут служить i
1
олекулы
равен нулю: μ
μi
0
. Примерами могут служить i
1
симметричные молекулы, связи в которых образованы гибридными орбиталями: BeF2, BF3, CH4, SF6 и др.
20
Молекулы, связи в которых образованы негибридными орбиталями с участием неподеленных пар электронов, не симметричны относительно линий связей. Дипольные моменты подобных молекул не равны нулю. Примерами полярных молекул являются H2S, NH3, H2O и др.
В полярной связи можно выделить две составляющие: ионную, обусловленную электростатическим притяжением, и ковалентную, обусловленную перекрыванием орбиталей. По мере увеличения разности электроотрицательностей валентная электронная пара все сильней смещается к более электроотрицательному атому, который приобретает более отрицательный эффективный заряд δ-. Увеличивается вклад в связь ионной составляющей. Эффективный заряд приближается к единице, т.е. к заряду электрона, количественные изменения переходят в качественные: можно считать, что образовались два иона: катион Kt+ и анион An-. Такая связь называется ионной. Ее можно рассматривать как крайний случай ковалентной полярной связи.
Электростатическое поле не имеет преимущественных направлений. Поэтому ионной связи в отличие от ковалентной, не свойственна направленность. Ион взаимодействуют с любым количеством ионов противоположного заряда. Этим обусловлено еще одно свойство ионной связи – отсутствие насыщаемости.
Граница между ковалентной полярной и ионной связями условна. Для молекул в газообразном состоянии считают, что при разности электроотрицательностей ∆Х > 2,5 связь между атомами ионная. В растворах полярных растворителей, а также в кристаллическом состоянии сильное влияние оказывают соответственнр молекулы растворителя и соседние частицы в узлах кристаллической решетки. Поэтому ионный характер связи проявляется при значительно меньшей разности электроотрицательностей. Связь между типичными металлами и неметаллами в кристаллах ионная.
Метод валентных связей не является универсальным. Например, в молекулярном ионе Н2 связь образуется одним электроном, а не парой, что
противоречит принципам метода ВС. В молекуле О2 по методу ВС все электроны должны быть спарены. Однако магнитные измерения указывают на наличие двух неспаренных электронов. Эти и многие другие противоречия объясняются методом молекулярных орбиталей (МО).
Более широкие возможности для объяснения химических связей и построения электронных структур предоставляет метод молекулярных орбиталей. Он достаточно сложен, особенно для многоатомных молекул, тем не менее широко используется в количественных расчетах с применением
21
компьютеров. Молекула в методе МО рассматривается как единая система из электронов и ядер. В молекуле, как в атоме, существуют орбитали, уровни и подуровни, соблюдается принцип наименьшей энергии, принцип Паули, и правило Хунда.
МО имеют более сложную форму, чем атомные орбитали АО. В простейшем приближении МО можно представить как линейные комбинации исходных АО. Число образующихся МО равно числу исходных АО.
3. Характер связи между частицами в кристалле определяет его химические и физические свойства. Кристаллические решетки подразделяют на ионную, атомную, молекулярную, металлическую.
узлах ионной кристаллической решетки (ИКР) чередуются катионы и анионы. Каждый ион окружен определенным числом ионов противоположного заряда (координационное число). Связь ионная. ИКР имеет большинство солей, образованных типичными металлами и неметаллами.
Вещества с ИКР весьма устойчивы. Разрушение прочной ИКР требует значительной затраты энергии, поэтому они плавятся при достаточно высокой температуре. Хрупкость таких веществ объясняется тем, что при деформации (смещении слоев) ближайшими соседями оказываются ионы одинакового знака, в результате чего преобладающими становятся силы отталкивания.
Атомную кристаллическую решетку (АКР) имеют кристаллы неметаллов, построенные из атомов одного элемента или элементов с близкой электроотрицательностью. Возникает ковалентная неполярная или малополярная связь. Типичным примером вещества с АКР является алмаз, построенный из атомов углерода. Алмаз является самым твердым из известных веществ, не растворяется ни в одном из растворителей, не окисляется при комнатной температуре даже фтором, горит на воздухе лишь при температуре выше 900 ºС. Прочность и равноценность ковалентных связей определяют химическую устойчивость и механическую прочность атомных кристаллов.
кристаллах с молекулярной кристаллической решеткой в узлах находятся молекулы, в которых связь между атомами имеет преимущественно ковалентный характер. Между молекулами связь осуществляется за счет слабого межмолекулярного взаимодействия – сил Ван-дер-Вальса, имеющих электростатическую природу. Молекулярной кристаллической решеткой обладают преимущественно органические соединения и конденсированные газы. Следствием слабой связи между
22
частицами в молекулярной кристаллической решетке являются низкие температуры плавления и кипения, хрупкость и низкая твердость.
К особому виду относят взаимодействие между молекулами, содержащими атом водорода, связанный с сильно электроотрицательным атомом (F, O, N, Cl). Такое взаимодействие классифицируют как особый вид связи, называемой водородной. На схемах ее обозначают тремя точками. Энергия связи значительно больше, чем при обычном межмолекулярном взаимодействии (до 40 кДж/моль), но меньше, чем энергия ковалентной связи. Образование такой аномально прочной межмолекулярной связи обусловлено тем, что в полярных молекулах поляризованный атом водорода обладает уникальными свойствами: отсутствием электронных оболочек, значительным сдвигом электронной пары к атому с высокой электроотрицательностью и очень малым размером. Поэтому водород глубоко внедряется в электронную оболочку соседнего отрицательно поляризованного атома. Вследствие этого наряду с электростатическим проявляется и донорно-акцепторное взаимодействие.
Образование водородных связей оказывает сильное влияние на свойства соединений. Например, наличие их в воде и фтористом водороде приводит к аномально высоким температурам кипения этих жидкостей по сравнению со своим аналогами H2S, H2Se, H2Te или HCl HBr HI.
Металлической кристаллической решеткой обладают элементарные металлы в твердом состоянии и некоторые сплавы. В узлах кристаллической решетки находятся катионы и атомы металлов. Связь между ними осуществляется за счет делокализованных электронов, принадлежавших не отдельным атомам и ионам, а кристаллу как целому. Электроны подвижны и обеспечивают специфические свойства металлов: высокую электропроводность, теплопроводность, характерный металлический блеск, пластичность, восстановительную способность. Химическую связь в металлах называют металлической связью.
Вопросы для контроля
1.Что называется химической связью? Какова природа сил, которые обусловливают химическую связь?
2. Какие существуют типы химической связи в веществах?
Литература
23
1. Коровин Н.В. Общая химия: Учебник для тех. направл. и спец. Вузов - 6-е изд., испр. – М.: Высш.шк., 2005. – С. 35-62.
Лекция 4
Химическая термодинамика
Основные вопросы:
Основные понятия термодинамики. Формы передачи энергии.
Первый закон термодинамики. Энтальпия. Закон Гесса.
Второй закон термодинамики. Энтропия. Третий закон термодинамики.
Энергия Гиббса и направление самопроизвольного протекания реакции.
Центральным в химии является учение о превращениях веществ, в том числе об энергетике и кинетике химических реакций.
1. Химическая термодинамика изучает энергетические эффекты химических процессов, переходы химической энергии в другие формы, а также направление и пределы самопроизвольного протекания процессов.
Основные понятия термодинамики (система, фаза, параметры состояния, функции состояния).
Под системой в химической термодинамике понимают отдельное вещество или совокупность веществ, физически или мысленно обособленных от окружающей среды. Например, атом водорода (система из ядра и электронов), водный раствор солей и т.д.. В зависимости от характера взаимодействия системы с окружающей средой различают изолированные, открытые и закрытые системы. Изолированной называется система, которая не обменивается с окружающей средой ни массой, ни энергией. Абсолютно изолированные системы являются лишь полезной абстракцией и реально не существуют. Для неизолированных систем (закрытых, открытых) возможна передача энергии через поверхность раздела.
Совокупность однородных частей системы, обладающих одинаковыми физическими и химическими свойствами, и отдельных от остальных частей системы поверхностью раздела называется фазой. Системы, состоящие из веществ, находящихся в одной фазе (смесь различных газов, ненасыщенный раствор соли в воде и др.), называются гомогенными, а содержащие несколько фаз (вода и плавающие в ней куски льда; керосин и вода; смесь различных кристаллических веществ) – гетерогенными.
24
Величины (температура, давление, объем, концентрация и др.) характеризующие свойства системы, называются параметрами состояния (физическими условиями существования вещества).
Свойства системы, которые зависят от параметров состояния, но не зависят от пути, по которому система пришла в данное состояние, называются функциями состояния.
Мерой и формами передачи энергии является теплота (Q) и работа (А). Они характеризуют процесс, а не состояние системы, поэтому не являются функциями состояния.
Полная энергия системы без учета потенциальной энергии, обусловленной положением системы в пространстве, и кинетической энергии как целого называется внутренней энергией. Она включает в себя все виды энергии взаимодействия между молекулами, атомами и внутриатомными частицами. Внутренняя энергия является функцией состояния, т.е. ее изменение однозначно определяется начальным и конечным состоянием системы и не зависит от пути, по которому протекает процесс: U 2 U1 U,
где U1 и U2 – внутренняя энергия системы в начальном и конечном состояниях. Изменение внутренней энергии системы U считают положительным (ΔU>0), если внутренняя энергия возрастает (U2>U1) и отрицательным (ΔU<0) если она уменьшается (U2<U1). Единицей внутренней энергии, как и энергии вообще, в СИ является джоуль (обозначение Дж). В химической практике, где расчеты идут на молярные количества реагентов и продуктов, более удобна кратная единица - килоджоуль (кДж). Абсолютная величина внутренней энергии неизвестна.
2. Первое начало термодинамики. Энтальпия. Для неизолированной системы, в которой обмен энергией происходит в форме теплоты, справедливо равенство Q=ΔU+A, выражающее закон сохранения энергии и представляющее собой математическую запись первого закона термодинамики, согласно которому теплота Q, подводимая к системе, расходуется на изменение внутренней энергии системы на величину U и на совершение работы А против сил, действующих на систему. В изохорно-изотермическом процессе (температура и объем постоянны) работа равна нулю и уравнение принимает вид QV = U.
Для химических процессов наиболее характерны изобарно-изотермические условия (температура и давление постоянны). За счет изменения объема V от V1 до V2 совершается работа А = p V, тогда
Q p U + p V.
25
Функция U + p V = Н называется энтальпией. Энтальпия зависит от параметров состояния, от природы процесса, фазового состояния и количества вещества. Абсолютную величину энтальпии определить невозможно.
Теплоту химических процессов, протекающих при V, T = const (Qv) или при p, T = const (Qp), называют тепловым эффектом. Его выражают в кДж (Дж) и относят к тем количествам веществ, которые указаны в уравнении реакции их коэффициентами. Уравнение, в котором указан тепловой эффект реакции, называется термохимическим.
Экзотермические реакции – реакции, протекающие с выделением теплоты. Для экзотермической реакции тепловой эффект отрицателен, так как выделение энергии в процессе приводит к передаче теплоты системой в окружающую среду (ΔН < 0).
Эндотермические реакции – реакции, протекающие с поглощением теплоты. Тепловой эффект эндотермической реакции считают положительным, так как процесс идет с поглощением теплоты, т.е. с передачей теплоты от среды к системе (ΔН > 0).
Тепловой эффект реакции в изохорно-изотермических условиях равен изменению функции состояния U, а в изобарно-изотермических – изменению функции состояния Н.
Закон Гесса (1840 г.): тепловой эффект реакции является функцией состояния, т.е. зависит только от природы и физического состояния исходных веществ и продуктов реакции и не зависит от ее пути. Это положение лежит в основе всех термохимических расчетов.
Тепловой эффект процесса образования одного моля соединения из простых веществ, устойчивых при данной температуре, называется теплотой образования. Теплоты образования, отнесенные к стандартным условиям (р = 101,3 кПа, Т = 298 К), называют стандартными, обозначают H 0f ,298 и
выражают в кДж/моль. Их значения приводятся в справочниках. Теплоты образования простых веществ, находящихся в наиболее устойчивом состоянии, приняты равными нулю.
Из закона Гесса вытекают следствия.
Тепловые эффекты прямого и обратного процессов равны по абсолютной величине, но противоположны по знаку.
С термодинамическими уравнениями можно проводить те же арифметические преобразования, что и с алгебраическими.
26
3. Тепловой эффект реакции равен разности между суммой теплот образования продуктов реакции и суммой теплот образования исходных веществ, взятых с учетом стехиометрических коэффициентов в уравнении реакции
Н (реакции) iH 0f ,298(прод.) iH 0f ,298(исх.) ,
где i – соответствующие стехиометрические коэффициенты.
Важной характеристикой многих веществ, в частности топлив, является энтальпия сгорания их в кислороде. Тепловой эффект реакции окисления кислородом элементов, входящих в состав вещества до образования высших окислов называется энтальпией сгорания этого вещества Н сг0 . В технических
расчетах используют удельную теплоту сгорания QТ , которая равна количеству теплоты, выделяющейся при сгорании 1 кг жидкого или твердого вещества и 1 м3 газообразного вещества до образования высших оксидов:
-
QТ
Нсг1000
или QТ
Нсг1000
.
М
22,4
3. Второй закон (второе начало) термодинамики и энтропия.
Многие химические реакции протекают самопроизвольно при комнатных условиях, без воздействия извне, а для осуществления других требуется изменение внешних условий (давление, температура и др.). Основываясь на первом законе термодинамики, нельзя определить возможность самопроизвольного протекания процесса.
Обратимым называют такой процесс, в результате которого система может вернуться в свое первоначальное состояние без каких-либо изменений в ней самой и окружающей среде. Реально обратимость процессов недостижима, однако многие процессы осуществляются в условиях, близких к обратимым.
Необратимым называют такой процесс, в результате которого в самой системе происходят необратимые изменения. Согласно второму закону термодинамики, в природе самопроизвольно протекают только необратимые процессы.
Клаузис ввел понятие некоторой функции состояния S – энтропии, которая является критерием и мерой необратимости процессов. Количество передаваемой теплоты Q можно представить как произведение фактора
27
интенсивности (температуры Т) на фактор емкости – изменение некоторой функции S, энтропии: Q = T S. Тогда изменение энтропии в обратимом изотермическом процессе равно
S = Q/T.
Для протекания необратимого процесса фактор интенсивности должен быть больше в связи с неизбежными потерями
S > Q/T.
Второй закон термодинамики указывает на энтропию как критерий самопроизвольного протекания процессов. Наиболее общая его формулировка: в изолированных системах могут совершаться только процессы с возрастанием энтропии (dS > 0), и процесс может идти самопроизвольно только до такого состояния, при котором энтропия достигает максимума для данных условий (dS = 0; S = Smax).
Состояние любой совокупности частиц можно характеризовать как микросостояние значениями среднестатистических, непосредственно измеряемых величин (температура, давление, объем). Другой характеристикой состояния является микросостояние, которое определяется значениями мгновенных параметров каждой частицы (положение в пространстве, скорость, направление перемещения, энергия и др.). Одно микросостояние может осуществляться огромным числом микросостояний, называемым термодинамической вероятностью W. Энтропия и термодинамическая вероятность связаны уравнением Больцмана (1872 г.):
S = k lnW,
где k – константа Больцмана (k = 1,38·10-23 Дж/К).
Практически, реализуются наиболее вероятные макросостояния, а наиболее вероятному макросостоянию соответствуют максимальная энтропия. Если считать идеальным порядком единственный способ осуществления макросостояния, а хаосом наличие максимального количества таких возможных способов, то энтропию можно считать мерой молекулярного хаоса, беспорядка в системе.
Чем более вероятным и более неупорядоченным является состояние, тем больше значение S. Нагревание, плавление, кипение, расширение, растворение приводят к увеличению энтропии, а охлаждение,
28
кристаллизация, конденсация, осаждение, сжатие – к ее уменьшению. Изменения энтропии при протекании химических реакций особенно велики при изменении числа молекул газов: их увеличение приводит к возрастанию энтропии, уменьшение – к понижению.
Стандартные энтропии S 0298 приводятся в справочниках.
Энтропия – функция состояния, поэтому для расчета S реакции применимо выражение, аналогичное по форме следствию из закона Гесса:
S(реакции) iS(прод.) iS(исх.) .
В отличие от других термодинамических функций, можно определить не только изменение, но и абсолютное значение энтропии. В 1911 г. немецкий физик Планк определил, что значения энтропии вещества являются абсолютными, поскольку статистически при термодинамической температуре, стремящейся к нулю, энтропия всех веществ, становящихся идеальными кристаллами, обращена в нуль. Этот постулат получил название третьего закона термодинамики.
4. Большинство химических процессов протекает в условиях, близких к изобарно-изотермическим. Здесь критерием является изменение функции ∆G. Функция ∆G называется энергией Гиббса, или изобарно-изотермическим потенциалом. Можно доказать, что в необратимом изобарно-изотермическом процессе изменение этой функции
G H TS 0 .
В изобарно-изотермических условиях самопроизвольно могут протекать только процессы с уменьшением энергии Гиббса (∆G < 0), и процесс может идти до такого состояния, при котором энергия Гиббса обладает минимальным для данных условием значений (∆G = 0; G = Gmin).
Энергия Гиббса – критерий самопроизвольного протекания химических реакций.
Энергия Гиббса зависит от характера реакции, а для многих реакций и от температуры (таблица 1).
Табл. 1 Влияние температуры на направление химических реакций
∆H |
∆S |
∆G |
Направление реакции |
Примеры реакций |
|
|
|
|
|

29
∆Н < 0 |
∆S > 0 |
∆G < 0 |
Прямая реакция |
может |
С(графит) 1/2О2 СО |
||
|
|
|
быть самопроизвольной |
|
|||
|
|
|
при любых температурах |
|
|||
∆Н > 0 |
∆S < 0 |
∆G > 0 |
Прямая |
реакция |
не |
|
|
|
|
|
может |
|
|
быть |
СО С(графит) 1/2О2 |
|
|
|
самопроизвольно |
|
при |
||
|
|
|
|
|
|||
|
|
|
любых температурах |
|
|||
∆Н < 0 |
∆S<0 |
∆G < 0 |
Самопроизвольно может |
СаО СО2 СаСО 3 |
|||
|
|
при |
идти прямая реакция при |
|
|||
|
|
Т < Тр |
низких температурах и |
|
|||
|
|
∆G > 0 |
обратная |
реакция |
при |
|
|
|
|
при |
высоких температурах |
|
|||
|
|
Т > Тр |
|
|
|
|
|
∆Н > 0 |
∆S>0 |
∆G > 0 |
Самопроизвольно может |
СН 4 2Н2О СО2 4Н2 |
|||
|
|
при |
протекать |
|
прямая |
|
|
|
|
Т < Тр |
реакция |
при высоких |
|
||
|
|
∆G < 0 |
температурах и обратная |
|
|||
|
|
при |
реакция |
при |
низких |
|
|
|
|
Т > Тр |
температурах |
|
|
|
|
|
|
|
|
|
|
|
|
Тр – температура при которой устанавливается равновесие, т.е. равновероятная возможность протекания прямой и обратной реакций.
Значения энергит Гиббса образования приводятся в справочниках. Для простых веществ в наиболее устойчивых формах G 0f ,298 приняты равными
нулю. Изменение энергии Гиббса системы при образовании 1 моль вещества из простых веществ, устойчивых при 298 К, называется энергией Гиббса образования вещества G 0f ,298 . Если вещество и исходные простые вещества
находятся в стандартных состояниях, то энергия Гиббса образования называется стандартной энергией Гиббса образования вещества. Энергия Гиббса – функция состояния, поэтому для расчета ∆G применимо выражение, аналогичное по форме следствию из закона Гесса
G(реакции) iG0f ,298(прод.) iG0f ,298(исх.) .
Максимальная работа, которую может совершить система при равновесном проведении процесса в изохорно-изотермических условиях,
30
равна изменению энергии Гельмгольца системы ∆F. Энергия Гельмгольца реакции равна
F U TS .
Критерием самопроизвольного протекания химических реакций при изохорно-изотермических условиях является отрицательное значение энергии Гельмгольца: ∆F<0.
Вопросы для контроля
Что такое термодинамическая система? Параметры состояния? Функции состояния?
Какие свойства системы характеризуют термодинамические функции системы?
Как определяется тепловой эффект реакции?
Какая термодинамическая функция указывает на возможность протекания реакции?
Литература
1. Коровин Н.В. Общая химия: Учебник для тех. направл. и спец. Вузов - 6-е
изд., испр. – М.: Высш.шк., 2005. – С. 115-135.
Лекция 5
Химическая кинетика. Химическое равновесие
Основные вопросы:
Понятие и определение скорости химической реакции.
Зависимость скорости реакции от концентрации. Закон Гульдберга-
Вааге.
Зависимость скорости реакции от температуры. Правило Вант-Гоффа.
Механизмы химических реакций. Молекулярность и порядок химической реакции.
Катализ.
Химическое равновесие. Основные условия и характеристики.
Принцип Ле-Шателье.
31
1.Термодинамически возможная реакция в различных условиях может протекать с различными скоростями. Для управления скоростью реакций следует знать закономерности протекания реакции во времени и механизм ее протекания. Этим занимается химическая кинетика. Под средней скоростью реакции в гомогенной системе понимают изменение количества вещества, отнесенное к единице объема, за единицу времени
|
|
|
|
|
c |
, |
|
|
|
v |
|||
|
|
|
|
|||
|
|
|
|
|
τ |
|
|
|
|
|
|||
где v – средняя скорость реакции |
в интервале времени τ ; c – |
|||||
соответствующее изменение концентрации, моль/л, моль/мл. |
||||||
|
|
Скорость гомогенных реакций |
измеряется в моль/(мл с) или в |
|||
м
оль/(л![]() с).
с).
Истинная скорость реакции v определяется пределом, к которому стремится средняя скорость при τ 0 , т.е. является производной концентрации во времени:
v dcdτ .
Гетерогенные реакции протекают на границе раздела фаз, а не во всем объеме, как гомогенные, поэтому в гетерогенных реакциях изменение количества реагирующих веществ ∆n относят не к единице объема системы, как в гомогенных, а к единице поверхности раздела фаз S. В этом случае средняя и истинная скорости реакции определяется выражениями
v nτ S1 , v dndτ S1 .
Скорость гетерогенных реакций измеряется в моль/(мл с).
Если измеряют скорость по исходным веществам, концентрация которых с течением реакции уменьшается ( с с2 с1 0 ), то в уравнениях определения скорости ставят знак минус, так как скорость реакции принято считать величиной положительной; если по продуктам – ставят знак плюс
( с с2 с1 0 ).
32
Скорость реакции зависит от природы реагирующих веществ и условий ее протекания: концентрации, температуры, присутствия и природы катализатора и некоторых других факторов.
Необходимым условием для осуществления химического элементарного акта является соударение молекул реагирующих веществ. Соударения молекул происходит тем чаще, чем больше их концентрация.
2. Зависимость скорости реакции от концентрации выражается основным законом химической кинетики – законом действия масс: при постоянной температуре скорость протекающих в одну стадию элементарных реакций прямо пропорциональна произведению концентраций реагирующих веществ, взятых в степенях, показателями которых являются стехиометрические коэффициенты в уравнении реакции.
В общем виде для гомогенной реакции
mA nB pD qE
закон действия масс выражается соотношением, которое называется кинетическим уравнением
v k c mA cBn ,
где k – константа скорости реакции; сА, сВ – концентрации веществ А и В; m, n – стехиометрические коэффициенты.
Например, для реакции
2NO + O2 2NO2
кинетическое уравнение имеет вид:
v k сNO2 сO2 ,
где k – константа скорости; сNO и сО2 – молярные концентрации реагирующих веществ, моль/л.
При сА =сВ = 1 моль/л следует: v = k, т.е. константа скорости равна скорости при единичных концентрациях реагирующих веществ. Таким образом, константа скорости зависит от температуры, природы реагирующих веществ и катализатора, но не зависит от концентрации.
33
В гетерогенных реакциях число частиц кристаллического вещества на единице поверхности постоянно, поэтому концентрация кристаллического вещества также постоянна. Она учитывается в константе скорости и не входит в выражение закона действия масс, например, для реакции
СаО(т) SO3(г) СаSO4(т)
скорость пропорциональна только концентрации газа:
v k cSO3 .
Необходимым условием химического акта является столкновение реагирующих частиц в одной точке. Вероятность столкновения трех частиц невелика, а более трех – пренебрежительно мала. Поэтому большинство реакций с суммой стехиометрических коэффициентов исходных веществ … m + n = 3 и все реакции, в которых m + n > 3, являются неэлементарными, т.е. протекают по стадиям. Поэтому в более общем виде закон действия масс выражается соотношением
v k c mA cBn
Сумма m n называется порядком реакции, а величины m и n называются порядком реакции, соответственно, по веществам А и В. Для элементарных реакций m m и n n , а для неэлементарных m m и n n . Порядок реакций определяют экспериментально и по результатам, судя о механизме их протекания.
Например, разложение окисла азота (V) выражается уравнением
2N2O5 4NO2 О2
Если бы реакция была элементарной, ей соответствовал бы второй порядок:
v k cN22O5 .
Однако экспериментально установлено, что это реакция первого порядка, поэтому она описывается кинетическим уравнением:
34
v k cN2O5
Это объясняется тем, что истинный механизм этой реакции включает две последовательные стадии:
N2O5 N 2O3 O2 ,
N2O5 N2O3 4NO2,
из которых первая стадия является наиболее медленной (лимитирующей). Лимитирующая стадия определяет суммарную скорость и порядок реакции.
Гидролиз этилацетата в водном растворе в присутствии сильных кислот
СН3СООС2Н5 Н2О СН3СООН С2Н5ОН
также относится к реакциям первого порядка, так как концентрация воды во много раз больше концентрации этилацетата и в процессе практически не изменяется: v k cCH3COOC2H5 .
Реакции с порядком выше трех неизвестны. В реакциях со сложным механизмом возможен и дробный порядок.
Порядок реакции характеризует особенности убывания концентрации исходных веществ во времени.
Для реакций первого порядка, например гомогенных реакций разложения типа А → В + D, выражение скорости реакции имеет вид:
v dcdτ k c .
После преобразования получаем
ln cc0 k τ ,
где с0 и с – исходная и конечная концентрация А в момент времени τ, моль/л; τ – время от начала процесса, с.
35
Следовательно, в реакциях первого порядка концентрация убывает логарифмически. Полученное выражение позволяет найти концентрацию (с)
в любой момент времени. Соотношение |
с0 |
не зависит от вида используемой |
с |
||
|
|
концентрации и может задаваться, например в %. Так если прореагировало
25 % исходного вещества, то это соотношение принимает значение ……...
100 % /75 % =1,33.
Для реакций второго порядка типа 2 А→… или А + В→… (при равных исходных концентрациях А и В) выражение скорости имеет вид
v dcdτ k c2 .
После преобразования получаем:
-
1
1
k τ
с
с0
Такая зависимость линейна в координатах 1/с – τ.
3. Влияние температуры на скорость реакции. Зависимость скорости реакции от температуры. Константа скорости реакции и, следовательно, скорость реакции интенсивно возрастают с увеличением температуры. Приближенная зависимость скорости реакции от температуры выражается правилом Вант-Гоффа: для большинства реакций повышение температуры на каждые 10 градусов приводит к увеличению скорости реакции в 2 – 4 раза
T2 T1
v2 k2 γ 10 ,
v1 k1
где v1 и v2 – скорости реакции (k1 и k2 – константы скорости реакции) при температурах) Т1 и Т2 соответственно; γ – температурный коэффициент Вант-Гоффа (γ=2 – 4).
Температурный коэффициент скорости химической реакции – это число, показывающее, во сколько раз возрастает скорость данной реакции при повышении температуры на 10 градусов. Уравнение Вант-Гоффа можно использовать лишь для приближенных расчетов, не имеющих высокой точности.
36
Согласно теории активных соударений Аррениуса, не всякое соударение молекул реагирующих веществ приводит к химическому акту. Для того чтобы произошла реакция, т.е. чтобы образовались новые молекулы, необходимо сначала разорвать или ослабить связи между атомами в молекулах исходных веществ, на что надо затратить определенную энергию. Если кинетическая энергия сталкивающихся молекул достаточна для этого, то столкновение может привести к химическому акту. Избыточная энергия (по сравнению со средней энергией при данной температуре), которой должны обладать молекулы для того, чтобы их столкновение привело к химическому взаимодействию, называется энергией активации данной реакции Еа (энергия, необходимая для перехода вещества в состояние активированного комплекса). Молекулы, обладающие такой энергией, называются активными молекулами. Активных молекул немного, но с увеличением температуры их доля резко возрастает, что приводит к резкому увеличению скорости реакции. Энергия активации зависит от природы реагирующих веществ. Чем она меньше, тем больше доля активных молекул и выше скорость реакции.
Зависимость константы скорости реакции от температуры выражается уравнением Аррениуса (1889 г.)
-
k Ae
Ea
Ea
RT
или
ln k ln A
,
RT
где А – константа для данной реакции (предэкспоненциальный множитель); R
– универсальная газовая постоянная, равная 8,341 Дж/(мольК); Еа – энергия активации, Дж/моль. Предэкспоненциальный множитель отражает частоту столкновения и ориентацию реагирующих частиц. Принципиально возможная реакция протекает при соблюдении двух условий: достаточной энергии и надлежащей ориентации частиц.
Константа скорости возрастает с увеличением температуры по экспоненциальному закону. В соответствии с уравнением Аррениуса, константа скорости реакции уменьшается с ростом энергии активации. Из уравнения Аррениуса также следует, что чем выше энергия активации, тем выше градиент скорости реакции по температуре. Уравнение Аррениуса позволяет рассчитывать константы скорости реакции при различных температурах.
4. Механизмы химических реакций. Выявление механизма химических реакций является важнейшей фундаментальной задачей химии. Все реакции можно подразделить на простые и сложные. Простые реакции протекают в
37
одну стадию и называются одностадийными. Сложные реакции идут либо последовательно (многостадийные), либо параллельно, либо последовательно-параллельно. В каждой стадии реакции может участвовать одна молекула (мономолекулярные реакции), две молекулы (бимолекулярные реакции) и три молекулы (тримолекулярны). Число молекул реагента, принимающих участие в простейшей (элементарной) стадии, называется ее молекулярностью. Реакции с молекулярностью больше трех неизвестны.
В последние годы уделяется большое внимание периодическим процессам, характеризующимся колебаниями концентраций некоторых промежуточных соединений и соответственно скоростей превращения (колебательным реакциям).
Практически все химические реакции идут через промежуточный активированный комплекс, образуемый исходными частицами, энергия которых не ниже некоторого значения, называемого энергией активации.
5. Катализ – изменение скорости реакции под воздействием веществ катализаторов. Явление катализа распространено в природе и широко применяется в технологии. Каталитическому воздействию подвержено большинство химических реакций и число веществ, каталитически активных по отношению к химическим реакциям, весьма велико.
Катализаторы – это вещества, не расходующиеся в процессе реакции, но влияющие на скорость.
Катализаторы ускоряют реакции, не изменяя термодинамику процесса. Катализаторы могут повышать выход тех или иных продуктов реакции. На активность катализатора влияют промоторы и каталитические яды.
Действие катализатора для большинства реакций сводится к снижению энергии активации. Любая реакция проходит через ряд промежуточных стадий с образованием активированных комплексов. В присутствии катализаторов изменяются промежуточные стадии, образуются более энергетически доступные активированные комплексы, т.е. комплексы, для образования которых требуется меньше энергии.
Катализаторы, которые находятся в системе в том же фазовом состоянии, что и реагенты, называются гомогенными. Гомогенные каталитические реакции протекают через образование промежуточных соединений, в которых участвует катализатор.
Если катализаторы и реагенты находятся в разных фазах, то катализ называется гетерогенным. Гетерогенные каталитические реакции начинаются со стадии адсорбции, в результате которой ослабляются или разрываются химические связи в молекулах реагентов. Для ускорения реакции
38
применяются катализаторы с развитой поверхностью. В настоящее время широко применяются различные катализаторы.
6. Химическое равновесие. Все химические реакции можно разделить на обратимые и необратимые. Обратимыми являются реакции, которые с заметной скоростью протекают в прямом и обратном направлениях. Они идут не до конца, ни одно из реагирующих веществ не расходуется полностью.
Химическим равновесием называют такое состояние реагирующей системы, при котом скорость прямой реакции равна скорости обратной реакции. Равенство скоростей прямой и обратной реакции является кинетическим условием химического равновесия. Основной характеристикой состояния химического равновесия является равенство скоростей прямой и обратной реакций, которые в состоянии равновесия непрерывно и одновременно протекают в обоих направлениях. Поэтому это состояние системы называется подвижным, или динамическим, равновесием. Это значит также, что система, находящаяся в равновесии, отзывается на всякое изменение условий тем, что равновесные концентрации изменяются, причем каждый раз устанавливается новое равновесие, соответствующее новым условиям существования системы.
Химическое равновесие характеризуется постоянством величины энергии Гиббса системы G. Равенство G=0 является термодинамическим условием химического равновесия.
Концентрации участников реакции в состоянии равновесия называются равновесными.
Положения обратимой реакции равновесия описывается с помощью закона химического равновесия, называемого, как и основной закон химической кинетики, законом действия масс (или действующих масс). По этому закону взаимосвязь концентраций веществ – участников реакции в равновесной смеси – определяется константой равновесия данной химической реакции КС – величиной, постоянной при неизменной температуре. Так для реакции, в общем виде выраженной уравнением:
mA+nB⇄pC+qD,
где m, n, p, q – стехиометрические коэффициенты в уравнении, константа равновесия данной обратимой реакции имеет вид:
39
-
Кс
[C] p [D]q
,
[ A]m [B]n
где [A], [B], [C], [D] – равновесные концентрации участвующих в реакции веществ (моль/л), m, n, p, q – показатели степеней, которые равны соответствующим стехиометрическим коэффициентам в уравнении реакции. Это уравнение является математическим выражением закона действующих масс Гульдберга-Вааге (1867): отношение произведения равновесных концентраций продуктов реакции в степенях, равных стехиометрическим коэффициентам, к произведению равновесных концентраций исходных веществ в степенях, равных стехиометрическим коэффициентам, при …… Т = const, является величиной постоянной. Константа равновесия не зависит от исходного состава реагирующей смеси, это одна из важнейших характеристик химической реакции. Величины констант равновесия приводятся в справочниках. Чем больше величина константы равновесия, тем полнее протекает прямая реакция, тем больше выход конечных продуктов.
Константа равновесия является функцией температуры. При изменении температуры она изменяется обычно значительно меньше, чем константа скорости реакции.
Рассмотрим несколько примеров записи выражений для константы равновесия:
-
1. СO+H2O⇄СO2+H2
К
С
[CO 2 ] [H 2 ]
.
[CO] [H2 O]
2. N2+3H2⇄2NH3
К С
[NH 3 ]2
.
[N
2
] [H
2
]3
Для реакций с участием газов помимо констант равновесия КС, выраженных через молярные концентрации, часто используются константы равновесия Кр, выраженные через парциальные давления газов:
-
2
H2 + I2 ⇄ 2HIК
р
рHI
,
pH
pI
2
2
где pHI , pH2 , pI2 – парциальные давления газов в равновесной смеси.
40
Если в реакции участвует одно или несколько твердых веществ и один или несколько газов, то выражение для константы равновесия содержит парциальные давления газообразных веществ при равновесии. Например, для реакции:
-
р 4
3 Fe(т) + 4 H2O(г) ⇄ Fe3O4(т) + 4H2(г)
К
р
H
2
.
p
4
H 2 O
этом случае система гетерогенная. Концентрация твердого вещества
паровой фазе определяется давлением насыщенного пара, которое имеет для каждого вещества постоянную величину при заданной температуре.
Концентрация Fe и Fe3O4 как величины постоянные включены в величину константы равновесия.
Константа равновесия связана со стандартной энергией Гиббса данной
реакции (G 0 ) следующим соотношением: |
|
|
|
Т |
|
|
|
G 0 |
= RТ ln K |
c |
, |
Т |
|
|
|
где R – универсальная газовая постоянная [8,31 Дж/(мольK)]; Т – абсолютная температура, K; КС – константа равновесия.
Химическое равновесие остается неизменным до тех пор, пока остаются постоянными параметры, при которых оно установилось. При изменении условий равновесие нарушается. Через некоторое время в системе вновь наступает равновесие, характеризующееся новым равенством скоростей и новыми равновесными концентрациями всех веществ.
Равновесие смещается в ту или иную сторону потому, что изменение условий по разному влияет на скорости прямой и обратной реакции. Равновесие смещается в сторону той реакции, скорость которой при нарушении равновесия становится больше. Например, если при изменении внешних условий равновесие нарушается так, что скорость прямой реакции становится больше скорости обратной реакции, то равновесие смещается вправо.
8 Смещение химического равновесия. Качественные задачи смещения химического равновесия могут быть решены с помощью принципа Ле-Шателье: если на систему, находящуюся в состоянии равновесия, оказать какое-либо внешнее воздействие, то в результате протекания процессов в
41
системе равновесие сместится в направлении приводящем к уменьшению оказанного воздействия.
Проиллюстрируем этот принцип некоторыми примерами.
Влияние концентраций (парциальных давлений) компонентов системы.
В равновесную систему N2 + 3 H2 ⇄ 2 NH3 ввели дополнительное количество водорода. Такое воздействие приведет к ускорению реакции взаимодействия водорода с азотом, в процессе которой концентрации водорода и азота будут уменьшаться, а концентрация аммиака увеличиваться. Увеличение концентрации аммиака, в свою очередь, приведет к ускорению реакции его разложения. В конце концов, устанавливается новое состояние равновесия, в котором концентрация аммиака больше, чем в первоначальном. Происходит сдвиг равновесия в сторону прямой реакции, т.е. увеличение выхода аммиака. Если в систему ввести дополнительное количество аммиака, то в соответствии с принципом Ле-Шателье, равновесие сместится влево, т.е. в сторону исходных веществ.
Если уменьшить концентрацию какого-либо из веществ, то результат будет обратным. Обе реакции начнут замедляться до тех пор, пока не будет достигнуто новое состояние равновесия.
Влияние общего давления в системе. Прежде всего, следует отметить,
что изменение давления может влиять только на равновесие реакций с участием газообразных веществ. Рассмотрим это влияние на примере
обратимого процесса синтеза аммиака из азота и водорода N2 + 3 H2 ⇄ 2 NH3. Как видно, в левой части уравнения – 4 молекулы, а в правой – 2. В соответствии с принципом Ле Шателье, при увеличении давления равновесие обратимых газовых реакций смещается в сторону образования меньшего числа молекул, при уменьшении давления – в сторону большего числа молекул. То есть при увеличении внешнего давления на рассматриваемую систему равновесие сместится в сторону образования аммиака, при уменьшении – в сторону его распада.
Влияние температуры. Согласно принципу Ле-Шателье, повышение температуры благоприятствует образованию того соединения, при получении которого поглощается тепло, т.е. ведет к сдвигу равновесия в сторону эндотермического процесса, понижение – в сторону экзотермического.
Влияние катализатора. Экспериментально установлено, что катализаторы не смещают равновесие и не меняют величины константы равновесия. Ускоряя прямую реакцию, они во столько же раз ускоряют и обратную. Катализаторы уменьшают время, необходимое для достижения равновесия, но не влияют на равновесные концентрации.
42
Следует отметить, что изучение влияния различных факторов на равновесные системы позволяет сознательно подходить к выбору оптимальных условий ведения того или иного процесса.
Знание константы равновесия позволяет вычислить равновесный выход продукта реакции, предсказать направление самопроизвольного протекания обратимой реакции при заданном исходном составе смеси и т.д.
Вопросы контроля
1.Что такое скорость химической реакции? От каких факторов она зависит?
В чем сущность закона действия масс? Сформулируйте правило Вант-Гоффа.
Что такое химическое равновесие?
Каким образом можно сместить химическое равновесие?
Литература
1. Коровин Н.В. Общая химия: Учебник для тех. направл. и спец. Вузов - 6-е изд., испр. – М.: Высш.шк., 2005. – С. 148-201.
Лекция 6
Растворы и другие дисперсные системы
Основные вопросы:
Понятие раствора. Способы выражения концентрации растворов.
Растворы неэлектролитов.
Растворы электролитов.
Водородный показатель среды.
Гидролиз солей.
Дисперсные системы.
Растворами называют гомогенные системы переменного состава, состоящие из двух или более независимых компонентов – растворителя и одного или нескольких растворенных веществ. Формой выражения количественного состава раствора является его концентрация. Способы выражения концентрации раствора:
43
Массовая доля ω(Х) – отношение массы данного компонента т(Х) к массе раствора т (сумме масс всех компонентов):
-
ω( Х )
т( Х )
т( Х )
т( Х )
,
т
т( Х ) т(Y ) ...т
ρ V
0
где т0 – масса растворителя, г; ρ – плотность раствора, г/мл; V – объем раствора, мл. Массовую долю часто выражают в процентах: ω(Х) % = ω(Х) 100 %. Сумма массовых долей всех компонентов равна 1 (100 %).
Молярная концентрация (молярность) сМ – количество молей растворенного вещества n в одном литре раствора (моль/л):
сМ Vn MmV ,
где m, M – масса и молярная масса растворенного вещества, г и г/моль соответственно; V – объем раствора, л.
Размерность молярности часто обозначают М, например 0,1 моль/л, или 0,1 М.
Молярная концентрация эквивалента сн (нормальность N) – количество молей эквивалентов растворенного вещества в одном литре раствора (моль/л):
-
сн
nэ
m
,
V
M Э V
где m, MЭ – масса и молярная масса эквивалентов растворенного вещества, г и г/моль соответственно; V – объем раствора, л.
Размерность нормальности часто обозначают “н.”, например: 0,2 моль/л, или 0,2 н.
Моляльная концентрация (моляльность) сm – число молей растворенного вещества n в одном килограмме растворителя (моль/кг):
-
ст
n
m / М
т 1000
,
т0
/1000
т0 /1000
М т0
где m, M – масса и молярная масса растворенного вещества, г и г/моль соответственно; т0 – масса растворителя, г.
44
Титр показывает, сколько граммов растворенного вещества содержится
1 мл раствора – Т, г/мл.
Разбавленные растворы неэлектролитов обладают рядом свойств, определяемых только количеством частиц (концентрацией) растворенного вещества, независимо от их природы. Такие свойства получили название коллигативных. Они могут проявляться в полной мере в идеальных растворах. Идеальным называют раствор, в котором не происходят химические реакции между компонентами, а силы межмолекулярного взаимодействия между компонентами одинаковы.
Закон Рауля
1. Понижение давления насыщенного пара растворителя А над раствором р А пропорционально молярной доле растворенного нелетучего вещества хВ :
0А р А р А р0А хВ ,
0А , р А – давления насыщенного пара растворителя соответственно над чистым растворителем и над раствором; р А – разность между давлением
насыщенного пара растворителя над раствором р А и растворителем р 0А , хВ
– молярная доля растворенного вещества.
Следствие из закона Рауля: температура замерзания (кристаллизации) раствора ниже температуры замерзания чистого растворителя. Понижение температуры замерзания пропорционально моляльности раствора:
Тзам Кк ст ,
где Кк – криоскопическая постоянная.
Второе следствие: температура кипения раствора выше температуры кипения растворителя. Повышение температуры кипения пропорционально моляльности раствора:
Ткип Кэ ст ,
где Кэ – эбулиоскопическая постоянная растворителя.
45
3. Электролиты. Электролитами называют вещества, растворы и расплавы которых проводят электрический ток, что вызвано диссоциацией их молекулы на ионы, несущие положительный (катионы) и отрицательный (анионы) заряды.
Автопротолиз. Константа автопротолиза. Самый распространенный неорганический растворитель – вода. Молекулы воды взаимодействуют и
образуют ионы: H2O + H2O ⇄ H3O+ + OН .Это же равновесие в сокращенном виде:
H2O ⇄ H+ + OH .
Реакции такого типа называют реакциями автопротолиза. Константу диссоциации воды записывают уравнением:
КН О Н ОН .
2![]() Н2О
Н2О
Концентрацию недиссоциированных молекул воды можно принять за постоянную величину при 25 ºС [H2O]= 100018 55,6 моль/ дм3. Тогда:
К Н2О ·[H2O] = Кw= [H+]· [ OH ].
Константу Кw называют ионным произведением воды. При 25 ºС она
равна 1,01·10 14. Ионное произведение воды широко используют при аналитических расчетах, например, для характеристики среды.
4. Водородный показатель среды. В большинстве расчетов, относящихся к кислотно-основному равновесию, концентрации (и другие величины) удобно выражать в виде отрицательных десятичных логарифмов концентрации водородных ионов:
pН= –lg[H+], pOH= –lg[ OH ],
где [H+], [ OH ] - равновесные молярные концентрации водородных и гидроксид ионов, моль/ дм3.
В термодинамических расчетах используется величина рН выраженная, через активную концентрацию ионов водорода: pН= –lg aH .
46
Ионное произведение воды можно выразить в логарифмическом виде:
pH + pOH = рК H2O = 14.
Чем выше концентрация ионов водорода, тем меньше рН. В кислой среде [H+] > 10 7моль/дм3 и рН < 7; в щелочной среде [H+] < 10 7моль/л и рН > 7.
Нейтральный раствор характеризуется [H+] = [ OH ] = 10 7моль/л и рН = рОН = 7.
Растворы сильных кислот и оснований. Сильные кислоты (НA) и сильные основания (МОН) в водных растворах практически полностью диссоциированы:
НA ⇄ Н+ + A ,
МОН ⇄ М+ + OH ,
поэтому концентрации ионов Н+ и OH в этих растворах правильнее выражать через активность ионов: а = f·c. Коэффициент активности (f) зависит (таблица 2) от ионной силы раствора (I), которую рассчитывают по формуле для электролита (1-1):
I 12 ci Zi2 ,
где ci – концентрации ионов катионов и анионов (моль/дм3), Zi – заряды ионов.
Табл. 2 Формулы для вычисления коэффициентов активности f при разной ионной силе раствора
|
|
I≤0,01 |
|
|
I≤0,5 |
|
|
|
|
|
|
|
|
|
0,5 Z2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
I |
|
|
|||
lgf |
i |
I |
lg fi 0,5 Z 2 |
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 I |
|
|
|||||
|
|
|
|
|
|
|
|
|||||
![]()
![]()
Для сильных электролитов только в очень разбавленных растворах (~0,0001 М) f = 1 и а = c. Коэффициент активности можно не учитывать также для недиссоциированных молекул.
47
Растворы слабых кислот и оснований. Для слабых кислот константа равновесия реакции диссоциации HА ⇄ H+ + А имеет вид:
-
K
[H
] [A-
]
K a ,
HA
где Ка – константа диссоциации кислоты HА (константа кислотности). Слабое основание, как и слабая кислота, в водных растворах
диссоциирует не полностью M(OH) ⇄ M+ + OH ,
M [OH ]
K MOH Kв ,
где Кв – константа диссоциации слабого основания (константа основности). Диссоциация слабых электролитов количественно характеризуется
степенью электролитической диссоциации (α). Степень диссоциации представляет собой отношение концентрации вещества, распавшихся на ионы, к общей концентрации в растворе. Между константой диссоциации электролита Кд и α существует следующая зависимость (закон разбавления Оствальда):
-
K д c
α 2
,
1 - α
где c – молярная концентрация слабого электролита. Если α мала (α<0,05), применительно приближенное уравнение Кд=c·α2, откуда
-
α
K д
.
c
![]()
Растворы многопротонных кислот и оснований. Многоосновные кислоты в водных растворах диссоциируют ступенчато:
-
H S ⇄ HS +H+
(I ступень)
2
HS ⇄ Н+ + S2
(II ступень)
H S ⇄ 2H+ + S2
(суммарный процесс)
2
48
Константы диссоциации по первой ступени всегда больше, чем по второй ступени. Если константа диссоциации по первой ступени во много раз (более чем в 103 раз) больше константы диссоциации по второй ступени, то концентрация ионов водорода определяется главным образом диссоциацией кислоты по первой ступени. Общая константа диссоциации равна произведению ступенчатых констант диссоциации.
5. Гидролиз это реакция обменного разложения растворенного вещества водой, результатом которой является образование малодиссоциированных молекул или ионов и как следствие – изменение рН
растворов таких солей накопление в них Н+ или ОН -ионов. Количественными характеристиками служат константа гидролиза Кг и степень гидролиза г.
Константа гидролиза – это константа равновесия гидролитической реакции, она выводится на основе закона действующих масс при условии, что концентрация воды постоянна.
Степень гидролиза выражает отношение гидролизованной части соли Х к общей ее концентрации: г=Х/собщ.
Примеры гидролиза солей в таблице 3.
Табл. 3 Примеры гидролиза солей
Формула, тип |
Уравнения реакций гидролиза |
рН |
|||||
гидролизующейся |
|
|
|
|
|
|
|
соли |
|
|
|
|
|
|
|
1. CH3COONa |
CH3COONa HOH CH3COOH NaOH |
pН>7 |
|||||
соль сильного осно- |
CH3COO HOH CH3COOH OH |
|
|||||
вания и слабой одно- |
|
|
|
|
|
|
|
основной кислоты |
|
|
|
|
|
|
|
2. Na2CO3 |
I ступень гидролиза: |
|
|
|
pН>7 |
||
соль сильного основа- |
Na2CO3 HOH NaHCO 3 NaOH |
||||||
|
|||||||
ния и слабой двухос- |
2- |
|
- |
|
|
|
|
новной кислоты |
CО3 |
HOH НСО |
3 OH |
|
|
|
|
|
II ступенью гидролиза пренебрегают |
|
|||||
3. NH4Cl |
NH4Cl HOH NH4OH HCl |
pН<7 |
|||||
соль слабого однокис- |
|
|
|
H |
|
|
|
лотного основания и |
NH 4 HOH NH4OH |
|
|
||||
|
|
|
|
|
|
|
|
сильной кислоты |
|
|
|
|
|
|
|
4. FeCl2 |
I ступень гидролиза: |
|
|
|
|
рН<7 |
|
|
|
|
|
|
|||
|
49 |
|
|
|
|
|
|
соль |
слабого |
FeCl2+HOH⇄ FeOHCl+HCl |
|
основания и |
сильной |
Fe2 HOH FeOH H |
|
кислоты |
|
|
|
|
II ступень |
|
|
|
|
|
|
|
|
FeOHCl + HOH ⇄ Fe(OH)2 + HCl |
|
|
|
FeOH+ + HOH ⇄Fe(OH)2 +HCl |
|
5. NH4CN |
|
NH4CN HOH NH4OH HCN |
рН 7 |
соль слабого |
основа- |
NH4 CN HOH NH4OH HCN |
|
ния и слабой кислоты |
|
рН 7 |
|
6. Al2S3 |
|
Al2S3 6HOH 2Al(OH)3 3H2S |
|
|
|
||
соль слабого |
основа- |
2Al3++6HOH Al(OH)3 +3H S |
|
ния и слабой кислоты |
2 |
|
|
|
|
||
7. NaCl, BaCl2, |
|
Гидролиз не происходит |
рН=7 |
Na2SO4 соли сильного |
|
|
|
основания и сильной |
|
|
|
кислоты |
|
|
|
6. Дисперсные системы. Дисперсные (раздробленные) системы являются гетерогенными. Они состоят из сплошной непрерывной фазы – дисперсионной среды и находящихся в этой среде раздробленных частиц того или иного размера и формы – дисперсионной фазы.
Если дисперсная (прерывная) фаза находится в виде отдельных небольших частиц, то дисперсные системы, в отличие от гетерогенных со сплошными фазами, называют микрогетерогенными, а коллоидно-дисперсные системы называют также ультрамикрогетерогенными.
Обязательным условием получения дисперсных систем является взаимная нерастворимость диспергируемого вещества и дисперсионной среды. Например, нельзя получить коллоидные растворы сахара или хлорида натрия в воде, но они могут быть получены в керосине или бензине, в которых эти вещества практически нерастворимы.
Дисперсные системы классифицируют по дисперсности, агрегатному состоянию дисперсной фазы и дисперсной среды, интенсивности взаимодействия между ними, отсутствию или образованию структур в дисперсных системах.
Количественной характеристикой дисперсности (раздробленности) вещества является степень дисперсности (степень раздробленности, D) – величина, обратная размеру (l) дисперсных частиц:
50
D 1l .
Здесь l равно либо диаметру сферических или волокнистых частиц, либо длине ребра кубических частиц, либо толщине пленок.
Степень дисперсности численно равно числу частиц, которые можно плотно уложить в ряд на протяжении одного сантиметра.
Если все частицы дисперсной фазы имеют одинаковые размеры, то такие системы называют монодисперсными. Частицы дисперсной фазы неодинакового размера образуют полидисперсные системы. С повышением дисперсности вещества все большее значение имеют его свойства, определяемые поверхностными явлениями, т.е. совокупностью процессов, происходящих в межфазовой поверхности. Своеобразие дисперсных систем определяется большой удельной поверхностью дисперсной фазы Sуд и физико-химическим взаимодействием дисперсной фазы и дисперсионной среды на границе раздела фаз. Удельная поверхность Sуд характеризует соотношение между поверхностью и объемом Sуд = S/V, которая для частиц сферической формы равна
S уд |
|
|
4π r 2 |
|
|
3 |
|
|
6 |
, |
|||
|
4 |
π r3 |
|
r |
d |
||||||||
|
|
|
3 |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
а для частиц кубической формы |
|
|
|
|
|
|
|
|
|
|
|
|
|
S уд |
|
6 l 2 |
|
|
6 |
, |
|
|
|||||
l 3 |
|
|
l |
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|||
где r – радиус шара; d – его диаметр, l – длина ребра куба.
Классификация корпускулярнодисперсных систем по степени дисперсности рассмотрена в таблице 4.
Табл. 4.Классификация корпускулярнодисперсных систем по степени дисперсности
Системы |
Раздробленн |
Радиус |
Степень |
Число |
|
ость |
частиц, см-1 |
дисперсност |
атомов в |
|
вещества |
|
и D, см-1 |
одной |
|
|
|
|
частице |
|
|
51 |
|
|
Грубодис- |
Макроскопи- |
1-10-2 |
1-102 |
>1018 |
персные |
ческая |
|
|
|
|
Микроскоп- |
10-2-10-5 |
102-105 |
>109 |
|
ическая |
|
|
|
Предельно- |
Коллоидная |
10-5-10-7 |
105-107 |
109-103 |
высокодис- |
|
|
|
|
персные |
|
|
|
|
Молекуляр- |
Молекуляр- |
10-7-10-8 |
>107 |
<103 |
ные и |
ная и ионная |
|
|
|
ионные |
|
|
|
|
Дисперсные системы также классифицируют по агрегатным состояниям дисперсной фазы и дисперсионной среды таблице 5.
Табл. 5. Классификация дисперсных систем по агрегатным состояниям дисперсной фазы и дисперсионной среды
Тип дисперсной |
Фазовое состояние |
Примеры |
|
системы |
Дисперсионной |
Дисперсионной |
|
|
среды |
фазы |
|
Аэрозоль |
газ |
жидкая |
туман, облака |
Аэрозоль |
газ |
твердая |
дым, пыль |
Пена |
жидкая |
газ |
взбитые сливки, |
|
|
|
мыльная пена |
Эмульсия |
жидкая |
жидкая |
молоко, майонез |
Золь |
жидкая |
твердая |
краски |
Твердая эмульсия |
твердая |
жидкая |
масло |
Гель |
твердая |
жидкая |
желе, агар-агар |
Дисперсионные системы с газообразной дисперсионной средой называют аэрозолями.
Пены – это дисперсия газа в жидкости.
Эмульсиями называют дисперсные системы, в которых одна жидкость раздроблена в другой, не растворяющей ее жидкости.
Низкодисперсные системы твердых частиц в жидкостях называют суспензиями, или взвесями, предельно-высокодисперсные – коллоидными растворами, или золями, часто лизолями, чтобы подчеркнуть, что дисперсионной средой является жидкость. Если дисперсионной средой
52
является вода, то такие золи называют гидрозолями, а если органическая жидкость – органозолями.
Дисперсные системы могут быть свободнодисперсными и связано дисперсными в зависимости от отсутствия или наличия взаимодействия между частицами дисперсной фазы. К свободнодисперсным системам относят аэрозоли, лизоли, разбавленные суспензии и эмульсии. Они текучи. В этих системах частицы дисперсной фазы не имеют контактов, участвуют в беспорядочном тепловом движении, свободно перемещаются под действием силы тяжести. Связанодисперсные системы – твердообразны; они возникают при контакте частиц дисперсионной фазы, приводящем к образованию структуры в виде каркаса или сетки. Такая структура ограничивает текучесть дисперсной системы и придает ей способность сохранять форму. Подобные структурированные коллоидные системы называют гелями. Переход золя в гель, происходящий в результате понижения устойчивости золя, называют гелеобразованием (желатинированием). Порошки, концентрированные эмульсии и суспензии – это примеры связанодисперсных систем.
Сплошную массу вещества могут пронизывать поры и капилляры, образующие капиллярно-дисперсные системы. К ним относятся древесина, разнообразные мембраны и диафрагмы, кожа, бумага, картон, ткани.
Высокодисперсное состояние вещества – качественно особая форма его существования. Поэтому область естествознания, изучающая объективные физические и химические закономерности поверхностных явлений и гетерогенных высокодисперсных систем, сформировалась в самостоятельную научную дисциплину – коллоидную химию. Коллоидное состояние характерно для многих веществ, если их частицы имеют размер от 1 до 500 нм. Характерной особенностью коллоидных частиц является наличие на их поверхности заряда, обусловленного избирательной адсорбцией ионов. Коллоидная частица имеет сложное строение. Она включает в себя ядро, адсорбированные ионы, противоионы и растворитель.
Существуют лиофильные (гидрофильные) коллоиды, в которых растворитель взаимодействует с ядрами частиц, и лиофобные (гидрофобные) коллоиды, в которых растворитель не взаимодействует с ядром частиц.
Коллоидные растворы в природе и технике. В природной воде содержится часть примесей в коллоидном состоянии. Поэтому воду, используемую для коммунальных нужд, электростанций, строительства, подвергают обработке, вызывающей коагуляцию коллоидных частиц. Дымовые газы электростанций, металлургических заводов и других предприятий представляют собой аэрозоли. Для их коагуляции применяется электрогазоочистка методом электрофореза при очень высоких напряжениях
53
поля. Можно разделить коллоидные частицы и ионы через мембрану, проницаемую для молекул и ионов и непроницаемую для коллоидных частиц. Такой метод разделения называется диализом. Он, например, лежит в основе аппарата «искусственная почка».
Вопросы для контроля
Какие способы выражения концентрации раствора существуют?
Чем отличаются растворы электролитов от неэлектролитов?
Что такое водородный показатель среды? Какая среда считается кислой, нейтральной, щелочной?
В чем сущность процесса гидролиза?
Какие дисперсные системы существуют?
Литература
1. Коровин Н.В. Общая химия: Учебник для тех. направл. и спец. Вузов - 6-е
изд., испр. – М.: Высш.шк., 2005. – С. 204-243.
Лекция 7
Окислительно-восстановительные реакции. Электрохимические процессы. Гальванический элемент и электролиз
Основные вопросы:
1. Окислительно- восстановительные реакции.
Виды электрохимических процессов.
Электродный потенциал.
Стандартный электродный потенциал.
Равновесный электродный потенциал. Уравнение Нернста.
Гальванический элемент Даниэля-Якоби. Электродвижущая сила.
Электролиз.
Последовательность электродных процессов при электролизе растворов.
Окислительно-восстановительные реакции являются наиболее распространенными химическими реакциями и играют большую роль в природе и технике. Сжигание топлива, электрохимическое осаждение металлов, получение ценных химических продуктов (аммиака, азотной и
54
серной кислот, щелочей и т.д.), коррозионные процессы, превращение химической энергии в электрическую – в гальванических элементах и аккумуляторах – основаны на окислительно-восстановительных реакциях. Окислительно-восстановительные реакции являются основой жизнедеятельности, так как с ними связаны дыхание и обмен веществ в живых организмах, фотосинтез у растений и многие другие процессы.
Окислительно-восстановительные реакции всегда характеризуются перемещением электронов от одних веществ (атомов, молекул или ионов) к другим и сопровождаются изменением степеней окисления атомов или ионов.
Для характеристики состояния элементов в соединениях введено понятие степени окисления. Степень окисления характеризует валентность и электроотрицаптельность атома в молекуле. Степенью окисления называется заряд, вычисленный из предположения, что соединение состоит только из ионов. Для расчета степеней окисления элемента в его соединениях выработаны простые и удобные эмпирические правила:
в простых веществах степень окисления элемента всегда равна нулю;
в сложных соединениях некоторые элементы проявляют всегда одну
ту же степень окисления. Постоянную степень окисления имеют щелочные металлы (+1), бериллий, магний, щелочноземельные элементы (+2), фтор … (-1). Для водорода в большинстве соединений характерна степень окисления (+1), а в его соединениях с s-элементами и в некоторых других соединениях она равна (-1). Степень окисления кислорода равна (-2); к важнейшим исключениям относятся пероксидные соединения (-1) и фторид кислорода
(+2);
сумма степеней окисления всех атомов в молекуле равна нулю;
степень окисления иона элемента равна заряду иона.
Любая окислительно-восстановительная реакция состоит из двух взаимосвязанных и пространственно неразделенных реакций: реакции окисления и реакции восстановления.
Окисление – это отдача веществом электронов, в результате которой степень окисления вещества повышается. Вещество, отдающее электроны в реакции, называется восстановителем, при этом вещество из восстановленной устойчивой формы превращается в окисленную форму. Рассмотрим реакцию окисления кальция: Ca - 2e =Ca+2. Нейтральный атом кальция, отдавая два электрона, превращается в двухзарядный положительный ион, степень окисления которого повышается от 0 до +2. В этом случае кальций является восстановителем.
55
Восстановление – это присоединение электронов к веществу, в результате чего степень окисления вещества понижается. Вещество, принимающие электроны, называется окислителем, оно из окисленной формы переходит в восстановленную. Например, реакции восстановления иона Fe3+ и нейтрального атома S0:
Fe+3 + e Fe+2; S0 + 2e S-2.
В этих реакциях ионы H+, Fe3+ и нейтральный атом серы, принимая электроны, понижают степень окисления от +1 до 0, от +3 до +2 и от 0 до -2 соответственно, они являются окислителями.
Окислители – простые вещества, элементы которых обладают высокой электроотрицательностью, т.е. неметаллы: F2, O2, Cl2, Br2. Особенно сильно их окислительная способность проявляется, когда такие вещества находятся в атомарном состоянии.
Соединения элементами в высшей степени окисления могут в окислительно-восстановительных реакциях выступать только в качестве окислителей, степень окисления элемента может в этом случае только
понижаться ( K |
|
6 |
|
7 |
|
, H |
|
6 |
, |
5 |
|
). |
|
2 |
Cr O |
7 |
, K M nO |
2 |
SO |
4 |
H N O |
|
|||||
|
2 |
|
4 |
|
|
|
|
3 |
|||||
Ионы металлов (и |
водорода) |
также |
являются окислителями, |
||||||||||
восстанавливаясь в свободные металлы или ионы промежуточного заряда:
Сu 2 2е Cu 0.
Восстановители – простые вещества, образованные элементами с низкой электроотрицательностью, т.е. все металлы и некоторые неметаллы
(Н2, В, С и др.).
Соединения с элементами в низшей степени окисления могут быть только восстановителями, степень окисления может только повышаться
2
( H2 S ).
Если же элемент в соединении находится в промежуточной степени окисления, то соединения обладают окислительно-восстановительной двойственностью – способностью вступать в реакции как с окислителями, так и с восстановителями.
4. Все реакции окисления-восстановления делятся на три типа: межмолекулярные, внутримолекулярные и реакции диспропорционирования.
В первом случае в реакции участвуют различные вещества, одно из которых является восстановителем, другое окислителем. Например, реакции
56
горения. Горением называется окислительно-восстановительная реакция, сопровождающаяся выделением теплоты и света.
Во втором случае атомы одной и той же молекулы могут быть окислителями, другие восстановителями.
В реакциях диспропорцианирования одни и те же атомы могут быть одновременно и окислителем, и восстановителем.
5. Для составления уравнений окислительно-восстановительных реакций используют метод электронно-ионных полуреакций или метод электронного баланса.
Метод электронно-ионного баланса применим для реакций, протекающих в растворах. Он основан на уравнивании в левой и правой частях уравнения суммарного числа зарядов реальных ионов, существующих в растворе, с помощью прибавления или вычитания необходимого числа электронов. В получающихся электронно-ионных уравнениях слабодиссоциирующие, летучие и малорастворимые вещества записывают в молекулярном виде. Более сложными уравнениями описывают окислительно-восстановительные реакции, результат которых зависит от рН раствора. В молекулярной и ионной схемах процесса указывают частицу, определяющую рН раствора: Н+, Н2О или ОН-.
При составлении электронно-ионных уравнений с участием среды необходимо учитывать:
на один атом кислорода, теряемый частицей (молекулой ионом) окислителя, в кислой среде затрачивается два иона Н+ и образуется одна молекула воды; в нейтральной и щелочной среде затрачивается одна молекула Н2О и образуется два иона ОН-;
на один атом кислорода, присоединяющийся к частице восстановителя, затрачивается в кислой и нейтральной среде одна молекула
Н2О и освобождаются два иона Н+; в щелочной среде – два иона ОН- и освобождается одна молекула Н2О. Приведем примеры использования метода электронно-ионного бланса при составлении окислительно-восстановительных реакций.
Пример 1. Составить уравнение реакции взаимодействия цинка с перманганатом калия в кислой среде.
1. Запишем схему уравнения реакции в молекулярной и ионной формах:
Zn + KMnO4 + H2SO4 ZnSO4 + MnSO4 + K2SO4 + H2O,
57
Zn 2 K MnO4 2H SO24 Zn 2 Mn 2 2K 3SO24 H2O.
2. Обозначим ионы одного и того же элемента, изменяющиеся в ходе реакции и запишем для них полуреакции восстановления и окисления. Полуреакция восстановления имеет вид:
.
MnO-4 + 8H+ Mn2+ + 4H2O.
Материальный баланс соблюден (схема уравнена по числу атомов). Далее уравниваем левую и правую часть полуреакции по числу зарядов. Сумма зарядов левой части равна (7+). Сумма зарядов правой части равна (2+). Для выполнения условия сохранения зарядов надо к левой части схемы прибавить пять электронов:
MnO-4 + 8H+ + 5е Mn2+ + 4H2O.
3. Составляем уравнение второй полуреакции – полуреакции окисления цинка:
Zn0 -2eZn2+.
4. Находим наименьшее общее кратное для числа отданных и принятых электронов и основные коэффициенты (множители) для исходных окислителя и восстановителя и продуктов окисления-восстановления:
-
Zn0 - 2e Zn2+
5
MnO + 8H ++ 5e Mn2+
+ 4H O
2
4
2
5. Умножаем обе части каждой полуреакции на соответствующие коэффициенты и суммируем, левые и правые части:
5Zn0+2MnO 4 +16H+5Zn2++2Mn2++8H2O.
58
Строго говоря, это уравнение и отражает все изменения, происходящие в растворе в результате окисления цинка перманганатом калия, и среду, в которой этот процесс возможен.
6. Уравнение в молекулярной форме имеет вид:
5Zn0 + 2KMnO4 + 8H2SO4 5ZnSO4 + 2MnSO4 + K2SO4 + 8H2O.
Процессы взаимного превращения химической и электрической энергии называют электрохимическими процессами. Электрохимические процессы можно разделить на две основные группы: 1) процессы превращения химической энергии в электрическую (в гальванических элементах); 2) процессы превращения электрической энергии в химическую (электролиз).
Реакции окисления-восстановления могут протекать пространственно разделенными, что реализуется в процессах электрохимической коррозии, гальванических элементах и электролизе. Если реакция протекает в гальваническом элементе или осуществляется путем электролиза, то каждая полуреакция протекает на соответствующем электроде; поэтому полуреакции называются электродными процессами. Простейшая электрохимическая система состоит из двух электродов и ионного проводника между ними. Электродами называют проводники, имеющие электронную проводимость (проводники 1-рода) и находящиеся в контакте с ионным проводником. Для обеспечения работы электрохимической системы электроды соединяют друг
другом металлическим проводником, называемым внешней цепью электрохимической системы. Рассмотрим электрохимические процессы, протекающие при погружении металла в раствор собственных ионов.
Атомы металла в пластине являются восстановленной формой, а его ионы в растворе – окисленной формой. При этом у поверхности раздела металла с раствором возникает потенциал вследствие образования двойного электрического слоя. В случае активного металла возможен переход его
атомов,легкотеряющихэлектроны, |
изметаллав |
раствор: |
М 0 (т) ne M n ( p) . За счет этого в |
растворе появляется |
избыток |
катионов, а в металле – электронов. Поверхностный слой металла заряжается отрицательно, а граничащий с ним слой раствора положительно.
Для пассивного металла, ионы которого энергично захватывают электроны М n (р) ne M 0 (т) , наблюдается процесс, который приводит к связыванию катионов поверхностью металла, заряжающейся положительно, и к появлению в растворе избытка анионов, притягивающихся к поверхности
59
металла. Возникает двойной электрический слой, характеризующийся противоположными зарядами на металле и в растворе.
Потенциал, установившийся в условиях равновесия электродной реакции, называется равновесным электродным потенциалом. Абсолютные значения электродных потенциалов экспериментально определить невозможно.
4. Значение электродного потенциала измеряют относительно какой-либо стандартной системы, в качестве нее обычно служит стандартный водородный электрод, потенциал которого принят равным нулю. Равновесие потенциалопределяющей стадии имеет вид: 2Н+ + 2е → Н2. Стандартный водородный электрод представляет собой платиновый электрод, покрытый платиновой чернью и погруженный в раствор кислоты (1,25 М НСl или 0,5 М H2SO4), aH =1 моль/л. Через раствор пропускается водород под давлением в
101,3 кПа при комнатной температуре. Если на водородном электроде происходит восстановление Н+ + е → 12 Н2, то потенциал исследуемой системы принимается за отрицательный. Если на водородном электроде идет процесс окисления 12 Н2 - е → Н+, то потенциал считается положительным.
Значения стандартных электродных потенциалов приводятся в справочных таблицах. Стандартный электродный потенциал – это потенциал данного электродного процесса при концентрации (точнее говоря, активностях) всех участвующих в нем веществ, равных 1 моль/л.
5. В результате изучения потенциалов различных электродных процессов установлено, что их величины зависят от следующих факторов: 1) от природы веществ – участников электродного процесса; 2) от соотношения между концентрациями этих веществ; 3) от температуры системы. Эта зависимость выражается уравнением Нернста (В. Нернст, 1889 г.). Для электродной реакции: a Ох + n e = b Red
-
E E 0
RT
ln
aOxa
.
b
nF
aRed
Здесь Е0 – разность потенциалов (В) между двумя электродами при стандартных условиях, когда активности всех веществ равны единицы; aOx, aRed – концентрации веществ (активности), участвующих в процессе окисленной и восстановленной форм, моль/л; R – газовая постоянная, равная
60
8,314 Дж/(К·моль); Т – абсолютная температура, F – число Фарадея, равное 96500 Кл; n – число электронов, участвующих в полуреакции; a и b – стехиометрические коэффициенты. Для разбавленных растворов активности могут быть заменены на равновесные концентрации. После подстановки числовых значений констант и перевода натуральных логарифмов в десятичные уравнение Нернста имеет вид:
-
0
0,059
a a
E E
lg
Ox .
n
b
aRed
Из уравнения Нернста ясно, что возрастание концентрации окисленной формы или уменьшение концентрации восстановленной формы вызывает увеличение электродного потенциала системы, и наоборот.
Рассмотрим, какой вид принимает общее уравнение электродного потенциала в важнейших случаях.
1. Электродный процесс выражается уравнением:
М n ne M .
![]()
Уравнение Нернста имеет вид:
-
E
n
E 0
0,059
lg a
n .
М
/ M
M
М n / M
n
2. Электродный процесс выражается уравнением:
2Н 2e Н2.
Уравнение Нернста:
E2Н / Н2 0,059 рН.
3. Электродный процесс выражается уравнением:
О2 4Н 4e 2Н2О .
61
Уравнение Нернста для электродного процесса:
ЕО2 4Н / 2Н2О 1,228 0,059рН.
6. Устройства, в которых химическая энергия окислительно-восстановительных реакций преобразуется в электрическую, называется гальваническими элементами. Их называют также химическими источниками электрической энергии, или химическими источниками тока (сокращенно ХИТ). Химические источник электрической энергии применяются в различных отраслях техники. В средствах связи (радио, телефон, телеграф) и в электроизмерительной аппаратуре они служат источниками электропитания, на автомобилях, самолетах, тракторах применяются для приведения в действие стартеров и других устройств, на транспорте, в переносных фонарях с их помощью производится освещение.
ХИТ состоят из двух электродов Э1 и Э2, погруженных в отдельные – Р1 и Р2 – или один общий раствор Р электролита. Схемы в этих случаях имеют вид:
Э1
Р1
![]() Р2
Э2
или Э1
Р Э2
.
Р2
Э2
или Э1
Р Э2
.
Здесь двойная черта означает пористую диафрагму, разделяющую разные по составу растворы или стенки отдельных сосудов, соединенных капилляром, называемым электролитическим ключом.
Гальванический элемент из электродов 1 рода и 2 рода. В электродах первого рода (металлы) носителями электричества являются электроны, в электродах второго рода (электролиты) – ионы. Рассмотрим гальванический элемент Даниэля-Якоби.
Погрузим цинковый электрод в сосуд с раствором сульфата цинка, а медный – с раствором сульфата меди ( сZn 2 cCu 2 = 1 моль/л). На
электродах в результате равновесий
() Zn Zn 2 2e
() Cu Cu 2 2e
62
устанавливаются стандартные потенциалы:E 0 |
0,76 Bи |
||||
|
|
|
Zn 2 |
/Zn |
|
E 0 |
0,34 B . |
Составим |
электрохимическую |
цепь: |
|
Cu 2 |
/Cu |
|
|
|
|
()Zn ZnSO4 CuSO4 Cu() . Для этого электроды соединим металлическим
проводником, а сосуды электролитическим ключом. Тогда электроны с отрицательно заряженного цинкового электрода по внешней цепи перемещаются на положительно заряженный медный электрод. На Zn-электроде концентрация электронов станет меньше равновесной, что по принципу Ле-Шателье вызовет смещение равновесия в прямом направлении. На Cu-электроде концентрация электронов станет больше равновесной, что соответственно, приведет к смещению равновесия в обратном направлении. Таким образом, на электродах протекают процессы:
Анод (-) Zn 2e Zn 2 – окисление; Катод (+) Cu 2 2e Cu – восстановление.
Процессы окисления в электрохимии получили название анодных процессов, а электроды, на которых идут процессы окисления, называются анодами.
Процессы восстановления в электрохимии получили название катодных процессов, а электроды, на которых идут процессы восстановления, называют катодами.
В результате суммирования уравнений анодного и катодного процессов получаем:
Zn Cu 2 Zn 2 Cu или Zn CuSO4 Cu ZnSO4 .
Вследствие этой химической реакции в гальваническом элементе возникает движение электронов по внешней цепи и ионов внутри элемента (оставшиеся в избытке сульфат-ионы мигрируют по электролитическому ключу из катодного пространства в анодное). Суммарная химическая реакция, протекающая в гальваническом элементе, называется токообразующей.
Схема элемента Даниэля-Якоби записывается в виде:
A()Zn
Zn
2
![]() Cu
2
Cu ()K
Cu
2
Cu ()K
63
Электрическая работа равна произведению разности потенциалов электродов на количество электричества. Максимальная разность потенциалов электродов, которая может быть получения при работе гальванического элемента, называется электродвижущей силой (ЭДС) элемента.
Электродвижущая сила (ЭДС) гальванического элемента есть разность между большим и меньшим потенциалами электродов. Если потенциалы электродов стандартные, то ЭДС также называется стандартной:
-
E 0 E 0
E 0
0,34 (0,76) 1,10 B .
Cu 2 /Cu
Zn 2
/Zn
При работе гальванического элемента концентрация Сu2+ уменьшается, а концентрация Zn2+ растет. Следовательно, согласно уравнению Нернста, потенциал катода уменьшается, анода – увеличивается, и ЭДС гальванического элемента уменьшается.
подобных гальванических элементах материал анода играет роль восстановленной формы и непосредственно участвует в химическом процессе. Это гальванические элементы с активными анодами.
Гальванические элементы с жидким электролитом неудобны в работе в связи с возможной утечкой электролита и громоздкостью. Они обладают большим внутренним сопротивлением. Практически гораздо чаще пользуются сухими элементами. В сухих элементах электролит находится в пастообразном состоянии. Роль анода может играть корпус из активного металла, например цинка, а катодом является инертный электрод – графитовый стержень.
технике ХИТ, в которых протекают практически обратимые реакции, называют аккумуляторами – их можно перезаряжать и использовать многократно. Широко распространены свинцовые и никелевые аккумуляторы. Аккумулятор в наиболее простом виде имеет два электрода и ионный проводник между ними. На аноде при разряде и при заряде протекают реакции окисления, на катоде реакции – восстановления. В процессе разряда и заряда изменяется состав активных масс аккумулятора и соответственно ЭДС и напряжение.
7. Электролиз. Электролизом называют совокупность электрохимических процессов раздельного окисления и восстановления, осуществляемых на электродах в растворах или расплавах электролитов при пропускании через них постоянного электрического тока от внешнего
64
источника. При электролизе электрическая энергия превращается в химическую. Ячейка для электролиза называется электролизером. Электрод, на котором идет реакции восстановления (катод), у электролизера подключен к отрицательному полюсу внешнего источника тока. Электрод, на котором протекает реакция окисления (анод), подключен к положительному полюсу источника тока. Электрохимические процессы, протекающие при электролизе, в принципе обратны процессам, идущим при работе гальванического элемента.
С количественной стороны процесс электролиза впервые был изучен в 30-х годах XIX в. М. Фарадеем, установившим следующие законы электролиза.
Масса образующегося при электролизе вещества пропорциональна количеству прошедшего через раствор электричества.
При электролизе различных химических соединений равные количества электричества приводят к электрохимическому превращению эквивалентных количеств веществ.
При превращении одного моля эквивалентов вещества на электроде через него проходит 96484, или округленно 96500 Кл (А*с). эта величина называется постоянной Фарадея F.
Первый и второй законы электролиза вместе описываются следующим выражением:
тВ М Э(В) I ,
F
где тВ – масса вещества, окисленного или восстановленного на электроде, г.; М Э(В) – масса моля эквивалентов, г/моль; I – ток, А; τ –
продолжительность электролиза, с; F – число Фарадея 96500 Кл/моль. Законы электролиза относятся к электролизу растворов, расплавов и
твердых электролитов с чисто ионной проводимостью.
Итак, под действием электрического тока на электродах происходят процессы, называемые электролизом. Последовательность электродных процессов при электролизе растворов определяется значениями потенциалов этих процессов и поляризацией при их протекании.
Минимальное напряжение электрического тока, приложенное к электродам электролизера, называется его потенциалом разложения. Однако на практике, чтобы начался электролиз, к электродам приходится прикладывать напряжение, заметно превышающее потенциал разложения вещества.
65
Рассмотрим процессы, протекающие при электролизе расплавленного вещества. В расплав вещества погружают инертные электроды. Один из них, катод, подключают к отрицательному полюсу генератора электрического тока, другой – анод – к положительному полюсу. К катоду притягиваются положительные ионы вещества (катионы), к аноду – отрицательные ионы (анионы). На катоде всегда происходит процесс восстановления, на аноде – процесс окисления. Схематически эти процессы на примере расплава NaOH можно представить следующим образом:
Ионный состав электролита:
NaOH → Na+ + OH-.
Электродные процессы:
K(-) Na+ + e = Na,
A(+) 4OH- - 4e = O2↑ + 2H2O↑.
3. Полное уравнение электролиза:
4NaOH = 4Na + O2↑ + 2H2O↑.
8. При электролизе растворов электролитов происходит конкуренция между растворенным веществом и растворителем за участие в электродном процессе. Поэтому состав продуктов окисления на аноде и восстановления на катоде зависит, в первую очередь, от концентрации раствора. Рассмотрим процессы электролиза разбавленных водных растворов электролитов, в которых концентрация ионов не превышает 1 моль/л.
Для объяснения электродных процессов, происходящих при электролизе разбавленных растворов, можно руководствоваться следующими правилами:
1) на катоде в первую очередь восстанавливаются катионы с наиболее высокими значениями Е0. Если значение Е0 восстановления значительно ниже чем -0,41 В (например -1 В и менее), то на катоде восстанавливаются молекулы воды с выделением водорода и накоплением в растворе гидроксид-ионов: 2H2O + 2e = H2↑ + 2OH-; при Е0восстановление значительно выше чем -0,41 В (например -015 В и более) на катоде восстанавливаются только ионы металла: М n ne M 0; при промежуточных значениях Е0 происходит восстановление и воды, и ионов металла;
66
2) на аноде в первую очередь окисляются простые анионы в порядке возрастания их Е0, не превышающих +1,5 В. Кислородсодержащие анионы, характеризующиеся слишком высокими для восстановителей значениями Е0, в водных растворах не окисляются; вместо них на аноде окисляется вода с выделением кислорода и накоплением в растворе ионов водорода. В щелочных растворах на аноде окисляются ионы OH-, как и в расплаве NaOH.
Рассмотрим электролиз растворов хлорида и сульфата натрия.
Электролиз раствора хлорида натрия NaCl
1. Ионный состав электролита:
NaCl → Na+ + Cl-,
H2O ⇄ H+ + OH-.
2. Электродные процессы:
K(-) 2H2O + 2e = H2↑ + 2OH-
A(+) 2Cl- - 2e = Cl2↑.
3. Полное уравнение электролиза:
2NaCl + 2H2O = 2H2↑ + 2NaOH + Cl2↑.
Электролиз раствора сульфата натрия Na2SO4
1. Ионный состав электролита:
Na2SO4 → 2Na+ + SO 24 ,
H2O ⇄ H+ + OH-.
2. Электродные процессы:
K(-) 2H2O + 2e = H2↑ + 2OH-,
A(+) 2H2O - 4e = O2↑ + 4H+.
3. Полное уравнение электролиза:
2Na2SO4 + 6H2O = 2H2↑ + 4NaOH + O2↑ + 2H2SO4.
Электролиз с растворимым анодом. Если атомы металла теряют электроны и из анода переходят в виде ионов в раствор, анод растворяется.
67
Ионы металла, перешедшие в раствор, притягиваются к катоду и восстанавливаются на нем. На катоде происходит отложение металла. Подобная разновидность электролиза называется электролизом с растворимым анодом. Электролиз с растворимым анодом широко применяется в технике; это электролитическая очистка металлов от примесей, т.е. электрорафинирование металлов, а также гальваностегия и гальванопластика, являющиеся разновидностями гальванотехники.
Отклонение потенциала электрода от его равновесного значения называется электрохимической поляризацией или просто поляризацией.
Поляризацию можно осуществить включением электрода в цепь постоянного тока. Для этого необходимо составить электролитическую ячейку из электролита и двух электродов – изучаемого и вспомогательного. Включая ее в цепь постоянного тока, можно сделать изучаемый электрод катодом или анодом. Такой способ поляризации называется поляризацией от внешнего источника электрической энергии. Поляризация может наблюдаться как на катоде, так и на аноде, поэтому различают катодную и анодную поляризации ∆Ек и ∆Еа. Изменение потенциала при прохождении тока также называется перенапряжением. Этот термин обычно употребляют, когда известна причина изменение потенциала. Его также относят к некоторым конкретным процессам, например к катодному восстановлению водорода (водородное перенапряжение). Возникновение поляризации обусловлено замедленностью отдельных стадий электрохимического процесса. Любая электрохимическая реакция протекает минимум в три стадии: а) подвод реагентов к электроду; б) собственно электрохимическая реакция; в) отвод продуктов реакции от электрода. Изменение потенциала электрода вследствие изменения концентрации реагентов в приэлектродном слое при прохождении тока называется концентрационной поляризацией. Изменение потенциала, обусловленное уменьшением скорости собственно электрохимических стадий реакций, называется электрохимической поляризацией (перенапряжением). В зависимости от типа замедленной стадии поляризация может быть снижена перемешиванием раствора, применением катализаторов, увеличением температуры, концентрации реагентов, площади поверхности электродов.
Вопросы для контроля
1. Какие реакции называют окислительно-восстановительными? Какое значение имеют окислительно-восстановительные реакции в природе, технике?
68
На какие две группы делятся электрохимические процессы?
Как возникает электродный потенциал?
Что такое гальванический элемент?
Что называют электролизом?
Какие факторы влияют на напряжение электролизера?
Какова последовательность электродных процессов на катоде электролизера?
Какова последовательность электродных процессов на аноде электролизера?
Литература
1. Коровин Н.В. Общая химия: Учебник для тех. направл. и спец. Вузов - 6-е
изд., испр. – М.: Высш.шк., 2005. – С. 260-300.
Лекция 8
Коррозия металлов. Защита металлов и сплавов от коррозии
Основные вопросы:
1.Коррозия металлов и сплавов.
2.Химическая коррозия.
3.Электрохимическая коррозия. Электрохимическая коррозия с кислородной и водородной деполяризацией.
4. Методы защиты металлов от коррозии
4.1. Защитные покрытия.
Изменение состава металла и среды.
Электрохимические методы.
1. Коррозия металлов – разрушение металлов вследствие химического или электрохимического взаимодействия их с внешней (коррозионной) средой.
В результате коррозии ежегодно теряется от 1 до 1,5 % всего металла, накопленного и эксплуатируемого человечеством.
Коррозионные процессы классифицируют:
а) по виду (геометрическому характеру) коррозионных разрушений на поверхности или в объеме металла;
б) по механизму реакций взаимодействия металла со средой (химическая и электрохимическая коррозия);
69
в) по типу коррозионной среды; г) по характеру дополнительных воздействий, которым подвергается
металл одновременно с действием коррозионной среды.
Коррозией называется самопроизвольный электрохимический процесс разрушения металлов и сплавов в результате их окисления под действием окружающей среды.
Коррозия является окислительно-восстановительным процессом, вероятность ее протекания тем больше, чем больше разность потенциалов систем окислителя и восстановителя Eоx Еred . Восстановителем при коррозии является металл. Коррозировать в первую очередь будет более активный металл, имеющий меньший потенциал. Окислителем (деполяризатором) в процессе коррозии выступает тот компонент окружающей среды, который имеет большой окислительно-восстановительный потенциал.
По механизму процессов коррозию подразделяют на химическую, электрохимическую, электрокоррозию, биокоррозию.
2. При химической коррозии происходит непосредственное взаимодействие металла с окислителем. Химическая коррозия протекает без участия влаги в среде сухих газов, например кислорода, сернистого газа, хлороводорода, сероводорода и др. Этот вид коррозии встречается сравнительно редко, например при высокотемпературной обработке металла, в двигателях внутреннего сгорания, в реактивных двигателях.
По условиям протекания коррозионного процесса различают газовую коррозию – в газах и парах без конденсации влаги на поверхности металла, обычно при высоких температурах; коррозию в неэлектролитах – агрессивных органических жидкостях.
Рассмотрим химическую коррозию в газах, в частности коррозию в атмосфере кислорода. В результате химической коррозии металл покрывается слоем продуктов его окисления – пленкой оксида или гидроксида. Образующаяся пленка препятствует диффузии окислителя к чистому металлу и тем самым замедляет коррозию металла. Скорость окисления металла зависит не только от его химической активности и температуры, но и от скорости диффузии кислорода сквозь образовавшуюся на поверхности металла пленку.
Способность такой пленки защищать металл от дальнейшей коррозии приближенно оценивают по ее характеристике – сплошости. Показателем сплошности пленки служит отношение объема продуктов коррозии к объему окисленного металла. Для оксидных пленок свинца, алюминия, хрома, железа эти показатели равны соответственно 1,15, 1,31, 2,02, 2,1. Считается,
70
что пленки, у которых показатели сплошности лежат в пределах от 1,2 до 1,6, препятствуют дальнейшей коррозии металла, так как являются сплошными (без разрывов) и плотными. При оценке качества пленки необходимо оценивать и структуру оксида металла. Строение оксидных пленок зависит от многих факторов: степени окисления металла, диффузии атомов кислорода в толщу металла и встречной диффузии атомов металла в толщу оксида, летучести оксида. Металлы, принимающие высокие степени окисления в продуктах коррозии, обычно не являются жаростойкими, оксидные пленки, представляющие собой смешанные оксиды дух металлов состава МеО Ме2О3 имеют плотную структуру и т.д.
Таким образом, скорость химической коррозии определяется прежде всего свойствами пленки, возникающей при коррозии на поверхности металла, характер ее определяется природой металла и окислителя, а также температурой.
3. Электрохимическая коррозия. Разрушение металла под воздействием возникающих в коррозионной среде гальванических элементов называют электрохимической коррозией. При электрохимической коррозии (наиболее частая форма коррозии) всегда требуется наличие электролита (конденсат, дождевая вода и т.д.), с которым соприкасаются электроды – либо различные элементы структуры материала, либо два различных соприкасающихся материала с различающимися окислительно-восстановительными потенциалами. Если в воде растворены ионы солей, кислот и т.п., электропроводность ее повышается и скорость процесса увеличивается.
Коррозионный элемент. При соприкосновении двух металлов с различными окислительно-восстановительными потенциалами и погружении их в раствор электролита, например дождевой воды с растворенным углекислым газом CO2, образуется гальванический элемент, так называемый коррозионный элемент. Он представляет собой не что иное, как замкнутую гальваническую ячейку. В ней происходит медленное растворение металлического материала с более низким окислительно-восстановительным потенциалом; второй электрод в паре, как правило, не корродирует. Этот вид коррозии особо присущ металлам с высокими отрицательными потенциалами. Так, совсем небольшого количества примеси на поверхности металла с большим редокспотенциалом уже достаточно для возникновения коррозионного элемента. Особо подвержены риску места соприкосновения металлов с различными потенциалами, например сварочные швы или заклепки.
Если растворяющийся электрод коррозионно-стоек, процесс коррозии замедляется. На этом основана, например, защита железных изделий от
71
коррозии путем оцинковки – цинк имеет более отрицательный потенциал, чем железо, поэтому в такой паре железо восстанавливается, а цинк должен корродировать. Однако в связи с образованием на поверхности цинка оксидной пленки процесс коррозии сильно замедляется.
Водородная и кислородная коррозия. Если происходит восстановление ионов H3O+ или молекул воды H2O, говорят о водородной коррозии, или коррозии с водородной деполяризацией. Восстановление ионов происходит по следующей схеме:
2H3O+ + 2e → 2H2O + H2
или
2H2O + 2e → 2OH− + H2 .
Если водород не выделяется, что часто происходит в нейтральной или сильно щелочной среде, происходит восстановление кислорода, и здесь говорят о кислородной коррозии, или коррозии с кислородной деполяризацией:
O2 + 2H2O + 4e → 4OH−.
Коррозионный элемент может образовываться не только при соприкосновении двух различных металлов, он образуется и в случае одного металла, если например структура поверхности неоднородна.
Значительно чаще встречается электрохимическая коррозия, которая протекает в среде электролитов или во влажных газах. В аэрируемых растворах и во влажной атмосфере присутствуют два возможных окислителя: ионы водорода и кислород. Окислительно-восстановительный потенциал кислорода в кислой, нейтральной, щелочной средах больше соответствующего потенциала иона водорода. Поэтому, в первую очередь, роль деполяризатора при коррозии выполняет кислород. Однако в кислых растворах из-за высокой скорости коррозии и малой растворимости кислорода в растворах роль деполяризатора выполняют ионы водорода.
Растворенный кислород и ионы водорода – важнейшие окислители, вызывающие электрохимическую коррозию. Так как окислители снимают избыточный отрицательный заряд с поверхности катода, уменьшая его деполяризацию, их называют в этом процессе деполяризаторами.
Коррозия с участием кислорода называется коррозией с поглощением кислорода (коррозией с кислородной деполяризацией). Коррозия с участием ионов водорода называется коррозией с выделением водорода (коррозией с водородной деполяризацией). Металлы, применяемые в технике, всегда содержат примеси других металлов. Поэтому при соприкосновении металла с раствором электролита на его поверхности возникает множество непрерывно
72
действующих коротокозамкнутых микрогальванических элементов, в которых окисляется более активный металл с меньшим электродным потенциалом.
Например, в техническом цинке в качестве примеси всегда присутствует медь, поэтому при коррозии возникает множество микрогальванических элементов типа ()Zn среда Cu(). На анодных
участках протекает окисление цинка, а на катодных (медь), не экранированных ионами цинка, окислитель беспрепятственно получает электроны и восстанавливается. Электроны перемещаются от цинка к меди.
Не только примеси металлов, но и включения других веществ, любая физическая или химическая неоднородность материалов и среды приводит к возникновению микрогальванических элементов.
Наиболее распространена атмосферная коррозия металлов. Например, при конденсации влаги на месте контакта железа с медью возникает гальванический элемент: ()Fe H2O, O2 Cu() . Исходя из значений
стандартных электродных потенциалов, железо является анодом, а медь катодом. При работе такого гальванического элемента железо окисляется и разрушается, а кислород, имеющийся в растворе, восстанавливается на поверхности меди:
-
() Fe 2е Fe2
2
() O2 2H2O
4e 4OH
1
2Fe O2 2H
2O 2Fe(OH) 2
Ионы Fe2+ соединяются с ионами ОН-, образуя малорастворимый дигидроксид железа Fe(OH)2, который окисляется кислородом воздуха до оксидгидроксида:
4Fe(OH)2 O2 4FeOOH 2H2O .
Если процесс коррозии протекает в кислой среде, то роль окислителя при Е2Н/Н2 EMen /Me выполняют ионы водорода: 2Н 2е Н2 .
Таким образом, электрохимическая коррозия протекает через сопряженные процессы анодного растворения металла и катодного восстановления окислителя, обычно молекул кислорода или ионов водорода. Основным отличием процессов электрохимической коррозии от процессов в гальваническом элементе является отсутствие внешней цепи. Электроны в
73
процессе коррозии не выходят из корродирующего металла, а двигаются внутри металла. Химическая энергия реакции окисления металла передается не в виде работы, а лишь в виде теплоты.
Таким образом, при наличии энергетической неоднородности поверхности металла коррозионный процесс заключается в работе огромного числа коррозионных микроэлементов. Коррозионный элемент в отличии от гальванического является короткозамкнутым.
Возможность протекания при коррозии того или иного катодного процесса определяется его потенциалом. Коррозия с поглощением кислорода лимитируется (контролируется) стадией диффузии кислорода возрастает с увеличением его концентрации при перемешивании, зависит от температуры. Коррозия с выделением водорода зависит от примесей в металле, от рН и возрастает с увеличением температуры.
Следует отметить, что окислители играют двойственную роль в коррозионных процессах. С одной стороны, они могут восстанавливаться и этим ускорять коррозию металлов, а с другой некоторые металлы имеют склонность к пассивации вследствие образования защитных слоев (Cr2O3 на хроме, TiO2 на титане, Al 2O3 на алюминии, Ta2O5 на тантале). Пассивностью металла называется состояние его повышенной коррозионной устойчивости, вызванное торможением анодного процесса.
Если потенциал металла положительнее потенциала кислородного электрода, то коррозия металла невозможна. Это металлы высокой стабильности: Hg, Pd, Ir, Pt, они устойчивы во влажной атмосфере в нейтральной среде. Металлом полной стабильности является золото, оно не может корродировать во всей области рН.
Если потенциал металла положительнее потенциала водородного электрода и отрицательнее потенциала кислородного электрода, то коррозия возможна с поглощением кислорода и невозможна с выделением водорода. Это металлы промежуточной термодинамической стабильности: Bi, Sb, Re, Tc, Cu, Ag, Rh, они устойчивы в любых кислых и нейтральных средах в отсутствии кислорода.
Если потенциал металла отрицательнее потенциала водородного электрода, то возможна коррозия как с поглощением кислорода, так и с выделением водорода. Это группа металлов повышенной термодинамической нестабильности: Li, Rb, Cs, Ba, Sr, Ca, Na, Mg, Al, Ti, Zr, Mn, Cr, Zn, Fe. Эти металлы могут корродировать даже в нейтральных средах. Термодинамически нестабильные металлы имеют значения стандартных электродных потенциалов больше чем металлы предшествующей группы, но меньше нуля: Cd, In, Tl, Co, Ni, Mo, Pb, W,
74
поэтому окисляться водой (рН = 7) они не могут, но будут неустойчивыми в кислых средах и в любых средах в присутствии кислорода. Восстановление деполяризатора при коррозии показано в таблице 5.
Табл.5 Восстановление дополяризатора при коррозии
Деполяризатор; |
|
Среда |
|
|
|||||
редокс-потенциал |
|
|
|
||||||
|
|
|
|
|
|
|
|
||
|
|
Кислая рН<7 |
|
|
|
Нейтральная |
Щелочная |
||
|
|
|
|
|
|
|
рН=7 |
|
рН>7 |
H |
0,059pH |
При Е2Н/Н2 |
EMen /Me |
||||||
E2H/H2 |
2Н 2е Н2 |
|
|
2Н2О 2е Н2 2ОО |
|||||
O2 |
|
При Е |
E |
Men /Me |
E |
2H/H2 |
|||
EO2/2H2O 1,23 0,059pH |
O2 4Н/Н2O |
|
|
|
|
||||
О2 4Н 4е 2Н2О |
О2 2Н2О 4е 4ОО |
||||||||
|
|
||||||||
Скорость коррозии с кислородной деполяризацией растет с увеличением коэффициента диффузии, растворимости кислорода и при перемешивании раствора. Для борьбы с коррозией металла, контролируемой скоростью восстановления кислорода, следует снижать концентрацию кислорода, введением восстановителя в раствор или снижением давления кислорода над раствором; изолировать металл от кислорода; металл не должен содержать примесей (особо чистые металлы).
Скорость коррозии с водородной деполяризацией возрастает с увеличением температуры и снижением рН. Скорость процесса заметно зависит от природы катодных участков. Некоторые металлы (платина, кобальт, никель) катализируют выделение водорода, и катодный процесс протекает с высокими скоростями.
Для борьбы с коррозией металла с водородной деполяризацией необходимо снижать температуру, уменьшать концентрацию ионов Н+, очищать металлы от примесей, катализирующих выделение водорода, изолировать поверхность металла.
Электрокоррозия протекает под действием блуждающих токов. Источником блуждающих токов является электрифицированный рельсовый транспорт и другие агрегаты, питающиеся электрическим током и имеющие контакт с грунтом. Часть потребляемого тока ответвляется и блуждает как по грунту, так и по металлическим сооружениям, находящимся на земле. Если на пути блуждающего тока встречается, например, металлическая труба, то
75
ток входит в нее, некоторое время протекает по ней, а потом снова выходит из нее в грунт. Место входа блуждающего тока является катодным участком, а место выхода тока из трубы – анодным. Разрушаются анодные участки.
4. Методы защиты |
металлов от |
коррозии |
разделяют |
на |
группы: |
а) защитные покрытия; |
б) изменение |
состава |
металла |
или |
среды; |
в) электрохимические методы; г) рациональное конструирование изделий. 4.1. Метод защитных покрытий заключается в изоляции защищаемого
металла от окислителя. Материалы, используемые для защитных покрытий разнообразны: лаки, краски, смазки и т.д.
Продукты коррозии, образующиеся на поверхности металла в виде плотной пленки, также могут играть роль покрытия. Например такие металлы, как алюминий, хром, бериллий, несмотря на их весьма отрицательные электродные потенциалы, корродируют очень медленно, так как надежно защищены плотными оксидными пленками.
Распространены металлические покрытия. Покрытие более активным металлом, обладающим меньшим потенциалом, чем защищаемый, называется анодным. Например, анодным является покрытие железа (-0,44 В) цинком (-0,76 В). При нарушении целостности покрытия возникает гальванический элемент, при работе которого корродирует анодное цинковое покрытие а на катоде – железе – протекает восстановление окислителя. Железо останется защищенным до тех пор, пока не будет разрушен весь цинковый слой.
Покрытие менее активным металлом, имеющим больший потенциал, чем защищаемый, называется катодным. Например, катодным является покрытие железа никелем (-0,25 В), оловом (-0.14 В), медью (+0,34 В). В месте нарушения целостности протекает коррозия защищаемого металла, железа, так как возникает гальванический элемент, где железо является анодом, а покрытие катодом. Поэтому при нанесении катодных покрытий особое внимание должно быть уделено их качеству.
В зависимости от метода нанесения защитного материала на металл различают следующие типы покрытий металлов:
поверхностное легирование (нанесение на защищаемую поверхность тонкого слоя металла из газовой фазы вакуумным или плазменным напылением с последующей термообработкой). Например, аллитирование (Al), хромирование (Cr), силицирование (Si) металлов;
плакирование (нанесение поверхностного слоя в процессе совместного проката листов защищаемого (сталь, дюралюмин) и защищающего (нержавсталь, алюминий) металла);
76
- оксидирование металлов (создание на поверхности плотных пленок оксидов металлов, что осуществляется химическим или электрохимическим путем).
4.2. При подборе материалов всегда учитывают их коррозионную устойчивость в условиях эксплуатации. Одним из эффективных методов повышения коррозионной стойкости металлов и сплавов является их легирование, введение компонентов, способствующих пассивации. Исходя из экономической и технологической целесообразности, заменяют металлы на пластмассы, керамику, металлокерамику и др. В особо коррозионно-агрессивных средах приходится применять дорогие материалы на основе ниобия, тантала, благородных металлов.
Изменение состава среды возможно, если объем среды конечный. Например, из воды, используемой в котлах теплоэлектростанций, предварительно удаляют активный окислитель – растворенный кислород. Кислые растворы подщелачивают, так как при этом уменьшается потенциал ионов водорода. Для защиты от коррозии широко применяются ингибиторы вещества, замедляющие коррозию.
К электрохимическим методам относится протекторная и катодная защита. Метод протекторной защиты заключается в том, что к защищаемой металлической конструкции в некоторых местах присоединяют протекторы – пластины из более активного металла. Образуются гальванические пары, где протектором является анод, а защищаемая металлическая конструкция – катодом. В результате работы гальванического элемента протектор разрушается, а защищаемая конструкция предохраняется от коррозии. Наиболее часто этот метод применяется для защиты от коррозии подводных частей судов, ворот шлюзов, лопастей турбин. В качестве материалов протектора для стальных конструкций чаще всего применяется цинк или сплавы магния.
Сущность катодной защиты состоит в том, что защищаемое сооружение присоединяют к катоду внешнего источника постоянного тока, а к его аноду дополнительную конструкцию из металлического лома. При достаточной разности потенциалов начинается электролиз, при котором материал дополнительной конструкции окисляется, а на защищаемом сооружении восстанавливается окислитель. Чаще всего этот метод используется для защиты протяженных сооружений, расположенных во влажном грунте или растворе: подземных и морских трубопроводов, кабелей, коммуникаций химических заводов.
Рациональное конструирование изделий должно исключать наличие особо опасных с точки зрения коррозии участков в изделиях или
77
конструкциях (сварных швов, узких щелей, контактов разнородных по электродным потенциалам металлов), а также предусматривать специальную защиту металла этих участков от коррозии.
Таким образом, к настоящему времени благодаря изучению механизма коррозии разработаны разнообразные методы защиты от коррозии, выбор которых определяется природой защищаемого металла, параметрами коррозионной среды и экономическими соображениями.
Вопросы для контроля
Что называют коррозией металлов?
В чем отличие электрохимической коррозии от химической?
Каковы причины возникновения коррозионных микрогальванических элементов?
Какие факторы влияют на скорость коррозии с выделением водорода?
Как можно замедлить скорость коррозии с поглощением кислорода?
Литература
1. Коровин Н.В. Общая химия: Учебник для тех. направл. и спец. Вузов - 6-е
изд., испр. – М.: Высш.шк., 2005. – С. 310-337.
Лекция 9 Раздел Избранные вопросы химии. Аналитическая химия.
Качественный и количественный анализ
Основные вопросы:
1 Аналитический сигнал. Предмет изучения аналитической химии.
2. Качественный анализ.
3 Количественный анализ.
Химические методы: гравиметрические, титриметрические
Инструментальные методы анализа (элетрохимические, хроматографические, оптические)
1. Химическая идентификация (качественный анализ) и измерения (количественный анализ) являются предметом специальной химической науки – аналитической химии. Практически все методы основаны на зависимости доступных измерению свойств веществ от их состава. При этом необходимо разработать способы регистрации свойства вещества –
78
аналитического сигнала. При количественном анализе величину аналитического сигнала переводят в единицы, характеризующие концентрацию компонентов. Измеряемыми свойствами могут быть: масса, объем, светопоглощение и др. При качественном анализе обычно фиксируют появление аналитического сигнала – образование окрашенного осадка, изменение окраски растворов и т.д. Таким образом, для получения аналитического сигнала в аналитической химии используют химические реакции (кислотно-основные, окислительно-восстановительные, комплексообразования, осаждения); разнообразные химические, физические, биологические свойства самих веществ.
В зависимости от массы или объема анализируемого образца метод разделения и определения иногда подразделяют на макро-, полумикро-, микро-, ультрамикро-, субмикро- и субультрамикрометоды (табл. 6).
Таблица 6. Классификация методов разделения и определения
|
Масса |
Объем |
|||||||||||||||
Наименование метода |
анализируемого |
раствора, мл |
|||||||||||||||
|
вещества, г |
|
|
|
|
|
|
|
|
||||||||
Макроанализ (грамм-метод) |
|
|
1-10 |
|
|
10–100 |
|||||||||||
Полумикроанализ (сантиграмм-метод) |
0,05-0,5 |
|
|
1-10 |
|
|
|||||||||||
Микроанализ (миллиграм-метод) |
10 |
3 |
-10 |
6 |
10 |
1 |
-10 |
4 |
|||||||||
|
|
|
|
|
|
|
|||||||||||
Ультрамикроанализ (микрограмм-метод) |
10 |
6 |
-10 |
9 |
10 |
4 |
-10 |
6 |
|||||||||
|
|
|
|
|
|
|
|||||||||||
Субмикроанализ (нанограмм-метод) |
10 |
9 |
-10 |
12 |
10 |
7 |
-10 |
10 |
|||||||||
|
|
|
|
|
|
|
|
|
|||||||||
Субультрамикроанализ (пикограмм- |
|
10 |
12 |
|
|
10 |
10 |
|
|||||||||
метод) |
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
2. Качественный анализ это установление вида и состояния фаз, молекул, атомов, ионов на основе сопоставления экспериментальных и соответствующих справочных данных для известных веществ.
Характеристиками эффективности метода анализа и аналитической реакции являются: предел обнаружения, чувствительность, избирательность (селективность).
Предел обнаружения (ПрО) – минимальная концентрация вещества которая может быть обнаружена данным методом с какой-то допустимой погрешностью ( сmin, P , где Р – доверительная вероятность). ПрО
химической реакции зависит от условий протекания реакции: кислотности среды, концентрации реагентов, присутствия посторонних веществ,
79
температуры, времени наблюдения. Для методов обнаружения применяют
реакции с пределом обнаружения 10 7 (0,1 мкг) в 1 мл раствора. Инструментальные методы позволяют обнаружить элементы в твердых
образцах с ПрО менее 10 15 г. Методом лазерной спектроскопии возможно обнаружение десятков отдельных атомов, с помощью электронного микроскопа можно обнаружить отдельные атомы.
Чувствительность характеризует измерение аналитического сигнала при изменении концентрации или количества вещества и выражается коэффициентом чувствительности (угол наклона градуировочного графика при линейной зависимости аналитического сигнала от концентрации определяемого вещества).
Избирательность. Различают специфические и избирательные (селективные) методы, реакции, реагенты. Специфическими называют те методы, реакции или реагенты, с помощью которых в данных условиях можно обнаружить только одно вещество; избирательными – методы, реакции и реагенты, позволяющие обнаружить небольшое число веществ.
3. Определение содержания (концентрации, массы) компонентов в анализируемом веществе называется количественным анализом. Его широко используют для изучения состава руд, металлов, неорганических и органических соединений, для определения токсичных веществ в воздухе, водоемах, почвах, в продуктах питания, различных материалах.
Все методы анализа можно разделить на две большие группы: химические и инструментальные. Методы классифицируют по измеряемому аналитическому сигналу. Если аналитический сигнал получают, используя химические реакции и измеряют его визуально – методы анализа называются химическими. Если получаемый аналитический сигнал фиксируют на физическом приборе – методы анализа называются инструментальными.
3.1. Химические методы подразделяются на гравиметрические и титриметрические.
Гравиметрией называют метод количественного анализа, заключающий в точном измерении массы определяемого компонента пробы, выделенного в виде соединения известного состава. Это наиболее точный из химических методов анализа, считается ‘эталонным.
Титриметрический метод основан на измерении количества реактива, расходуемого на реакцию с определяемым веществом. Способом измерения количества реактива может быть – измерение объема раствора точно
80
известной концентрации, израсходованного на реакции. В титриметрическом методе используются следующие типы химических реакций: кислотно-основное взаимодействие, реакции окисления-восстановления, реакции осаждения и комплексообразования. По типу реакций, лежащих в основе метода титриметрические методы классифицируют на 4 большие группы: кислотно-основное,окислительно-восстановительное, комплексонометрическое, осадительное титрование. В каждой из этих групп выделяют частные методы, связанные с применением того или иного титранта.
Титриметрические методы используются при химическом контроле за показателями качества теплоносителя в энергетике (определение щелочности, жесткости вод).
3.2. В современной практике анализа используют в основном инструментальные методы, обладающие многими достоинствами: быстротой анализа, высокой чувствительностью, возможностью одновременного определения нескольких компонентов, автоматизации и использования компьютерной обработки результатов анализа. Инструментальные методы делятся на две группы: физико-химические и физические. В группу физико-химических методов входят наиболее распространенные и широко применяемые в практике анализа методы: электрохимические, хроматографические, оптические.
Электрохимические методы анализа основаны на использовании ионообменных или электронообменных процессов, протекающих на поверхности электрода или в приэлектродном пространстве. Эти процессы могут быть равновесными или неравновесными в зависимости от условий эксперимента и давать информацию о скорости химических реакций, природе участвующих в них соединений, термодинамике. Аналитическим сигналом служит любой электрический параметр (потенциал, сила тока и др.) функционально связанный с составом и концентрацией раствора.
Электрохимические методы анализа играют важную роль в современной аналитической химии, поскольку характеризуются высокой чувствительностью, низкими пределами обнаружения, широким интервалом определяемых содержаний, простотой и невысокой стоимостью аппаратуры. К числу существенных достоинств электроаналитических методов относится широкий спектр их практического применения – от определения следов токсичных металлов в водах, атмосферных осадках, других объектах окружающей среды до идентификации и количественного определения сложных органических веществ (лекарственные препараты, белки, нуклеиновые кислоты, пестициды)..
81
Хроматографические методы анализа. Анализ основан на хроматографии позволяющей разделять двух-и многокомпонентные смеси газов, жидкостей и растворенных веществ методами сорбции в динамических условиях.
Оптические методы анализа основаны на измерении оптических свойств веществ и излучений, взаимодействия электромагнитного излучения с атомами или молекулами анализируемого вещества, вызывающего излучение, поглощение или отражение лучей.
На самостоятельное изучение выносятся потенциометрический, полярографический, эмиссионный спектральный, абсорбционный спектральный, спектрофотометрический и фотоколометрический методы.
Вопросы для контроля
Какой раздел химии имеет задачу идентификации веществ?
Что такое аналитический сигнал? Какие свойства веществ определяются при идентификации?
Сравните гравиметрический и титриметрический методы анализа.
Какие принципы лежат в основе потенциометрического и полярографического методов анализа?
В чем различие принципов эмиссионного спектрального и абсорбционного спектрального методов анализа?
В чем заключается разница в спектрофотометрии и фотоклориметрии?
Литература
Коровин, Н.В. Общая химия: Учеб.для технических направл. и спец. Вузов- 6-е изд., испр. – М.: Высш.шк., 2005. – 556 с
Будяк Е.В. Общая химия: учебно-методическое пособие [электронный ресурс]. 3-е изд., перераб. и доп.– СПб.: Издательство «Лань», 2011. - 384 с.
– Режим доступа: http://e.lanbook.com
Гибадуллина Х.В. Химия: методические указания к практическим занятиям. - Казань: КГЭУ, 2010. - 59 с.
Химия: программа, методические указания для самостоятельной работы студентов на основе балльно-рейтинговой системы обучения / сост. Д.Ф. Гайнутдинова. – Казань: КГЭУ, 2010. – 44 с.
Урядова Л.Ф., Чичирова Н.Д. Химия: учебно-практическое пособие. –
Казань: КГЭУ, 2001. – 200 с.
82
СОДЕРЖАНИЕ |
|
Введение |
3 |
Лекция 1. Предмет химии. Первые модели строения атома |
5 |
Лекция 2. Квантово-механическая модель атома водорода. Периодический закон. Периодическая система Д.И. Менделеева. Электронные формулы
элементов |
|
9 |
Лекция 3. |
Строение вещества. Химическая связь |
17 |
Лекция 4 Химическая термодинамика |
……24 |
|
Лекция 5. Химическая кинетика. Химическое равновесие |
31 |
|
Лекция 6. Растворы и другие дисперсные системы |
43 |
|
Лекция 7. Окислительно-восстановительные реакции. Электрохимические
процессы. Гальванический элемент и электролиз |
54 |
Лекция 8. Коррозия металлов. Защита металлов и сплавов от коррозии ……69 Лекция 9. Раздел Избранные вопросы химии. Аналитическая химия.
Качественный и количественный анализ |
78 |
Литература |
84 |
83
Лекции 1, 2
ОСНОВНЫЕ ПОНЯТИЯ И ЗАКОНЫ ХИМИИ
(4 час)
Цель:
Ознакомление с теоретическими представлениями и концепциями, со-ставляющими фундамент системы современных химических знаний. Обозна-чить роль химии для будущей профессиональной деятельности студентов, а именно при решении задач связанных с инженерной защитой объектов окру-жающей среды.
План:
Предмет и объект химии. Место химии в ряду естественнонаучных дисци-плин.
Материя, вещество, поле. Представление о фазах постоянного и перемен-ного химического состава.
Основы количественных расчетов в химии.
Стехиометрические законы химии.
Газовые законы химии. Уравнение Клапейрона-Менделеева.
Литература:
Коровин Н.В. Общая химия. – М.: ВШ, 2005, Введение, §§ 1-3, С.10-16.
Урядова Л.Ф., Чичирова Н.Д. Химия. – Казань.: КГЭУ, 2002. Ч.1, §§ 1.1-
1.3, С. 11-15.
Будяк Е.В. Общая химия: учебно-методическое пособие [электронный ре-сурс]. 3-е изд., перераб. и доп.– СПб.: Издательство «Лань», 2011. - 384 с. –
Режим доступа: http://e.lanbook.com.
Лекционный материал:
Современная химия – это система из многих химических наук: общей неорганической, аналитической, органической, физической химий, электро-химии и т.д.
Химия – наука, изучающая процессы превращения веществ, сопровож-дающиеся изменением состава и структуры, а также взаимные переходы между этими процессами и другими формами движения материи.
Предметом изучения химии как науки является химическая форма движения материи, а центральным объектом являются вещества и их пре-вращения.
Общая химия изучает теоретические представления и концепции, со-ставляющие фундамент всей системы химических знаний.
Основные понятия:
Материя есть объективная реальность, которая существует независимо от нас, вызывает в нас ощущения, ее можно изучать, характеризовать коли-чественно. Материя проявляет себя в форме вещества и поля.
Вещество – это форма материи, которая обладает массой покоя и со-стоит из элементарных частиц: протонов, нейтронов, электронов, мезонов и др.
Поле – форма материи, проявляющая себя энергией, а не массой. Поле
– это материальная среда, в которой осуществляется взаимодействие частиц. Масса – мера инерции материи, энергия – мера ее движения. Масса и
энергия неотделимы от материи, но они не эквивалентны и не превращаются друг в друга.
Закон сохранения массы и закон сохранения энергии – это первые за-коны сохранения, установленные в науке.
Закон сохранения массы веществ (Ломоносов, 1760 г.): суммарная мас-
са веществ, вступивших в реакцию, равна суммарной массе продуктов реак-ции. При этом до и после реакции сохраняются массы каждого сорта атомов (дополнение Гей-Люсссака).
Между массой и энергией существует взаимосвязь, количественно вы-ражаемая уравнением (соотношение Эйнштейна,1905 г): Е = m ∙ c2.
К микрочастицам относятся: элементарные частицы (протоны, нейтро-ны, электроны), атомные ядра, атомы, ионы, молекулы.
Молекула – наименьшая частица индивидуального вещества, способная к самостоятельному существованию и сохраняющая основные химические свойства вещества.
Атом – наименьшая частица химического элемента, сохраняющая все его химические свойства.
Химический элемент – это вид атомов с одинаковым зарядом ядер.
Относительная атомная (Аr) (молекулярная – Mr) масса показывает,
во сколько раз абсолютная масса атома (или молекулы) больше массы 1/12 абсолютной массы изотопа атома 12С.
Простое вещество образовано молекулами, состоящими из одинако-
вых атомов (He, Ar, Kr, O2, Cl2, N2, S8, P4 и т.д.).
Сложное вещество образуют молекулы, состоящие из атомов разных элементов (H2O, SO2, NH3,H2SO4, CH4, C2H5OH, и т.д.).
Дальтониды – химические соединения, состав которых не зависит от условий и метода получений (соединения постоянного состава).
Бертоллиды – химические соединения, включающие кристаллические фазы переменного состава, а также все другие химические соединения, при образовании которых нарушается закон постоянства состава.
Для соединений переменного состава, не имеющих молекулярной структуры, вместо молекулярной массы введена формульная масса – сумма атомных масс элементов в соединении, умноженная на фактический индекс каждого элемента в формуле химического соединения.
Для веществ с молекулярной структурой формульная масса совпадает с молекулярной массой.
Моль (ν) – это количество вещества системы, содержащей столько же структурных элементов (6,022 ∙ 1023), сколько содержится атомов в изотопе 12С массой 0,012 кг. Массу одного моля вещества называют молярной массой (М, г/моль).
Эквивалент (Э) – это реальная или условная частица, соответствующая одному иону водорода в кислотно-основных или ионообменных реакциях или одному электрону в окислительно-восстановительных реакциях.
Массу одного моля эквивалентов называют молярной массой эквива-
лентов вещества (Мэ, г/моль).
Число, обозначающее, какая доля от реальной частицы эквивалентна одному иону водорода или одному электрону, называют фактором эквива-
лентности (fэ).
Для окислительно-восстановительных реакций используют понятие «эквивалентное число» (Z), которое равно числу электронов, присоединен-ных одной молекулой окислителя или отданных одной молекулой восстано-вителя.
Стехиометрические законы химии:
Закон постоянства состава (Пруст, 1806 г.): для индивидуальных хи-мических соединений молекулярной структуры качественный и количе-ственный состав остается постоянным независимо от способа его получения.
Закон не соблюдается для веществ, имеющих разный изотопный состав и для бертоллидов.
Закон эквивалентов – вещества взаимодействуют в количествах прямо пропорциональных их молярным массам эквивалентов (или молярным объе-мам эквивалентов в случае газообразных веществ):
m1 / m2 = Mэ1 / Mэ2, m1 / m2 = Vэ1 / Vэ2.
Газовые законы. Закон Авогадро: в равных объемах разных газов при одинаковых условиях (давлении и температуре) содержится равное количе-ство молекул. Для расчетов важны следствия из закона Авогадро:
Один моль любого газа при нормальных условиях (н.у. – 0 °С и 1,0133 ∙ 105 Па) занимает объем Vм = 22,416 л (мольный объем).
2. Число, показывающее, во сколько раз данный газ тяжелее второго газа, называется относительной плотностью первого газа по второму (D = 1 / 2); по закону Авогадро массы (m) равных объемов двух различных газов в одинаковых условиях и их молекулярные массы (M) связаны соотношением: m1 / m2 = M1 / M2. Так как D = m1/m2, то D = m1/m2 = M1/M2, D = M1/M2,
отсюда M1 = D M2: молекулярная масса газа или пара равна произведению его плотности по отношению к любому другому газу на молекулярную массу последнего. Газом сравнения может быть любой газ, молекулярная масса М которого известна. Если сравнивают плотность по отношению к водороду, то M1 = D 2. Если газом сравнения является воздух, М которого равна 29, то формула примет вид M1 = D 29.
Объединенный газовый закон. Уравнение газового состояния:
P V / T = P0 V0 / T0,
где P, V, T – давление, объем и температура для 1 моль газа, соответственно, в условиях опыта и нормальных условиях (н.у.).
Для ν моль газа: P V / T = ν P0 V0 / T0. Если учесть, что ν = m / M, то
-
P V / T = (m / M) P0 V0 / T0 или P V = (m/M) R T
(1)
где Р – давление (Па), V – объем (м3), T – температура (К), m – масса (г), М – мольная масса (г/моль), R – газовая постоянная, равная 8,314 Дж/моль К. Если объем V выражен в литрах, то уравнение (1) примет вид:
-
P V = 1000(m/M) R T
(2)
Выражения (1) и (2) называют уравнением Клапейрона–Менделеева. Это объединенный газовый закон применительно к химии, используется при пересчетах от нормальных условий (н.у.) к реальным. Если газ собран над водой, вводится поправка на давление водяных паров (р) при данной темпе-ратуре; р – табличная величина. С учетом этой поправки уравнения примут вид:
(P - р) V / T = P0 V0 / T0 , (P - р) V / T = (m / M) P0 V0 / T0, (P - р) V = (m / M) R T
Расчеты с использованием газовых законов носят приближенный ха-рактер, так как выведены для идеальных газов.
Лекции 3, 4
СТРОЕНИЕ АТОМА. ПЕРИОДИЧЕСКИЙ ЗАКОН И
ПЕРИИОДИЧЕСКАЯ СИСТЕМА ХИМИЧЕСКИХ ЭЛЕМЕНТОВ Д.И. МЕНДЕЛЕЕВА.
(4 часа)
Цель:
Ознакомить студентов с современными представлениями и моделями описывающими строение атома, показать связь между электронным строением атомов и свойствами химических веществ, которые они образуют.
План:
Развитие представлений о строении атома (модель Резерфорда, закон Планка, теория Бора).
Квантово-механическая модель атома. Квантовые числа и атомные орбитали.
Правила построения электронной структуры атомов.
Периодическая система Д.И. Менделеева. Периодичность свойств элементов.
Литература:
Коровин Н.В. Общая химия. – М.: ВШ, 2005, Гл. 1, §§ 1.1-1.5, С.17-34.
Урядова Л.Ф., Чичирова Н.Д. Химия. – Казань.: КГЭУ, 2002. Ч.1, §§ 2.1-
2.4, С. 15-28.
Будяк Е.В. Общая химия: учебно-методическое пособие [электронный ресурс]. 3-е изд., перераб. и доп.– СПб.: Издательство «Лань», 2011. - 384 с.
– Режим доступа: http://e.lanbook.com.
Лекционный материал:
Атом – это электронейтральная микросистема, состоящая из положительно заряженного ядра и электронной оболочки, заряженной отрицательно.
Носителем положительного заряда ядра являются протоны. Их число определяет величину заряда ядра и совпадает с порядковым номером химического элемента.
Основные характеристики элементарных частиц, образующих атом – протона, нейтрона и электрона, приведены в таблице 1.
Таблица 1 Основные характеристики элементарных частиц, образующих атом
|
|
|
|
Масса покоя |
|
|
частица |
символ |
кг |
|
относительная |
заряд |
|
|
|
|
|
|
масса |
|
протон |
1 |
1,673 |
10 |
−27 |
1 |
+1 |
|
||||||
|
1р |
|
|
|
||
нейтрон |
01 n |
1,675 |
10−27 |
1 |
0 |
|
электрон |
ē |
9,109 |
10−31 |
0 |
−1 |
|
Масса электрона почти в 2000 раз меньше массы протона и нейтрона. Поэтому масса атома практически равна массе ядра – сумме масс протонов и нейтронов (нуклонов).
Важной характеристикой ядра является массовое число A, которое равно общему числу нуклонов – сумме протонов (Z) и нейтронов (N), входящих в состав ядра: A = Z + N.
Разновидности одного и того же химического элемента, отличающиеся массой атомов называются изотопами. Ядра атомов изотопов различаются числом нейтронов. Так, изотопами водорода являются атомы с массовыми числами соответственно 1, 2 и 3:
-
1H
(2H) 2D
(3H) 3T
11p (ē)
(11p + 01 n ) ē
(11 p + 2 01 n ) ē
протий
дейтерий
тритий
Протий и дейтерий – стабильные изотопы, тритий – радиоактивен. Превращение химических элементов осуществляется в результате
ядерных реакций. Состав ядра в результате химической реакции не изменяется.
Состояние электронов в поле ядра атома. Квантовые числа.
Современная теория строения атома основана на законах, описывающих движение микрочастиц. В 20-е годы XX века возник раздел физики, описывающий движение и взаимодействие микрочастиц, – квантовая (или волновая) механика. Она основывается на представлении о квантовании энергии, волновом характере движения микрочастиц и вероятностном методе описания микрообъектов, т.к. для микрочастиц невозможно
одновременно точно определить координаты местоположения и скорость движения (принцип неопределенности, В. Гейзенберг, 1927 г.).
Из-за этих особенностей используется термин «атомная орбиталь электрона», так как понятие «электронная орбита как траектория точного местоположения электрона» в квантовой механике не имеет смысла. Математическое описание движения электронов в атоме вокруг ядра в трехмерной системе координат дает уравнение Шредингера:
2ψ + 8π2m0/h2(E - En)ψ = 0;
здесь E – полная энергия системы, En – потенциальная энергия, m0 – масса движущегося электрона, ψ(x,y,z) – волновая функция, зависящая от пространственных координат электрона, 2 – оператор Лапласа (набла в
квадрате), показывающий, что необходимо найти вторые производные от функции ψ: 2 = 2/x2 + 2/y2 + 2/z2.
Основной характеристикой, определяющей состояние электрона в поле ядра атома, в квантовой механике является энергия. Энергия электрона принимает не любые, а лишь определенные дискретные (квантующиеся) значения. Для полной характеристики энергетического состояния каждого электрона в атоме разработана система четырех параметров, называемых квантовыми числами.
Главное квантовое число (n) с точки зрения волновых представлений об электроне определяет, насколько удалено данное электронное облако от ядра. Чем больше значение n, тем слабее связан электрон с ядром, тем на более высоком энергетическом уровне он находится и тем большей энергией он обладает. Главное квантовое число может принимать значения целых чисел от 1 до +∞, но для электронов в невозбужденных атомах, открытых до сих пор элементов, оно изменяется реально от 1 до 7. Энергетические уровни, для которых n=1,2,3,4,5,6,7, называются, соответственно, K, L, M, N, O, P, Q – уровнями, и характеризуют, практически, номер слоя электронов.
Орбитальное, или побочное, квантовое число (ℓ) характеризует различное энергетическое состояние электронов в пределах данного энергетического уровня, его значение определяет распределение электронов по подуровням данного уровня. С точки зрения волновых представлений орбитальное квантовое число ℓ характеризует форму электронного облака, пространственную область его наиболее вероятного нахождения. Для электронов, находящихся на энергетическом уровне с главным квантовым числом n, орбитальные квантовые числа ℓ могут принимать значения 0, 1, 2, 3…до (n − 1). Для s-электронов (ℓ = 0) характерна форма шара, для р-электронов – форма гантели, для d-электронов – четырехлопастного винта, для f-электронов эта форма еще сложнее.
Электроны, которым отвечают значения побочного квантового числа ℓ = 0,1,2 и 3, называются, соответственно, s-, p-, d- и f- электронами.
Электронное облако, форма которого определяется значением побочного квантового числа ℓ, а общий запас энергии – значением главного квантового числа n, называется атомной орбиталью (АО).
Магнитное квантовое число (mℓ) характеризует пространственное расположение орбиталей. Оно определяет ориентацию электронного облака относительно произвольно выбранных направлений (взаимно перпендикулярных осей x, y, z) или относительно внешнего магнитного поля. Магнитное квантовое число принимает целочисленные значения от − ℓ до + ℓ, включая и нуль, т.е. всего (2ℓ + 1) значений, которым отвечают (2ℓ + 1) энергетических состояний, возможных для электронов данного подуровня. Например, для f-электрона ℓ = 3, mℓ может иметь (2∙3 + 1) значений, а именно mℓ = −3, −2, −1, 0, +1, +2, +3.
Спиновое квантовое число (ms) характеризует внутреннее движение электрона, приводящее к возникновению у последнего собственного магнитного поля. Спиновое квантовое число может принимать только два значения: + ½ и −½ в зависимости от того, параллельно или антипараллельно ориентируется собственное магнитное поле электрона относительно магнитного поля, обусловленного вращением электрона вокруг ядра.
Два электрона с одинаковыми значениями n, ℓ, mℓ , но с различными значениями ms называют спаренными или неподеленной парой электронов
(↑↓).
Распределение электронов в атомах элементов определяется тремя основными положениями: принципом наименьшей энергии, принципом запрета Паули, а также правилом Гунда.
Согласно принципу наименьшей энергии: размещение электронов должно отвечать наибольшей связи их с ядром, т.е. электрон, прежде всего, занимает такие положения, при которых он будет обладать наименьшим запасом энергии.
Так как энергия электрона, в основном, определяется значением главного квантового числа n и, в меньшей степени, значением побочного числа ℓ, согласно правилам В.М. Клечковского:
1. По мере увеличения заряда ядра атома заполнение подуровней осуществляется в таком порядке, при котором электрон в первую очередь поступает на подуровень, для которого значение суммы (n + ℓ) оказывается меньшим.
2. Если сумма (n + ℓ) оказывается одинаковой для двух или трех подуровней, то электрон в первую очередь поступает на подуровень с меньшим значением главного квантового числа.
Например, энергия электрона на подуровне 4s (n + ℓ = 4 + 0 = 4) меньше, чем на 3d (n + ℓ = 3 + 2 = 5). На подуровне 5s (n + ℓ = 5 + 0 = 5) энергия меньше, чем на 4d (n + ℓ = 4 + 2 = 6); на 5p (n + ℓ = 5 + 1 = 6) энергия меньше, чем на 4f (n + ℓ = 4 + 3 = 7).
Согласно принципу запрета Паули (1925): в атоме не может быть двух электронов с одинаковыми значениями всех четырех квантовых чисел.
Паули установил, что максимальное количество электронов, которое может находиться на n-ном уровне, определяется по формуле x = 2n2. Так, на первом уровне может находиться не более двух электронов, на втором − 8, на третьем − 18, на четвертом − 32, на пятом − 50.
Третье теоретическое положение, которым руководствуются для выяснения электронной конфигурации атома – правило Гунда: электроны в пределах данного подуровня (s-, p-, d- или f-) располагаются так, чтобы сумма абсолютных значений спиновых квантовых чисел была максимальной.
Например, для 2p3 возможны два варианта распределения трех электронов по ячейкам:
-
↑↓
↑
∑ ms = +½ или ∑ ms = −½;
↑
↑
↑
∑ ms = +1½ или ∑ ms = −1½.
По правилу Гунда минимуму энергии будет отвечать второй вариант размещения электронов.
Состояние электронной оболочки можно выражать следующими
способами:
схема электронного строения электронная формула
Э ) ) ) ) … Э 1s22s22p63s23p63d104s2…
2 8 18 2
В зависимости от того, на какой подуровень поступил последний электрон при заполнении электронной оболочки атома, различают s-, p-, d-, f-элементы.
Периодический закон и периодическая система (ПС) химических элементов Д.И. Менделеева. Периодический закон – один из важнейших законов природы, открыт Д.И. Менделеевым (март 1869 г.). Менделеев сформулировал закон так: свойства простых тел, также формы и свойства
соединений элементов находятся в периодической зависимости от атомных масс элементов.
Современная формулировка периодического закона: свойства элементов, а также формы и свойства соединений этих элементов, находятся в периодической зависимости от величины заряда ядра их атомов.
Периодическая зависимость выражается периодическими кривыми, имеющими ряд максимумов и минимумов. Ни атомная масса, ни заряд ядра атомов при возрастании порядкового номера элементов в Периодической системе не изменяются периодически. Истинная причина периодичности свойств элементов и их соединений заключается в периодически возобновляющихся (повторяющихся) сходных электронных оболочках атомов при последовательном возрастании положительного заряда их ядер.
Период − это последовательность элементов, атомы которых имеют одинаковое число электронных слоев (энергетических уровней), это число равно номеру периода. Заканчивается каждый период 8-электронной оболочкой со структурой ns2np6.
В пределах одного периода у s- и p-элементов идет заполнение внешнего слоя, у d-элементов – предвнешнего, у f-элементов – третьего снаружи. Поэтому наиболее сильно отличаются по свойствам соседние s-(p-) элементы. У d-и f-элементов отличия в свойствах проявляются незначительно. d- и f-Элементы одного периода объединяются в семейства по 10 и 14 элементов, соответственно. Семейство 4f-элементов называют лантаноидами, семейство 5f-элементов называют актиноидами.
Группа – вертикальный ряд элементов, состоящий из двух подгрупп. Положение в группах s-и p-элементов определяется общим числом
электронов внешнего слоя. Эти элементы составляют главную подгруппу (подгруппа А). Положение в группах d-элементов определяется общим числом s –электронов внешнего и d – электронов предвнешнего слоев. d-Элементы образуют побочную, или подгруппу В. Таким образом, для элементов А и В групп существует соответствие между общей формулой валентных электронов и типом семейства.
Поведение простых веществ и химических соединений в различных реакциях зависит от размеров (радиусов) их атомов. Радиус атома – это расстояние от ядра атома до области наибольшей электронной плотности внешнего слоя.
Атомные радиусы в подгруппах периодической системы возрастают с увеличением порядкового номера элемента, поскольку увеличивается число электронных уровней (оболочек), а с ним и эффективный радиус атома, эта
закономерность сохраняется и для d-элементов. В периодах с увеличением порядкового номера радиусы атомов, как правило, уменьшаются. Объясняется это тем, что в пределах одного периода число электронных уровней не изменяется, в то время как заряд ядра увеличивается, и облака внешних электронов все с возрастающей силой притягиваются к ядру.
У переходных элементов, лантаноидов и актиноидов (т.е. у всех d- и f-элементов) радиус атома в пределах периода изменяется мало. Объясняется это тем, что нарастание заряда у атомов этих элементов в значительной степени компенсируется соответствующим увеличением отрицательного заряда в предвнешнем слое электронов и внешние электроны не испытывают притяжения со стороны ядра по мере усложнения структуры атома.
От размера атома или образованного им иона в неменьшей степени, чем от заряда ядра, зависит легкость, с которой этот атом или ион теряет или приобретает электроны. Критерием этой легкости являются энергия (потенциал) ионизации, сродство к электрону и электроотрицательность.
Энергией ионизации ( I ) называется количество энергии, необходимое для отрыва электрона от невозбужденного атома и превращения его в ион Э+:
Э0 + I = Э+ + ē.
Энергия ионизации атома сильно зависит от его электронной конфигурации. Завершенные слои проявляют повышенную устойчивость. Наименьшими значениями энергий ионизации обладают s-элементы первой группы (Li, Na, K). Энергии ионизации в пределах главных подгрупп с увеличением порядкового номера уменьшаются, а восстановительная способность (металличность) нейтральных атомов увеличивается. В периодах значение энергии ионизации, с возрастанием порядкового номера, увеличивается, а восстановительная способность нейтральных атомов уменьшается. Таким образом, среди s- и p-элементов наиболее сильные восстановители находятся слева внизу (лучший из них – франций), а наиболее слабые – справа вверху (особенно фтор, окислить который вообще не удается). В пределах декад переходных элементов (d-элементов), а также у лантаноидов и актиноидов (f- элементов) значение энергии ионизации с возрастанием порядкового номера постепенно увеличиваются, но незначительно, поскольку с возрастанием порядкового номера мало изменяются радиусы атома.
Сродством к электрону (F) называется энергия присоединения
электрона к нейтральному атому Э0 с превращением его в отрицательный ион Э−
Э0 + ē = Э− + F.
Чем больше сродство к электрону, тем выше окислительная способность (неметалличность) данного атома. Наибольшим сродством к электрону обладают р-элементы VII группы. Наименьшее сродство к электрону имеют атомы с конфигурацией …s2 (Be, Mg, Zn) и …s 2p6 (Ne, Ar, Kr). Это служит доказательством повышенной устойчивости указанных электронных конфигураций.
Понятие электроотрицательности (ЭО) позволяет оценить способность атома данного элемента к оттягиванию на себя электронной плотности по сравнению с другими элементами соединения. Эта способность зависит от энергии ионизации и его сродства к электрону. Мерой электроотрицательности, по Малликену, является полусумма его энергии ионизации и сродства к электрону:
ЭО = ½ (I + F).
Чем выше значение ЭО для данного элемента, тем более выражены его неметаллические свойства, тем больше способность его атомов притягивать облака связевых электронов и приобретать отрицательный эффективный заряд.
Из вышесказанного следует, что в периоде, вследствие увеличения заряда ядра, усиливается притяжение электронов ядром, что приводит к:
уменьшению радиуса атома,
увеличению энергии ионизации,
увеличению сродства к электрону,
увеличению электроотрицательности, −уменьшению металличности,
увеличению неметалличности.
группе (сверху вниз) увеличение заряда ядра и увеличение количества электронных слоев приводит к увеличению атомного радиуса и ослаблению силы притяжения ядром электронов, что приводит к:
уменьшению энергии ионизации,
уменьшению сродства к электрону,
уменьшению электротрицательности,
увеличению металличности,
уменьшению неметалличности.
Лекции 5,6
ХИМИЧЕСКАЯ СВЯЗЬ
(4 часа)
Цель:
Ознакомить студентов с видами и характеристиками химической свя-зи, с различными методами описания химической связи для предсказания свойств химических соединений и их реакционной способности.
План:
Химическая связь. Причины образования химической связи.
Виды и характеристики химической связи. Метод валентных связей (ВС). 3.Ковалентная связь. Механизмы образования ковалентной связи.
Гибридизация атомных орбиталей.
Метод молекулярных орбиталей (МО ЛКАО).
Межмолекулярные взаимодействия. Водородная связь.
Металлическая связь.
Литература:
Коровин Н.В. Общая химия. – М.: ВШ, 2005, Гл. 2, §§ 2.1-2.5, С.35-64.
Урядова Л.Ф., Чичирова Н.Д. Химия. – Казань.: КГЭУ, 2002. Ч.1, §§ 3.1-
3.16, С. 29-43.
3. Будяк Е.В. Общая химия: учебно-методическое пособие [электронный ре-сурс]. 3-е изд., перераб. и доп.– СПб.: Издательство «Лань», 2011. - 384 с. –
Режим доступа: http://e.lanbook.com.
Лекционный материал:
Химическая связь. При образовании устойчивой многоатомной системы
– молекулы – между атомами возникает химическая связь. Под химической связью понимают явление взаимодействия атомов, обусловленное перекры-ванием электронных облаков связывающихся частиц, которое сопровождает-ся уменьшением энергии системы, а уменьшение энергии системы приводит к увеличению ее устойчивости.
Энергия связи (Есвязи; кДж/моль) – мера ее прочности, определяется работой, необходимой для разрыва связей или тепловым эффектом при обра-зовании молекулы:
H + H → H2 + 435 кДж/моль или H2 → H + H – 435 кДж/моль.
Под длиной связи (ℓ; нм, Ǻ ) понимают расстояние между центрами ядер атома в молекуле или кристалле. Угол между воображаемыми лини-ями, проходящими через ядра химически связанных атомов, называется
валентным.
Химическая связь образуется в том случае, когда электроны оказыва-ются вблизи двух или нескольких ядер, благодаря взаимодействию электри-ческих полей, создаваемых электронами и ядрами атомов. Процесс этого вза-имодействия может протекать различным образом. Поэтому в настоящее время различают три основных типа химических связей: ковалентную, ион-ную и металлическую. Ионную связь можно рассматривать как крайний слу-чай полярной ковалентной связи.
Химическая связь, осуществляемая общими электронными парами, называется ковалентной. Различают две разновидности ковалентной связи: неполярную и полярную. В случае ковалентной неполярной связи электрон-ное облако, образованное общей электронной парой, распределяется в про-странстве симметрично относительно ядер обоих атомов (например, в моле-
кулах Н2, О2 , Cl2, N2, F2):
-
H∙ + ∙H → H : H
( H + H → H-H);
:Cl∙ + ∙Cl: → :Cl : Cl:
( Cl + Cl → Cl−Cl ).
В случае ковалентной полярной связи электронное облако смещено к атому с большей электроотрицательностью (∆ЭO < 1,7). Например, в моле-кулах HCl, CO, SO2, электронная пара связи смещается в сторону более элек-троотрицательного атома, у которого возникает частичный отрицательный заряд (δ-). У менее электроотрицательного атома возникает частичный поло-жительный заряд (δ+). Между центрами зарядов в молекуле возникает неко-торое расстояние. Такие молекулы называют дипольными или диполями, а смещение электронов связи называется поляризацией связи.
H∙ + ∙Cl → H : Cl ∆ЭO = ЭO(Cl) − ЭO(Н) = 3,0 − 2,1 = 0,9.
Для многоатомных молекул следует различать понятия «полярность связи» и «полярность молекулы» и их количественные характеристики «электрический момент диполя связи» и «электрический момент диполя мо-лекулы». Это обусловлено тем, что при наличии нескольких связей в молеку-лах вывод о полярности молекулы как целого делается на основании величи-ны результирующего электрического момента диполя. Его величину получа-ют путем суммирования векторов электрических моментов диполей отдель-ных связей (по правилу параллелограмма). Результирующий вектор может быть равен нулю (например, для высокосимметричных молекул CO2, BF3,
SF6), несмотря на значительную полярность отдельных связей. В решении вопроса о полярности молекул это подтверждает роль такого фактора, как геометрия молекул. Например, в молекулах CO2 (имеет линейное строение) и SO2 (имеет угловое строение) каждая связь ЭО является полярной, однако из-за особенностей геометрии молекул CO2 – неполярная молекула (результи-рующий электрический момент диполя равен нулю), а для SO2 эта характе-ристика отличается от нуля, подтверждая, что молекула – диполь.
Способ образования ковалентной связи, когда каждый из взаимодей-ствующих атомов предоставляет электроны для образования общих элек-тронных пар, называется обменным. Механизм образования ковалентной свя-зи за счет неподеленной электронной пары донора и свободной орбитали ак-цептора называется донорно-акцепторным. Например, при образовании ка-тиона аммония атом азота (донор) предоставляет готовую (неподеленную) электронную пару, а атом водорода (акцептор) – свободную орбиталь.
Ковалентная связь характеризуется направленностью и насыщаемо-стью. Насыщаемость обусловлена тем, что число перекрываний электрон-ных облаков ограничено числом неспаренных электронов. Поскольку элек-тронные облака направлены в пространстве (кроме s-), то и химические свя-зи, образованные с их участием, пространственно направлены.
Электронные облака имеют различную форму, их взаимное перекрыва-ние может осуществляться разными способами. Различают σ(сигма)- и π(пи)-связи.
σ−связь осуществляется при перекрывании атомных орбиталей вдоль линии соединения атомов.
π-связь осуществляется при перекрывании атомных орбиталей по обе стороны от линии соединения центров атомов. Степень перекрывания π-связи значительно меньше, чем σ-связи, и, следовательно, π-связь менее прочная, чем σ-связь. Одинарные связи образуются только за счет σ-связей, а кратные связи могут иметь и π-связи. Кратные связи – это ковалентные свя-зи, образованные более чем одной парой электронов. Увеличение кратности связи приводит к упрочению межатомной связи и к уменьшению длины свя-зи:
-
 Н3С−СН3
Н3С−СН3Есвязи(С−С) = 85 кДж/моль;
ℓ (С−С) = 1,543 Ǻ;
Н2С=СН2
Есвязи (С=С) = 145 кДж/моль;
ℓ (С=С) = 1,353 Ǻ;
НС≡СН Есвязи (С≡С) = 198 кДж/моль; ℓ (С≡С) = 1,205 Ǻ.
Для объяснения пространственного строения молекул используется представление о гибридизации валентных орбиталей в атоме в процессе обра-
зования им химических связей. Под гибридизацией понимают процесс пере-стройки неравноценных по форме и энергии электронных облаков, приводя-щий к образованию гибридных облаков, одинаковых по форме и энергии. При этом число гибридных орбиталей равно числу исходных.
Причины возникновения гибридизации. При гибридизации электронная энергия атома увеличивается (энергетически невыгодный процесс), но затра-та энергии компенсируется с избытком по следующим причинам:
Гибридные орбитали из-за своей большей вытянутости в направле-нии связей полнее и глубже перекрываются с орбиталями связываемого ато-ма, поэтому связи получаются более прочными и достигается бóльший выиг-рыш в энергии.
При возбуждении один из спаренных валентных электронов перехо-дит на другую гибридную орбиталь, то есть увеличивается ковалентность атома (количество неспаренных электронов) и, следовательно, количество химических связей (энергетически выгодный процесс).
В зависимости от числа участвующих в гибридизации орбиталей раз-
личают следующие типы: sp-, sp2-, sp3-, sp3d, sp3d2.
Основные положения метода валентных связей (ВС):
– образование химической связи в молекуле сопровождается уменьше-нием потенциальной энергии системы;
– принято, что молекула образуется из атомов;
– при образовании молекулы атомы сохраняют свою индивидуаль-ность, изменяется состояние только валентных электронов;
– ковалентная связь образуется между соседними атомами с образова-нием общих электронных пар из неспаренных электронов с антипараллель-ными спинами; механизмы – обменный, донорно-акцепторный, дативный;
– образуемые связи в молекуле 2-хцентровые 2-хэлектронные;
– количество образуемых связей определяется числом неспаренных валентных электронов у атомов с учетом возбужденного состояния;
– при образовании ковалентной связи происходит перекрывание элек-тронных облаков с противоположными направлениями спинов и между вза-имодействующими атомами увеличивается плотность электронного облака. Положительные ядра атомов притягиваются к этой области повышенного от-рицательного заряда, что приводит к уменьшению энергии системы;
– ковалентная связь образуется в направлении максимального пере-крывания электронных облаков взаимодействующих атомов;
– прочность связи зависит от степени перекрывания электронных обла-ков: чем она больше, тем выше концентрация отрицательного заряда, стяги-вающего положительные ядра атомов;
– для описания ковалентной связи с участием валентных неспаренных электронов разной формы и энергии метод ВС использует представление о гибридизации АО.
Метод молекулярных орбиталей. Метод молекулярных орбиталей
(МО) – это более общий метод описания ковалентной связи, позволяющий объяснить ряд явлений и фактов, необъяснимых с позиций метода ВС.
1. В методе МО принято, что молекула образуется из атомов, при этом изменяется состояние не только валентных, а всех электронов, которые рас-пределяются на новых, так называемых молекулярных орбиталях. Молеку-лярные орбитали – это местонахождение электронов с определенными зна-чениями энергий и форм, охватывающие не один атом (центр), а всю молеку-лу. Поэтому МО могут быть 2-хцентровыми (если молекула 2-хатомная) или многоцентровыми.
При образовании 2-хатомных молекул, МО рассматривается как ли-нейная комбинация АО. Поэтому метод называется – метод молекулярных орбиталей как линейная комбинация атомных орбиталей (метод МО ЛКАО).
Под линейной комбинацией атомных орбиталей понимаются два дей-ствия – сложение и вычитание волновых функций (ψ1 и ψ2), которые описы-вают состояние электронов двух взаимодействующих атомов. Образующиеся две новые волновые функции – это характеристики 2-х молекулярных орби-талей.
Одна из МО (волновая функция которой есть результат сложения ψ1 + ψ2) соответствует такому состоянию электронов в молекуле, при котором концентрация электронной плотности сосредоточена в пространстве между ядрами и способствует их стяжению. Такая МО получила название «связы-вающей»: (ψсв. = ψ1 + ψ2). Энергия связывающих МО всегда ниже, чем ис-ходных АО, поэтому размещение на них приводит к выигрышу энергии.
Вторая МО (волновая функция которой результат вычитания 1 – ψ2) соответствует такому состоянию электронов в молекуле, при котором кон-центрация электронной плотности сосредоточена в пространстве за ядрами и не способствует их стяжению. Такую МО называют «разрыхляющей»: (ψ* =
ψ1 – ψ2) или (ψразр. = ψ1 – ψ2). Энергия разрыхляющих МО всегда выше, чем исходных АО, поэтому размещение на них электронов не способствует устойчивости системы.
Третий тип МО – это несвязывающие МО, они не вносят вклад в обра-зование связи, т.к. состояние электронов на таких орбиталях не отличается от исходного (атомного) состояния. Поэтому размещение электронов на таких МО или удаление с них не влияет на прочность связи в молекуле.
2. При линейной комбинации атомных орбиталей количество образу-ющихся МО подчиняется правилу: сумма связывающих и разрыхляющих МО равна общему числу АО, из которых комбинируются МОсв. и МОразр., при этом число связывающих МО = числу разрыхляющих МО. По симмет-рии МО (как связывающие, так и разрыхляющие) могут быть - и -типа.
3. Для случая образования 2-хатомных молекул и ионов из атомов эле-ментов I периода ПС – (от каждого атома в комбинировании участвуют 1s-АО, в сумме – две АО) – разрешены следующие две МО: σ св.1s и σ*1s, энер-гия которых возрастает в ряду: Еσсв.1s Еσ*1s;
При образовании 2-х-атомных молекул и ионов из атомов элементов II периода ПС от каждого атома в комбинировании участвуют 1s-, 2s-, 2p-АО (всего 10 АО) – набор разрешенных десяти МО следующий: σсв.1 s, σ*1s,
σсв.2s, σ*2s, σсв.2px, πсв.2pz, πсв.2py, π*св.2pz, π*св.2pz, σ*2px.
Расположение МО в порядке возрастания их энергии определяется со-ставом молекулы:
– для двухатомных молекул из атомов элементов II периода ПС до азо-та (например, Be2, B2, C2) энергия МО возрастает в следующем порядке:
Еσсв.1s Еσ*1s Еσсв.2s Еσ*2s Еπсв.2pz = Еπсв.2py Еσсв.2px Еπ*2pz =
Еπ*2pz Еσ*2px.
– для молекул, образованных из атомов элементов II периода ПС от азота до фтора (N2, O2, F2), или молекулярных частиц, содержащих сильно ЭО элемент (CN–, CO, NO+ и т.п.), последовательность МО по возрастанию энергии отличается от предыдущей положением σсв.2px – МО (ее энергия становится ниже, чем у πсв.2pz и πсв.2py): Еσсв.1s Еσ*1s Еσсв.2s Еσ*2s
Еσсв.2px Еπсв.2pz = Еπсв.2py Еπ*2pz = Еπ*2pz Еσ*2px.
Оба ряда содержат МО одного типа с практически равными значения-ми энергии. Такие МО, как и АО, называются вырожденными.
Электроны заполняют МО в соответствии с такими же правилами, какие действуют при заполнении АО:
− от МО с меньшей энергией в порядке возрастания их энергии; − в соответствии с запретом Паули – на каждой МО – не более двух
электронов с противоположно направленными спинами; − на вырожденных МО электроны распределяются сначала по одному,
затем по второму электрону в соответствии с правилом Гунда.
Условием образования химической связи является выигрыш в энер-гии при образовании молекулы. Количественно на это указывает величина порядка связи n. Формула для расчета порядка связи:
n = (число электронов на всех МОсв. – число электронов на всех МО*) / 2. n может иметь целочисленные, дробные значения и 0. Если n = 0, выигрыша в энергии нет, молекулярная частица не может образоваться. Если n 0 (це-лое или дробное значение), молекулярная частица образуется и, следователь-но, есть выигрыш в энергии системы при переходе из атомарного в молеку-лярное состояние.
Образование химической связи по методу МО ЛКАО изображают с помощью энергетических диаграмм, а также электронными формулами, пе-редающими электронную конфигурацию молекул (подобно электронным формулам атомов).
Энергетические диаграммы или электронные формулы молекул со-держат информацию о магнитных свойствах молекулярных частиц: если на МО любого типа содержится один или больше неспаренных электронов, мо-лекулярная частица обладает магнитными свойствами, является парамагнит-ной, об этом же свидетельствует дробное значение величины порядка связи. При отсутствии у молекулярной частицы неспаренных электронов она не об-ладает магнитными свойствами и является диамагнитной.
Достоинством метода МО ЛКАО является возможность объяснить существование молекулярных частиц, содержащих на АО, предоставляемых для линейной комбинации, нечетного числа валентных электронов (напри-
мер, Н 2 , He 2 и т.п.). Метод ВС не объясняет их образования, т.к. не выпол-няется исходное условие метода, заключающееся в том, что все связи 2-хэлектронные.

Схема образования молекулярных орбиталей из атомов 2-го периода
а б
Диаграмма МО для молекул фтора (а) и кислорода (б)
Ионная связь – химическая связь между атомами, резко отличающими-ся по электроотрицательности (∆ЭO > 1,7). Взаимодействие сопровождается переносом электрона от менее электроотрицательного атома к более электро-отрицательному атому.
Na∙ + ∙Cl → Nа :Cl ( Na+Cl− ); ∆ЭО = ЭО(Cl) − ЭО(Na) = 3,0 – 0,9 = 2,1.
Атом, присоединивший электрон, становится отрицательно заряжен-ным ионом (Cl−) − анионом; атом, отдавший электрон, становится положи-тельно заряженным ионом (Na+) – катионом. Между ионами возникает элек-тростатическое взаимодействие с образованием нейтральных молекул. Ион является точечным электрическим зарядом, у которого, по закону Фарадея, электрическое поле по всем направлениям одинаково. Поэтому ионная связь обладает ненаправленностью и ненасыщаемостью, в отличие от ковалент-
ной связи.
Металлическая связь. Металлы характеризуются рядом особых свойств
– это тепло- и электропроводность, ковкость, пластичность, металлический блеск, отличающих металлы от соединений с ионной и ковалентной связями. Перечисленные отличия металлов обусловлены проявлением в них особого типа химической связи, получившей название металлической. При обычных условиях большинство металлов существует в кристаллическом состоянии, между частицами в узлах решетки реализуется металлическая связь, поэтому такие решетки называют металлическими.
Металлическая связь – химическая связь между атомами или ионами металла и относительно свободными общими валентными электронами («электронным газом»). В кристалле металла постоянно происходит обмен электронами потому, что у атомов много свободных валентных орбиталей и электроны слабо притягиваются ядрами атомов. Эти орбитали отдельных атомов перекрываются друг с другом, обеспечивая электронам способность свободно перемещаться между ядрами во всем объёме металла. В кристалли-ческой решетке металлов электроны обобществлены. Они непрерывно пере-мещаются между положительно заряженными ионами, которые расположены в узлах кристаллической решетки. При этом сравнительно небольшое число обобществленных электронов связывает большое число ионов.
Водородная связь – химическая связь между положительно поляризо-ванным атомом водорода одной молекулы и отрицательно поляризованным атомом с неподеленной электронной парой (F, O, N) другой молекулы. Эта связь является разновидностью донорно-акцепторного взаимодействия, кото-рая обуславливает ее направленность.
Энергия водородной связи составляет 8−40 кДж/моль, т.е. на порядок меньше энергии ковалентной связи, но этой энергии достаточно для ассоциа-ции молекул.
Ассоциированные молекулы воды при низких температурах образуют полимерные агрегаты. Такая ажурная пространственная структура характер-на для воды в твердом состоянии (льда).
Возникновение водородных связей приводит, например, к аномально за-вышенным температурам кипения веществ, изменению агрегатного состоя-ния и некоторых других характеристик, которые выпадают из общей законо-мерности для однотипных соединений.
Водородная связь может быть не только межмолекулярной, но и внут-римолекулярной. Особенно большую роль внутримолекулярная водородная связь играет в образовании ряда высокомолекулярных веществ, в частности белков и нуклеиновых кислот.
Лекции 7, 8
ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
(4 часа)
Цель:
Ознакомить студентов с основными понятиями и законами химической термодинамики. Ознакомить с методикой термохимических расчетов и уста-новления критериев возможности или невозможности самопроизвольного протекания химических процессов с целью практического применения теоре-тических знаний для управления химическими процессами
План:
Энергетика химических процессов. Основные понятия термодинамики.
Внутренняя энергия и энтальпия. Первый закон термодинамики.
Стандартные энтальпии образования веществ и стандартные энтальпии сгорания веществ.
4.Термохимические законы и термохимические расчеты.
Энтропия. Второй и третий законы термодинамики.
Свободная энергия Гиббса (или изобарно-изотермический потенциал). Направление и предел протекания химических процессов.
Энергия Гельмгольца (или изохорно-изотермический потенциал).
Литература:
Коровин Н.В. Общая химия. – М.: ВШ, 2005, Гл. 5, §§ 5.1-5.4, С.115-142.
Урядова Л.Ф., Чичирова Н.Д. Химия. – Казань.: КГЭУ, 2002. Ч.1, §§ 4.1-
4.10, С. 44-53.
Будяк Е.В. Общая химия: учебно-методическое пособие [электронный ре-сурс]. 3-е изд., перераб. и доп.– СПб.: Издательство «Лань», 2011. - 384 с. –
Режим доступа: http://e.lanbook.com.
Лекционный материал:
Каждое отдельное химическое вещество или их совокупность пред-ставляет собой термодинамическую систему. Если термодинамическая си-стема не обменивается с окружающей средой ни веществом, ни энергией, ее называют изолированной. Система, обменивающаяся с окружающей средой только энергией, называется закрытой. Если же возможен энергетический и материальный обмен – система открытая.
Состояние системы определяется термодинамическими параметрами состояния – температурой (Т), давлением (р), концентрацией (с), объемом
(V).
К важнейшим величинам, характеризующим химические системы, от-носятся внутренняя энергия U, энтальпия H, энтропия S и энергия Гиббса (изобарно-изотермический потенциал) G. Все эти величины представляют собой функции состояния, то есть зависят только от состояния системы, но не от способа, которым это состояние достигнуто.
Внутренняя энергия системы U складывается из энергии движения и взаимодействия молекул, энергии связи в молекулах, энергии движения и взаимодействия электронов и ядер.
Абсолютная величина внутренней энергии не может быть определена, но можно рассчитать ее изменение при переходе системы из начального со-стояния в конечное в результате осуществления химического процесса си-стема поглотила количество теплоты Q и совершила работу А, то изменение внутренней энергии U определяется уравнением:
U = Q − А.
Функции U, Q и А выражаются в Дж или кДж/моль.
В изобарно-изотермических условиях (р = const, Т = const), когда един-ственным видом работы является работа расширения газа изменение энталь-пии равно тепловому эффекту химической реакции Н. Если Н < 0, процесс идет с выделением теплоты в окружающую среду (экзотермическая реакция), если Н > 0, процесс идет с поглощением теплоты (эндотермическая реак-ция).
Тепловой эффект химического процесса, протекающего в условиях р, Т = const; А = р V и V = const, Т = const не зависит от пути его протекания, а зависит от природы и физического состояния исходных веществ и продуктов реакции (закон Гесса).
В основе термодинамических расчетов лежит следствие из закона Гес-са: тепловой эффект химической реакции равен сумме теплот образования продуктов реакции за вычетом суммы теплот образования исходных веществ с учетом стехиометрических коэффициентов реакции
Нхр = Σ fН0(прод. реак.) – Σ fН0(исх. вещ.).
Стандартные теплоты образования различных веществ при Т = 298 К ( fН0) приведены в таблицах термодинамических констант. Стандартные
теплоты образования простых веществ равны нулю. Энтальпия образования соединения – мера его термодинамической устойчивости.
В технических расчетах используют удельную теплоту сгорания Qт, которая равна количеству теплоты, выделяющейся при сгорании 1 кг жидко-го или твердого вещества и 1 м3 газообразного вещества до образования высших оксидов:
Qт = − Нсг ∙ 1000 / М
или
Qт = − Нсг ∙ 1000 / 22,4,
где М – масса 1 моль вещества; 22,4 л – объем 1 моль газа. Если расчет теп-лоты сгорания ведется с учетом образования жидкой воды, то удельная теп-лота сгорания называется высшей, а для реакции с образованием газообраз-ной воды – низшей.
Энтропия (S) является мерой неупорядоченности системы:
S = R ∙ lnW,
где R − молярная газовая постоянная, 8,31 Дж/(моль ∙ К); W - термодинамиче-ская вероятность состояния системы, то есть число микросостояний, с помо-щью которых осуществляется данное макросостояние. Единица измерения энтропии − Дж/(моль ∙ К). Энтропия увеличивается с ростом температуры и при понижении давления. Энтропия возрастает при переходе вещества из кристаллического состояния в жидкое и из жидкого в газообразное.
Изменение энтропии в процессе химической реакции можно рассчи-тать, используя следствие из закона Гесса:
Sхр = Σ S0(прод. реак.) – Σ S0(исх. вещ.).
Стандартные энтропии веществ (S0) при Т = 298 К приведены в табли-цах термодинамических величин.
Для изобарно-изотермических процессов энтальпийный и энтропийный факторы объединяются функцией, называемой энергией Гиббса (ΔG), кото-рая равна:
G = Н − T S.
Энергия Гиббса (изобарно-изотермический потенциал) является крите-рием, определяющим направление самопроизвольного протекания химиче-
ских процессов. Химическая реакция термодинамически возможна, если энергия Гиббса уменьшается, то есть G < 0.
Если G = 0, то Н = T S, что является условием равновесного состоя-ния системы, из которого ее можно вывести только путем внешнего воздей-ствия.
Стандартную энергию Гиббса реакции рассчитывают по следствию из закона Гесса:
Gхр = Σ fG0(прод. реак.) – Σ fG0(исх. вещ.).
Стандартные значения энергий Гиббса образования простых веществ равны нулю. fG0 веществ при Т = 298 К приведены в таблицах термодина-мических величин.
Если реагенты находятся в состояниях, отличных от стандартного, то изменение энергии Гиббса рассчитывается по уравнению, которое называет-ся изотерма Вант-Гоффа. Например, для реакции
aA + bB = dD + eЕ
записывается в виде
-
p d
pe
Gхр Gхр0
RT ln
D
E
,
p a
pb
A
B
где рD, pE, pA, pB – относительные парциальные давления соответствующих веществ.
Лекция 9
Тема: ХИМИЧЕСКОЕ РАВНОВЕСИЕ
(2 часа)
Цель:
Ознакомить студентов с основными понятиями и законами химическо-го равновесия. Рассмотреть влияние различных факторов на установление и смещение химического равновесия в обратимых реакциях.
План:
Кинетическое и термодинамическое условия равновесности систем.
Константа равновесия гомогенных и гетерогенных процессов.
Принцип Ле-Шателье, условия его применимости.
Литература:
1. Коровин Н.В. Общая химия. – М.: ВШ, 2005, Гл. 6, §§ 6.1-6.3, С.148-
165.
Урядова Л.Ф., Чичирова Н.Д. Химия. – Казань.: КГЭУ, 2002. Ч.1, §§
5.1-5.6, С. 54-59.
Заббарова Р.С. Общая и неорганическая химия. Ч.1. - Казань: КГЭУ, 2009. 152 с.
Будяк Е.В. Общая химия: учебно-методическое пособие [электрон-ный ресурс]. 3-е изд., перераб. и доп.– СПб.: Издательство «Лань», 2011. -
384 с. – Режим доступа: http://e.lanbook.com.
Лекционный материал [3]:
По признаку обратимости химические реакции делятся на необратимые и обратимые. При протекании химической реакции концентрации исходных веществ уменьшаются вследствие образования продуктов реакции. Если при этом исходные вещества расходуются практически полностью (их концен-трация уменьшается до нуля) с образованием продуктов реакции, реакцию называют идущей до конца или необратимой. Этому способствуют, напри-мер, образование газообразных продуктов, осадка трудно растворимых со-единений, слабых (мало диссоциирующих) электролитов типа воды, уксус-ной кислоты или прочных комплексов и др.
Большинство реакций не протекает до конца, они относятся к обрати-мым реакциям, то есть реакциям, одновременно протекающим в двух взаим-но противоположных направлениях ().
k1
Запись H2 + J2 2HJ
k2
означает, что реакция водорода с йодом (исходные вещества) с образованием йодоводорода (его концентрация в начальный момент была равна нулю), ко-торая записана слева направо (), называется прямой реакцией, ее
cкорость: = k1 СH2 СJ2. Графическое выражение этого процесса во време-ни – ниспадающая кривая до определенного уровня, так как, по мере накоп-ления HJ, начнет протекать обратная реакция (справа налево) () со скоро-стью:
= k2 С2HJ. Скорость этого процесса во времени растет и наступит момент времени, когда скорости прямого и обратного процессов будут равными:
= .
Состояние системы, в которой два взаимно протекающих процесса происходят с одинаковой скоростью, называется химическим равновесием.
Химическое равновесие является частным случаем динамического рав-новесия, которое характерно для многих физико-химических процессов, та-ких, как испарение – конденсация (фазовое равновесие), адсорбция – десорб-ция и т.д.
Кинетическое условие химического равновесия. Кинетическим услови-ем химического равновесия является равенство скоростей процессов обрати-мой реакции. После преобразований этого равенства получим соотношение
констант k1 / k2 = С2HJ / СH2 СJ2, где k1 и k2 – константы скорости прямой и обратной реакций, СHJ, СH2, СJ2 – концентрации компонентов обратимого
процесса в состоянии химического равновесия, которые называются равно-весными концентрациями; чтобы их отличать от текущих концентраций, вве-ли обозначение HJ, H2, J2.
Отношение констант k1 / k2 = Кс = HJ2 / H2 J2, где Кс – концентрацион-ная константа равновесия, показывает соотношение концентраций компонен-тов обратимого процесса в состоянии динамического равновесия.
Особенности при записи выражения константы равновесия. Для обра-
тимой реакции в общем виде aA + bB + … dD + eE + … концентрационная константа равновесия Кс = Dd Ee/ Aa Bb равна отношению, где чис-
литель – это произведение равновесных концентраций продуктов реакции в степенях, равных стехиометрическим коэффициентам, а знаменатель – это
произведение равновесных концентраций исходных веществ в соответству-ющих степенях.
В выражение концентрационной константы равновесия Кс гетероген-ной реакции, как и в выражение закона действия масс, входят только концен-трации веществ, находящихся в жидкой или газообразной фазе, так как кон-центрации твердых веществ остаются, как правило, постоянными. Например, для обратимой реакции
C(графит) + H2O(г) CO(г) + H2(г) выражение Кс = CO ∙ H2 / H2O не содер-жит концентрации твердого компонента графита.
Если обратимая реакция с участием газообразных веществ протекает при невысоких давлениях (условие подчинения газа законам идеальных га-зов), можно вместо концентраций использовать значения соответствующих парциальных давлений (p): Кp = pCO pH2/pH2O.
На величину константы равновесия влияют: природа исходных веществ и продуктов реакции, температура, давление (при определенных условиях). Изменения концентрации реагентов не влияют на ее величину, если сохра-няются неизменными условия реакции (Т, Р).
Константа равновесия – важная количественная характеристика обра-тимого процесса. Ее величина характеризует степень превращения исходных веществ в продукты реакции: чем больше константа равновесия, тем больше выход ее продуктов, т.е. на ее основании можно оценить теоретически воз-можный выход продуктов реакции.
Величину константы равновесия находят путем расчетов на основании экспериментальных данных.
Термодинамический критерий состояния химического равновесия – это равенство G = 0 (или H – TS = 0, или H = TS). Последнее равенство означает, что энтальпийная и энтропийная составляющие являются движу-щими силами двух одновременно протекающих реакций в химически обра-тимом процессе. Константа равновесия химической реакции Кс связана со стандартным изменением энергии Гиббса этой реакции G уравнением: G = – R ∙ T ln Кс или G = – 2,3R ∙ T lg Кс . При 298 К (25 С) это уравнение
имеет вид G298 = – 5,69 lg Кс(298), где G298 выражено в кДж/моль. Вывод из этих уравнений:
– отрицательное значение G означает, что равновесие смещено в направле-
нии прямой реакции и выход продуктов реакции сравнительно велик; это возможно, если lg Кс 0, т.е. Кс 1;
– при положительном знаке G равновесие смещено в сторону обратной ре-акции и выход продуктов прямой реакции мал; этому будет способствовать
lg Кс 0, т.е. Кс 1.
Состояние равновесия характеризуется неизменностью состава реакци-онной смеси во времени. Сместить положение равновесия можно изменени-ем температуры, концентраций реагентов, общим давлением. Смещение рав-новесия подчиняется принципу Ле Шателье: при всяком внешнем воздей-ствии на систему, находящуюся в состоянии химического равновесия, равно-весие смещается в таком направлении, которое приводит к ослаблению этого воздействия.
Основные выводы из принципа Ле Шателье:
Равновесие смещается вправо при увеличении концентраций исход-ных веществ и уменьшении концентраций продуктов реакции. Равновесие смещается влево при уменьшении концентрации исходных веществ и увели-чении концентрации продуктов реакции.
При повышении температуры равновесие смещается в сторону эндо-термической реакции, а при понижении – в сторону экзотермической реак-ции.
При увеличении давления равновесие смещается в сторону умень-шения объема, то есть уменьшения числа молей газа, а при уменьшении дав-ления – в сторону увеличения числа молей газа.
Лекция 10
ХИМИЧЕСКАЯ КИНЕТИКА
(2 часа)
Цель:
Ознакомить студентов с основными понятиями и законами химиче-ской кинетики, показать зависимость скорости химической реакции от усло-вий проведения процесса с целью предсказания возможности осуществления химических реакций, а также определения пределов их протекания.
План:
1.Скорость химических реакций и методы ее регулирования. 2. Закон действующих масс. Правило Вант-Гоффа. 3.Уравнение Аррениуса. Энергия активации.
4. Гомогенный и гетерогенный катализ.
Литература:
Коровин Н.В. Общая химия. – М.: ВШ, 2005, Гл. 7, §§ 7.1-7.5, С.166-201.
Урядова Л.Ф., Чичирова Н.Д. Химия. – Казань.: КГЭУ, 2002. Ч.1, §§ 6.1-
6.6, С. 60-65.
Будяк Е.В. Общая химия: учебно-методическое пособие [электронный ре-сурс]. 3-е изд., перераб. и доп.– СПб.: Издательство «Лань», 2011. - 384 с. –
Режим доступа: http://e.lanbook.com.
Лекционный материал:
Химическая кинетика изучает механизм превращений, и закономерно-сти протекания реакций во времени. Количественной характеристикой хими-ческой реакции является скорость реакции.
Скоростью химической реакции (v) называют изменение количества вещества (N) в единицу времени (t) в единице реакционного пространства (R). Различают среднюю скорость за какой-то промежуток времени между t1
и t2:
v
1 N
2N
1
R t2 t1
и мгновенную скорость, если промежуток времени бесконечно мал
v 1 dN ∙ R dT
Скорость гомогенной химической реакции – это изменение концентра-ции какого-либо из исходных веществ или продуктов реакции в единицу времени
v = + (dc / dt ),
где c – молярная концентрация реагента (моль/л).
Скорость реакции в СИ имеет единицу измерения [моль ∙ м−3 ∙ с−1], од-
нако используются и другие единицы измерения [моль ∙ л−1 ∙ с−1], [моль ∙ см−3 ∙ с−1], [моль ∙ см−3 ∙ мин−1].
Смысл химической реакции состоит в превращении исходных веществ
(А, В) или реагентов в продукты реакции (D, E): |
|
|
aA + bB |
dD + eЕ. |
(1) |
![]()
В ходе реакции концентрации исходных веществ уменьшаются, а про-дуктов реакции – увеличиваются. График зависимости концентрации от вре-мени называется кинетической кривой и представлен на рис. 1.

CD
СA
C2
C1
-
0
t
0
t1
t2
t
а
б
Рис. 1. Кинетические кривые: а – для исходного вещества А; б – для продукта реакции D.
Все реакции можно подразделить на простые и сложные. Простые ре-акции протекают в одну стадию и называются одностадийными (элементар-ными). Сложные реакции представляют собой многостадийный процесс.
В зависимости от числа исходных частиц, участвующих в элементар-ном акте, говорят о разной молекулярности реакции.
Молекулярностью реакции называют число молекул или других ча-стиц, одновременным взаимодействием которых осуществляется элементар-ный акт химического превращения; она может характеризоваться только це-лыми числами.
Если в элементарном акте реакции принимает участие одна молекула, превращающаяся в одну или несколько молекул других веществ, то такая ре-
акция называется одномолекулярной или мономолекулярной. Схемы реакций:
А → В или А → В + С.
Примером таких реакций может служить термический распад гидро-карбоната кальция или превращение тиоцианата аммония в тиомочевину:
Са(НСО3)2 → СаСО3 + Н2О + СО2,
NH4CNS
![]() (NH2)2CS.
(NH2)2CS.
Единовременным участием в элементарном акте химической реакции двух молекул характеризуются бимолекулярные реакции:
А + В → С,
2А → В.
Примерами бимолекулярных реакций могут быть взаимодействие мо-нооксида углерода и хлора с образованием фосгена, или взаимодействие йо-да и водорода с образованием йодоводорода:
СО + Cl2 = COCl2,
I2 + H2 = 2HI.
Реакции, элементарный акт которых сводится к одновременному столкновению трех молекул, называют тримолекулярными. Статистически этот акт мало вероятен. Реакции с молекулярностью выше трех в химической практике неизвестны.
На скорость химических реакций влияют следующие факторы: природа реагирующих веществ, концентрация реагентов, внешние условия: темпера-тура, давление, катализатор (ингибитор), излучения. В случае гетерогенной реакции реакционным пространством можно считать поверхность раздела фаз. Скорость гетерогенной реакции зависит от площади поверхности разде-ла фаз.
Формулировка основного закона кинетики или закона действующих масс может быть дана в следующем виде: скорость химической реакции пропорциональна произведению концентраций реагирующих веществ в сте-пенях их стехиометрических коэффициентов.
В общем виде для реакции (1) кинетическое уравнение записыва-ется следующим образом:
-
v = k ca(A) ∙ cb(B),
(2)
где v – скорость реакции; k – константа скорости реакции; c(A), c(B) – кон-центрации реагирующих веществ А и В, соответственно; а – порядок реакции по веществу А; b – порядок реакции по веществу В. Сумма порядков реакции по реагентам − есть общий порядок реакции:
a + b = n.
Уравнение (2) называется основным кинетическим уравнением хими-ческой реакции. Для элементарных реакций порядок и молекулярность сов-падают.
Скорость реакции первого порядка характеризуется кинетическим уравнением
v = k ∙ c.
Скорость реакции второго порядка для двух реагентов А и В подчиня-ется кинетическому уравнению
v = k ∙ cА ∙ сВ.
Если cА = сВ или реагируют одинаковые частицы, то
v = k ∙ c2.
Уравнение (2) отражает физический смысл константы скорости. При концентрации реагирующих веществ, равных 1 моль/л, константа скорости численно равна скорости данной реакции. Константа скорости реакции k не зависит от концентрации регентов, но зависит от их природы, температуры и присутствия катализатора. Размерность k зависит от порядка реакции и опре-деляется из анализа размерностей величин, входящих в основное кинетиче-ское уравнение.
Зависимость скорости реакции от температуры приближенно выража-ется правилом Вант-Гоффа:
|
|
|
|
|
kT |
|
T2 T1 |
, |
|
|
|
|
|
γ 10 |
|||
|
|
|
|
|
1 |
|||
|
|
|
|
kT |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
где |
k |
T1 |
и k |
- константы скорости при температурах Т1 и Т 2 , γ – темпера- |
||||
|
|
T2 |
|
|
|
|||
турный коэффициент Вант-Гоффа. |
|
|
|
|||||
|
|
Аналогично для скорости реакции справедливо выражение |
||||||
|
|
|
|
|
v2 = v1 ∙ γ |
Т /10, |
||
где v1, v2 –скорости реакции при температурах Т1 (начальная) и Т2 (конеч-
ная), Т = Т2 − Т1.
С повышением температуры скорость реакции увеличивается не столь-ко за счёт увеличения числа соударений, сколько из-за роста числа частиц с достаточно высокой энергией, так называемых активных частиц.
На основании экспериментальных данных С. Аррениус показал, что скорость и константа скорости возрастает с температурой по экспоненциаль-ному закону. Выведенная им зависимость называется уравнением Аррениуса:
k = k0 ∙ е –Eа/(RT),
где k0 – предэкспоненциальный множитель; R – универсальная газовая по-стоянная (8,314 Дж/(моль ∙ К)); Т – температура, К; Eа – энергия активации.
Энергетические изменения в реагирующей системе можно представить схемой (рис.2):
Е
А…В
Еа
А, В
Н АВ
Координата реакции
Рис. 2. Энергетическая диаграмма, иллюстрирующая изменение энергии в реакции: А + В = АВ.
На рис. 2. высшая точка отвечает состоянию активированного ком-плекса [A…B], когда реагирующие молекулы настолько сближены, что ста-новится возможным образование продуктов реакции. Энергия, необходимая для перехода вещества в состояние активированного комплекса, называется энергией активации Еа. Н – тепловой эффект реакции (в данном случае эк-зотермический).
Уравнение Аррениуса позволяет рассчитать константы скорости реак-ций при различных температурах, а также энергию активации по формулам:
E |
a |
|
R (T2 T1) |
ln |
k2 |
||||
|
|
|
|
||||||
|
|
T2 T1 |
|
k1 |
|||||
|
|
|
|
||||||
и |
|
|
|
|
|
|
|
|
|
ln k0 |
ln k |
Ea |
|
1 |
. |
||||
|
|
||||||||
|
|
|
|
R |
T |
||||
Катализ – явление ускорения скорости реакции под воздействием спе-цифических веществ, количество которых в ходе реакции не изменяется. Эти вещества называются катализаторами. Действие катализатора сводится к снижению энергии активации за счёт образования промежуточных нестойких соединений, которые в дальнейшем распадаются на продукты реакции с вы-делением катализатора в химически неизменном виде (рис.4). Согласно этой теории катализатор (К) сначала образует с одним из веществ промежуточное соединение (АК). Затем последнее реагирует с другим исходным веществом
восстановлением катализатора. Схематически это можно представить так:
+ В = АВ (протекает медленно).
Катализатор разбивает процесс на две стадии:
А + К = АК (протекает быстро);
АК + В = АВ + К (протекает быстро).
Освободившийся катализатор образует промежуточные соединения с новыми количествами одного из реагирующих веществ.
Различают гомогенный (однородный) и гетерогенный (неоднородный) катализ. При гомогенном катализе реагирующие вещества и катализатор об-разуют однофазную систему – газовую или жидкую. В этом случае между катализатором и реагирующими веществами отсутствует поверхность разде-ла.
Лекция 11
РАСТВОРЫ. ОБЩИЕ СВОЙСТВА РАСТВОРОВ НЕЭЛЕКТРОЛИТОВ
(2 часа)
Цель:
Ознакомить студентов с физико-химической природой процесса растворе-ния химических веществ. Обозначить актуальность рассматриваемых вопросов, а именно коллигативных свойств растворов неэлектролитов для решении задач свя-занных с инженерной защитой объектов окружающей среды.
План:
Растворы – общие понятия.
Растворимость.
Теплота растворения.
Произведение растворимости.
Идеальный раствор.
Общие (коллигативные) свойства растворов неэлектролитов.
Литература:
Коровин Н.В. Общая химия. – М.: ВШ, 2005, Гл. 8, §§ 8.1-8.3, С.204-210.
Урядова Л.Ф., Чичирова Н.Д. Химия. – Казань.: КГЭУ, 2002. Ч.1, §§ 7.1-8.4, С.
66-78.
Будяк Е.В. Общая химия: учебно-методическое пособие [электронный ресурс]. 3-е изд., перераб. и доп.– СПб.: Издательство «Лань», 2011. - 384 с. – Режим до-
ступа: http://e.lanbook.com.
Лекционный материал:
Растворы – это гомогенные системы, состоящие из двух и более компонен-тов и продуктов их взаимодействия.
Растворы классифицируют: по агрегатному состоянию на жидкие (вода + спирт; вода + уксусная кислота), твердые (сплавы металлов) и газообразные (воз-дух: кислород, азот и др. газы).
Для характеристики жидких растворов используют понятия растворитель и растворенное вещество. При образовании раствора из двух смешивающихся жидкостей растворителем называют ту жидкость, которая присутствует в боль-шем количестве. Если же раствор образуется путем растворения в жидкости ве-щества, находящегося в твердом или газообразном состоянии, то растворителем называют вещество, агрегатное состояние которого при образовании раствора не изменяется.
Понятия «растворитель» и «растворенное вещество» не применяют по от-ношению к твердым и газовым смесям, независимо от того, в каких соотношениях находятся в этих системах компоненты.
Растворимостью называют способность данного вещества растворяться в воде или ином растворителе. Растворимость зависит от природы растворенного вещества и растворителя, температуры, внешнего давления.
Количественно растворимость характеризуется концентрацией насыщенно-го раствора при определенной температуре и давлении, или числом граммов рас-творенного вещества, которое может растворяться в 100 г растворителя. Различа-
ют: легкорастворимые вещества (хорошо), растворимость которых при 200С пре-вышает 10г на 100г растворителя; труднорастворимые (малорастворимые) – ес-
ли эта растворимость в пределах 0,01 – 1,00г на 100г растворителя; практически нерастворимые – при растворимости менее 0,01г. Абсолютно не растворимых веществ нет.
Теплота растворения
Количество теплоты. Поглощающейся (или выделяющейся) при растворе-нии одного доля вещества, называется теплотой растворения этого вещества. Теп-лоты растворения имеет отрицательное значение. Если при растворении теплота поглощается, и положительное, если выделяется. Например, теплота растворения
(Q) NH4NO3 = - 26,4 кДж/моль, Q (KOH) = + 55,5 кДж/моль.
Растворение жидкостей друг в друге обычно сопровождается поглощением теплоты, в системах жидкость – жидкость наблюдается рост взаимной раствори-мости с увеличением температуры. Кроме того, для жидкостей справедливо пра-вило: подобное растворяется в подобном. Например: полярные жидкости воду и уксусную кислоту (спирт этиловый) можно смешивать в любых соотношениях. Но, вода - полярная молекула и бензол (С6Н6) – не полярная молекула не смеши-ваются, т.е. взаимно не растворимы.
Растворение в жидкостях газов почти всегда связано с выделением теплоты, чему отвечает снижение растворимости с ростом температуры
Растворение газа в жидкости (или в твердом веществе) сопровождается зна-чительным уменьшением объема системы. Поэтому растворимость газов в жидко-стях заметно возрастает по мере увеличения давления. Согласно закону Генри при постоянной температуре растворимость газа прямо пропорциональна его парци-альному давлению над раствором:
Сж = к∙Р, где Сж – растворимость газа в жидкости, к – коэффициент пропорциональ-
ности, Р – парциальное давление, которое производило бы имеющееся в смеси ко-
личество данного газа, если бы оно занимало при той же температуре весь объем занимаемый системой.
Закон Генри соблюдается относительно точно лишь в случае разбавленных растворов, при небольших парциальных давлениях газа и при отсутствии химиче-ского взаимодействия газа с растворителем. Природная вода – это не идеальный раствор, растворенные в воде соли, и другие вещества влияют на процесс раство-рения газов.
Растворимость газов в растворах электролитов и других веществ, склонных к гидратации, всегда меньше, чем в чистой воде. Русский естествоиспытатель, фи-зиолог И.М.Сеченов (1859) установил взаимосвязь между растворимостью газа в воде и растворимостью его в растворе электролита:
Сэл. = Сж∙℮к·С,
где Сэл. – растворимость газа в растворе электролита; Сж – растворимость га-за в воде; ℮ – основание натурального логарифма; С– концентрация электролита; к – константа Сеченова, значение которой зависит от природы газа, электролита и температуры.
В природной воде прежде всего растворяются газы атмосферы: O2, N2 и
CO2.

Рис.1. Растворимость газов атмосферы в природных водах
Растворимость кислорода в два раза больше растворимости азота, но из-за большего парциального давления (78 %) в природной (дождевой) воде азота рас-творено в два раза больше, чем кислорода. Минерализация воды приводит к уменьшению растворимости воздуха. Так, при 0 0С растворимость кислорода (чи-стого) составляет 49 мл/л, а морской воде только 15 мл/л.
Процесс растворения твердого вещества в жидкости начинается с отделения его молекул, атомов или ионов от поверхности кристаллов под влиянием их соб-ственных колебаний и молекул растворителя, а также за счет силового взаимодей-
ствия между всеми частицами. Далее отделившиеся от поверхности кристаллов частицы постепенно распределяются во всем объеме раствора за счет диффузии.
Раствор, для которого устанавливается равновесие между скоростью пере-хода частиц в раствор и скоростью их обратного осаждения на поверхности кри-сталлов, называют насыщенным.
При растворении в жидкостях твердых веществ с увеличением температуры растворимость большинства веществ увеличивается. Если твердое вещество при растворении не образует с растворителем прочных химических соединений, про-цесс растворения будет сопровождаться поглощением теплоты. Если же происхо-дит выделение теплоты, то это свидетельствует о том, что одновременно с рас-творение происходит взаимодействие растворителя с растворенным веществом в виде теплоты больше энергии чем её расходуется на разрушение кристаллической решетки. При растворении многих веществ их молекулы (или ионы) связываются с молекулами растворителя, образуя сольваты (в случае воды – гидраты). H2SO4∙H2O или H2SO4∙2H2O – моно и дигидрат.
После образования гидратов многие вещества при кристаллизации связы-вают воду образуя кристаллогидраты. Например:
Железный купорос (семиводный сульфат железа (II)) FeSO4 ∙ H2O Кристаллическая сода (десятиводный карбонат натрия) Na2CO3∙ 10H2O Гипс (двуводный сульфат кальция) CaSO4 ∙ 2 H2O
Бура (десятиводный тетраборат натрия) Na2B4O7∙ 10H2O.
Идеальный раствор
Общие свойства растворов зависят только от концентрации растворенных веществ и не зависят от природы веществ – это коллигативные свойства раство-ров.
Коллигативные свойства проявляются в идеальных растворах или разбав-ленных растворах, которые приближаются по свойствам к идеальным.
Идеальным называют раствор, в котором не происходят химические реак-ции между компонентами (растворителем и растворенным веществом), а силы межмолекулярного взаимодействия между компонентами одинаковы. Образова-ние идеальных растворов не сопровождается тепловым эффектом (∆Н=0) и каж-дый компонент ведет себя в растворе независимо от других компонентов.
Коллигативные свойства растворов
Общими или коллигативными свойствами растворов называются свойства растворов неэлектролитов, которые не зависят от природы растворенного веще-ства, а только от числа растворенных частиц в определенном количестве данного растворителя. Такими свойствами являются осмотическое давление, понижение
давления насыщенного пара над раствором, повышение температуры кипения и понижение температуры замерзания раствора.
Давление пара растворов. При данной температуре давление насыщенно-го пара над каждой жидкостью – величина постоянная. При растворении в жидко-сти какого-либо вещества давление насыщенного пара этой жидкости понижает-ся.
Таким образом, давление пара над раствором нелетучего вещества в раство-рителе всегда ниже давления пара над чистым растворителем при той же темпера-туре. Согласно закону Рауля относительное понижение давления пара раствори-теля над раствором равно мольной доле растворенного вещества и выражается соотношением:
-
p0 p
n
p0
N n
где po – давление пара над чистым растворителем; р – давление пара растворителя над раствором; n - количество растворенного вещества, моль; N – количество рас-творителя, моль.
Замерзание и кипение растворов. Индивидуальные вещества характери-зуются строго определенными температурами переходов из одного агрегатного состояния в другое. Так, вода при нормальном атмосферном давлении (101,3 кПа) кристаллизуется при 0 ºС и кипит при 100 ºС. Иначе обстоит дело с растворами. Присутствие растворенного вещества повышает температуру кипения и понижает температуру замерзания растворителя.
Рауль установил, что для разбавленных растворов неэлектролитов повыше-ние температуры кипения и понижение температуры замерзания пропорциональ-ны концентрации раствора:
tкип = КЕ ∙ сm,
где tкип – повышение температуры кипения раствора, то есть разность между температурами кипения раствора и чистого растворителя; сm – моляльная концен-трация раствора (моль/кг), КЕ – эбуллиоскопическая константа растворителя. Она численно равна повышению температуры кипения раствора, содержащего 1 моль растворенного вещества в 1 кг растворителя.
tзам = КК ∙ сm,
где tзам − понижение температуры замерзания раствора в сравнении с температу-рой замерзания чистого растворителя, КК – криоскопическая константа раствори-
теля. Она численно равна понижению температуры замерзания раствора, содер-жащего 1 моль растворенного вещества в 1 кг растворителя.
Константы КЕ и КК зависят только от природы растворителя, но не зависят от природы растворенного вещества.
Осмос. Осмотическое давление. В гомогенном растворе частицы раство-ренного вещества и растворителя равномерно распределяются по всему объему раствора за счет теплового движения. Если поместить в цилиндр концентриро-ванный раствор сахара, а поверх него осторожно налить чистую воду, то вначале сахар и вода будут распределены в объеме раствора не равномерно. Через некото-рое время вследствие теплового движения – диффузии сахар и вода равномерно распределятся по всему объему жидкости.
Вместо цилиндра используем U-образную трубку, снабженную внизу мем-браной (полупроницаемой перегородкой), через которую могут проникать только молекулы воды (растворителя), но не молекулы (ионы) растворенного вещества. Заполним правое колено водой (чистым растворителем), а левое – раствором са-харозы в воде так, чтобы уровни жидкости в обоих коленах были одинаковыми. Выравнивание концентраций осуществляется в одностороннем порядке, только за счет перехода части молекул воды из правой части прибора в левую, где концен-трация сахарозы выше. В итоге уровень жидкости в правом колене начнет сни-жаться, а в левом возрастать, пока не установится некоторая разность столбов, от-вечающая давлению, при котором односторонняя диффузия прекратится.
Процесс самопроизвольного перехода (диффузии) растворителя через полу-проницаемую перегородку из той части системы, где концентрация растворенного вещества ниже, в другую, где она выше, называется осмосом. Давление, которое нужно приложить к раствору, чтобы прекратить осмос, то есть проникновение в него через полупроницаемую перегородку чистого растворителя, называется ос-
мотическим давлением (росм).
Для растворов неэлектролитов зависимость осмотического давления от кон-центрации и температуры раствора выражается законом Вант-Гоффа: осмотиче-ское давление раствора равно тому давлению, которое производило бы раство-ренное вещество, если бы оно при той же температуре находилось в газообразном состоянии и занимало объем, равный объему раствора.
Математическое выражение осмотического закона Вант-Гоффа:
росм = см ∙ R ∙ Т,
где росм – осмотическое давление (Па); см – молярная концентрация (моль/л); R − универсальная газовая постоянная, равная 8,314 Дж/моль∙К; Т – абсолютная тем-пература раствора (К).
Лекция 12
СВОЙСТВА РАСТВОРОВ ЭЛЕКТРОЛИТОВ
(2 часа)
Цель:
Познакомить студентов с основными понятиями темы «Свойства растворов электролитов: классификацией веществ на сильные, слабые электролиты и неэлектролиты; особенностями их поведения в водном растворе. Обозначить актуальность рассматриваемых вопросов для будущей профессиональной деятельности студентов, а именно при решении задач связанных с инженерной защитой объектов гидросферы.
План:
Свойства растворов электролитов: основные понятия теории кислот и оснований С. Аррениуса, гидратная теория Д.И. Менделеева;
Количественные характеристики диссоциации электролитов (степень и константа диссоциации), закон разведения Оствальда; активность, коэффициент активности.
Диссоциация воды. Водородный (рН) и гидроксильный (рОН) показатели.
Гидролиз солей.
Литература:
Коровин Н.В. Общая химия. – М.: ВШ, 2005, Гл. 8, §§ 8.4-8.6, С.218-232.
Урядова Л.Ф., Чичирова Н.Д. Химия. – Казань.: КГЭУ, 2002. Ч.1, §§ 9.1-
9.10, С. 79-88.
Будяк Е.В. Общая химия: учебно-методическое пособие [электронный ресурс]. 3-е изд., перераб. и доп.– СПб.: Издательство «Лань», 2011. - 384 с.
– Режим доступа: http://e.lanbook.com.
Лекционный материал:
Электролитами называются вещества, водные растворы или расплавы которых проводят электрический ток.
Электролиты при растворении в воде диссоциируют, то есть распадаются на положительно и отрицательно заряженные ионы. Диссоциация – обратимый процесс, так как одновременно идет распад молекул на ионы и процесс соединения ионов в молекулы (моляризация).
Ионы в растворе связаны с молекулами воды, то есть гидратированы. Гидратация ионов – основная причина диссоциации. Гидратированные ионы могут включать как постоянное, так и переменное число молекул воды. При диссоциации в воде различных кислот в растворе образуются не свободные ионы водорода Н+, а гидратированные из которых наибольшей прочностью обладает ион гидроксония H3O+:
Н+ + H2O → H3O+.
Cильные и слабые электролиты. Количественно процесс электролитической диссоциации характеризуется степенью и константой диссоциации (табл. 5 и 6 Приложения).
Степенью диссоциации α электролита называется отношение числа молекул, распавшихся на ионы (n), к общему числу молекул электролита в растворе (N):
= ( n / N ) · 100 %.
зависимости от степени диссоциации все электролиты подразделяются на три группы: сильные, слабые и средней силы.
Сильные электролиты при растворении в воде практически полностью диссоциируют на ионы (α > 30 %). К ним относятся:
все растворимые соли (NaCl, CuSO4, MgCl2);
сильные кислоты (H2SO4, HNO3, HCl);
основания щелочных и щелочно-земельных металлов (NaOH,
Ba(OH)2).
Слабые электролиты при растворении диссоциируют лишь частично (α < 3 %). К ним относятся:
многие основания металлов (Cu(OH)2, Al(OH)3, Mg(OH)2, NH4OH);
некоторые соли (HgCl2, Fe(CNS)3, Hg(CN)2);
кислоты (CH3COOH, H2CO3, HNO2);
вода.
Электролиты средней силы имеют степень диссоциации от 3 до 30 %. К ним относятся: H3PO4, H2SO3, H2C2O4.
Степень диссоциации уменьшается с увеличением концентрации электролита, с повышением температуры. Диссоциация воды усиливается с повышением температуры.
Для сильных электролитов наряду с истинной концентрацией различают эффективную, или активную, концентрацию (активность). Для определения активной концентрации электролита необходимо знать
моляльность его раствора, ионную силу раствора и коэффициенты активности. Значения коэффициентов активности ионов электролита определяются ионной силой раствора.
В растворах слабых электролитов устанавливается равновесие между недиссоциированными молекулами и продуктами их диссоциации – ионами. Константа диссоциации – отношение произведения равновесных концентраций ионов электролита и равновесной концентрации недиссоциированных молекул его. Например, в водном растворе уксусной
кислоты устанавливается равновесие: |
CH3COO− + H+, |
CH3COOH |
к![]() онстанта
которого (константа диссоциации) связана
с концентрациями соответствующих частиц
соотношением:
онстанта
которого (константа диссоциации) связана
с концентрациями соответствующих частиц
соотношением:
K Д [CH3COO ] [H ] . [CH3COOH]
Константа диссоциации характеризует способность электролита диссоциировать на ионы, чем эта величина выше, тем больше ионов в растворе. Она не зависит от концентрации электролита, а зависит от природы электролита, природы растворителя и температуры.
Константа и степень диссоциации связаны соотношением, называемым законом разведения Оствальда:
2 с
К Д 1 М
Здесь см – молярная концентрация электролита, моль/л.
Для слабых электролитов степень диссоциации значительно меньше единицы. Тогда выражение закона разбавления упрощается:
К Д 2 сМ
откуда
-
К Д
∙
сМ
![]()
Многоосновные кислоты и основания диссоциируют обратимо, по ступеням. Например, угольная кислота:
первая ступень:
H2CO3 |
|
|
|
|
|
|
|
H+ + HCO3 |
−; |
||
|
|
|
|||||||||
|
|
|
|
|
|
||||||
вторая ступень: |
|
|
|
|
|
|
|
|
|
|
|
HCO3 |
− |
|
|
|
|
|
|
|
H+ + CO3 |
2−. |
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
||||
Д![]() ля
расчетов, связанных с диссоциацией
кислот удобно пользоваться не константой
К, а так называемым показателем константы
диссоциации рК, который определяется
соотношением:
ля
расчетов, связанных с диссоциацией
кислот удобно пользоваться не константой
К, а так называемым показателем константы
диссоциации рК, который определяется
соотношением:
рК = − lg K.
Очевидно, что с возрастанием К, то есть с увеличением силы кислоты, значение рК уменьшается, следовательно, чем больше рК, тем слабее кислота.
Ионные уравнения реакций. Все реакции в водных растворах электролитов являются реакциями между ионами. При написании ионных уравнений вещества слабодиссоциирующие, малорастворимые и газообразные следует записывать в виде молекул. Только сильные электролиты пишут в виде ионов. При этом пользуются таблицей растворимости. Например: уравнение в молекулярной форме:
CuCl2 + 2NaOH → Cu (OH)2↓+ 2NaCl,
полное ионное уравнение:
Cu2+ + 2Cl− + 2Na+ + 2OH− → Fe(OH)2↓+ 2Na+ + 2Cl−;
краткое ионное уравнение:
Cu2+ + 2OH− → Cu (OH)2↓.
Ионообменные реакции протекают без изменения степени окисления. Условиями необратимости ионообменных реакций являются
образование в результате реакции:
малорастворимого вещества (осадок);
газообразного вещества;
слабодиссоциирующего вещества (вода, комплексные соединения и др.).
Ионное произведение воды. Водородный показатель.
Вода – слабый электролит, который в незначительной степени
подвергается
диссоциации по схеме (без учёта гидратации
ионов Н+):
Н2О
![]()
![]() Н+
+ ОН−.
Н+
+ ОН−.
Применяя закон действующих масс можно записать выражение константы диссоциации:
КД = [H+] · [OH–] / [H2O].
Из экспериментальных данных при 25 ºС КД = 1,86 ∙ 10−16. Так как концентрация ионов воды очень мала, концентрацию недиссоциированной части молекул воды можно считать постоянной величиной. В 1 л воды массой 1000 г содержится 55,56 молей воды. Подставив эту величину в выражение КД, получим, так называемое, ионное произведение воды (Kв)
Kв = Кд · [H2O] = [H+] · [OH−] = 1,86 · 10–16 · 55,56 = 1·10–14.
С повышением температуры ионное произведение воды возрастает, так как процесс диссоциации воды эндотермический. Например, при 100 ºС Kв = 74 · 10–14. Ионное произведение воды даёт возможность вычислить концентрацию гидроксид-ионов для любого водного раствора, если известна
концентрация ионов водорода, и наоборот. |
|
|
||
Водородным |
показателем |
рН |
называется |
отрицательный |
десятичный логарифм концентрации |
ионов |
водорода: |
|
|
pH = – lg [H+].
Из уравнения электролитической диссоциации воды видно, что концентрации водородных и гидроксильных ионов в чистой воде равны [H+] = [OH–] = 10–7 моль/л, рН = 7 – среда нейтральная. Если [H+] > 10–7 , рН < 7
– среда кислая и при [H+] < 10–7 рН > 7 – среда щелочная.
Для расчета рН сильных кислот находят активность ионов водорода по формуле:
aH γH [H ] ,
тогда
pH lg aH ,
где [H+] – равновесная молярная концентрация Н+ с учетом полной диссоциации кислоты, γ Н – коэффициент активности ионов водорода,
определяемый по правилу ионной силы из табл. 7 Приложения. Для расчета рН щелочей используют соотношение
рКв = рН + рОН =14,
где
рКв = − lg Kв.
рОН щелочей определяют аналогично соответствующим параметрам для кислот:
pOH lg aOH .
При расчете рН слабого электролита (кислота или основание), как правило учитывают только первую ступень диссоциации.
Гидролиз солей
Водные растворы солей имеют разные значения рН и показывают различную реакцию среды – кислую, щелочную или нейтральную.
Например, водный раствор сульфата алюминия Al2(SO4)3 имеет кислую среду (рН < 7), раствор карбоната калия К2CO3 – щелочную среду (рН > 7), растворы хлорида натрия NaCl и ацетата аммония СН3СООNH4 – нейтральную среду (рН = 7). Эти соли не содержат в своем составе ионы водорода Н+ или гидроксид-ионы ОН−, которые определяют среду раствора. Изменение рН объясняется тем, что в водных растворах соли подвергаются гидролизу.
Гидролизом соли называется взаимодействие ионов соли с водой, в результате которого образуются слабые электролиты.
Например, раствор ацетата натрия CH3COONa имеет щелочную реакцию среды. Эта соль, как сильный электролит, при растворении в воде полностью диссоциирует на ионы Na+ и СН3СОО–, которые взаимодействуют с Н+ и ОН–-ионами воды. При этом ионы Na+ не могут связывать ионы ОН– в молекулы, так как NaOH является сильным электролитом и существует в растворе только в виде ионов. В то же время ацетат-ионы связывают ионы Н+ с образованием молекул слабого электролита – уксусной кислоты, в результате чего новые молекулы Н2О диссоциируют на Н+ и ОН−-ионы. Эти процессы протекают до тех пор, пока не установится равновесие:
СН3СОО− + Н+ |
|
|
|
|
|
|
СН3СООН, |
||||||
|
|
|
|
|
|||||||||
|
|
|
|
|
|
||||||||
Н2О |
|
|
|
|
|
|
|
Н+ + ОН–. |
|||||
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
||||||
Суммарное уравнение одновременно протекающих процессов имеет вид |
|||||||||||||
СН3СОО− + Н2О |
|
|
|
|
СН3СООН + ОН−. |
||||||||
|
|
||||||||||||
|
|
|
|
|
|
|
|||||||
Это уравнение показывает, что |
|
в результате образования слабого |
|||||||||||
![]()
![]()
электролита (уксусной кислоты) смещается ионное равновесие диссоциации воды и создается избыток ОН−-ионов, а раствор приобретает щелочную реакцию. Применив к данному обратимому процессу закон действующих масс получим:
КД = [CH3COOH] ∙ [OH−] / [CH3COO−] ∙ [H2O].
Концентрация воды в растворе соли постоянна, поэтому [Н2О] объединяем с КД:
КД ∙ [Н2О] = [СН3СООН] ∙ [ОН−] / [СН3СОО−] = КГ , где КГ – константа гидролиза. Чем больше КГ, тем сильнее соль подвергается гидролизу.
Гидролиз – процесс обратимый для многих солей. В состоянии равновесия только часть молекул соли гидролизуется. Количественно гидролиз характеризуется степенью гидролиза (β):
β = (сг / с0) · 100% ,
где сг – число молекул, подвергшихся гидролизу; с0 – исходная концентрация ионов соли, подвергшихся гидролизу.
Степень гидролиза можно выразить через ионное произведение воды КВ, концентрацию раствора см, константу диссоциации образующегося слабого электролита КД:

К
β В .
сМ К Д
Гидролиз усиливается при повышении температуры и разбавлении раствора. Подавить гидролиз можно сместить гидролитическое равновесие по принципу Ле Шателье. Если в результате гидролиза соли среда кислая, гидролиз подавляют введением в раствор кислоты, если среда щелочная – введением щелочи.
Любую соль можно представить как продукт взаимодействия кислоты с основанием. Гидролизу подвергаются только те соли, в состав которых входят ионы слабой кислоты или ионы слабого основания. Если ионы растворённой соли не взаимодействуют с ионами воды и не смещают равновесие ее диссоциации, то гидролизу соль не подвергается, при этом среда остается нейтральной.
Гидролизу не подвергаются соли, образованные сильными основаниями и сильными кислотами (NaCl, КNO3, Na2SO4, KMnO4 и др.). Например,
КNO3 + HOH КOH + HNO3.
В ионной форме это уравнение будет иметь вид:
К+ + NO3– + HOH К+ + OH– + H+ + NO3–,
или сокращенно
НОН
![]() Н+
+ ОН−,
рН = 7.
Н+
+ ОН−,
рН = 7.
Гидролизу подвергаются следующие типы солей:
1. Соли, образованные сильным основанием и слабой кислотой.
Гидролиз идет по аниону и обусловлен связыванием ионов водорода воды в слабую кислоту или анион кислой соли:
NaCN + HOH HCN + NaOH,
в ионном виде
Na+ + CN− + HOH HCN + Na+ + OH−,
CN− + HOH HCN + OH−, pH > 7.
2. Соли, образованные слабым основанием и сильной кислотой.
Гидролиз идет по катиону и обусловлен связыванием гидроксид-ионов воды в слабое с основание или катион основной соли. В растворе накапливаются ионы водорода, среда становится кислой. Например:
NH4Cl + HOH NH4OH + HCl,
в ионном виде
NH4+ + Cl− + HOH NH4OH + H+ + Cl−,
NH4+ + HOH NH4OH + H+, pH < 7.
3. Соли, образованные слабым основанием и слабой кислотой.
Гидролиз протекает сильнее, так как их ионы – катион и анион одновременно связывают водородные Н+ и гидроксильные ОН– ионы воды, сдвигая равновесие диссоциации её. Например:
CH3COONH4 + HOH CH3COOH + NH4OH, pH ≈ 7;
в ионной форме
CH3COO– + NH4+ + HOH CH3COOH + NH4OH.
При гидролизе солей, образованных слабым основанием и слабой кислотой, реакция среды может быть нейтральной, если образующиеся кислота и основание одинаковы по силе, – слабокислой, если кислота сильнее основания, – слабощелочной, если основание сильнее кислоты.
Соли подвергаются необратимому (полному) гидролизу если они образованные слабым нерастворимым или летучим основанием и слабой летучей или нерастворимой кислотой. Такие соли не могут существовать в водных растворах. К ним относятся: Al2S3, Cr2S3, Fe2(CO3)3, (NH4)2SiO3, Al(CN)3 и другие. Например, гидролиз сульфида алюминия Al2S3 протекает практически полностью до образования гидроксида алюминия Al(OH)3 и сероводорода H2S:
Al2S3 + 6HOH → 2Al(OH)3↓ + 3H2S↑.
Здесь продукты гидролиза уходят из сферы реакции в виде осадка Al(OH)3 и в виде газа H2S, равновесие полностью смещается слева направо.
Лекция 13
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ И
ЭЛЕКТРОХИМИЧЕСКИЕ ПРОЦЕССЫ
(2 часа)
Цель:
Ознакомить студентов с классификацией и природой ОВР, показать значение различных факторов на направление и полноту протекания элек-трохимических процессов, а также практическую значимость рассматривае-мых вопросов (ГЭ, ХИТ) для будущей профессиональной деятельности с сфере инженерной защиты окружающей среды.
План:
1.Окислительно-восстановительные реакции.
Электрохимические процессы и системы. Двойной электрический слой.
Электродные потенциалы. Уравнение Нернста.
Гальванические элементы и электродвижущие силы.
Литература:
Коровин Н.В. Общая химия. – М.: ВШ, 2005, Гл. 9, §§ 9.1-9.5, С.251-278.
Урядова Л.Ф., Чичирова Н.Д. Химия. – Казань.: КГЭУ, 2002. Ч.1, §§ 10.1-
11.4, С. 89-101.
Будяк Е.В. Общая химия: учебно-методическое пособие [электронный ре-сурс]. 3-е изд., перераб. и доп.– СПб.: Издательство «Лань», 2011. - 384 с. –
Режим доступа: http://e.lanbook.com.
Лекционный материал:
Металлы и сплавы во многом являются фундаментом цивилизации. По-вышение эффективности использования металлов и сплавов на их основе, а так же готовых изделий, является одной из главнейших задач современного производства.
В процессе производства тех или иных материалов, а также эксплуата-ции готовых изделий протекают различные химические процессы, связанные с окислением-восстановлением металл-компонентов материала. Причиной и инициатором окислительно-восстановительных процессов является не всегда и не только кислород воздуха, но и возникающие в самом материале окисли-тельно-восстановительные пары.
ОВР. Химические реакции классифицируются по различным призна-кам. Реакции, в ходе которых степени окисления атомов или ионов изменя-
ются, называются окислительно-восстановительными.
При переходе электронов от одних атомов к другим происходит изменение степени окисления. Под степенью окисления понимают услов-ный заряд атома элемента в соединении, который вычисляется из предполо-жения, что молекула состоит из ионов и её суммарный заряд равен нулю.
Процесс отдачи электронов называется окислением, а процесс присо-единения электронов – восстановлением. В реакциях эти два процесса протекают одновременно: одни частицы окисляются, другие – восстанавли-ваются. Вещества, присоединяющие электроны, называются окислителями, а вещества, отдающие электроны, – восстановителями.
Различают следующие типы окислительно-восстановительных реак-
ций:
Межмолекулярные реакции – реакции, которые идут с изменением
степеней окисления атомов в различных молекулах. Например: 2KMn+7O4 + 10KI–1 + 8H2SO4 = 2Mn+2SO4 + 5I20 + 6K2SO4 + 8H2O.
Внутримолекулярные реакции – реакции, в которых атомы, изменя-ющие свои степени окисления, находятся в одной молекуле. Например:
2KCl+5O3–2 = 2KCl–1 + 3O20.
Реакции диспропорционирования (самоокисления-самовосстановления)
– реакции, которые идут одновременно с увеличением и уменьшением степе-ни окисления атомов одного и того же элемента. Например:
Сl20 + HOH = HCl–1 + HCl+1O.
Эквивалентом окислителя называют такое массовое количество окислителя, которое соответствует одному присоединённому электрону в данной окислительно-восстановительной реакции. Чтобы определить экви-валент окислителя, надо его молярную массу разделить на число элек-тронов, присоединённых одной молекулой окислителя.
Эквивалентом восстановителя называют такое массовое количество восстановителя, которое соответствует одному отданному электрону в данной окислительно-восстановительной реакции. Для определения эквива-лента восстановителя надо его молярную массу разделить на число электро-нов, отданных одной молекулой восстановителя. В общем виде эквива-лент определяется по формуле:
Э = М / n ,
где Э – эквивалент окислителя или восстановителя, М – молярная масса окислителя или восстановителя, г/моль; n – число электронов, полученных
или отданных атомами или ионами каждой молекулы окислителя или вос-становителя.
Эквивалент окислителя (восстановителя) не является постоянной вели-чиной (как, например, молярная масса), а может принимать различные значе-ния в зависимости от реакции. Например, перманганат калия KMnO4 . В за-висимости от условий проведения реакции марганец восстанавливается в различной мере.
кислой среде процесс восстановления характеризуется следующим электронным уравнением: Mn+7 + 5ē = Mn+2. Эквивалент окислителя для этой реакции определяется: ЭMn = М/5. Влияние кислоты на характер про-цесса можно объяснить тем, что ионы Н+ связывают кислород иона MnO4¯ в малодиссоциированные молекулы воды.
нейтральной и слабощелочной средах процесс восстановления ха-рактеризуется следующим электронным уравнением: Mn+7 + 3ē = Mn+4. Эк-вивалент KMnO4 в этой реакции определяется: ЭMn = М/3.
сильнощелочной среде степень окисления марганца изменяется в соответствии с уравнением: Mn+7 + 1ē = Mn+6; эквивалент KMnO4 определя-
ется: ЭMn = M/1.
Электродные потенциалы. Гальванические элементы:
Переход ионов металла с электрода в раствор приводит к равновесию реакции окисления-восстановления. Этому равновесному состоянию отвеча-ет потенциал, называемый электродным потенциалом, величина которого за-висит от свойств металла, активности его ионов в растворе (приближённо концентрации [Mn+]) и температуры.
Величина
равновесного электродного потенциала
для различных элек-трохимических систем
рассчитывается по уравнению Нернста.
Для металли-ческого электрода (Mn+
+ nē
![]()
![]() M0)
имеет вид:
M0)
имеет вид:
EP(Mn+/ M0) = E0(Mn+/ M0) + (RT/nF) ∙ ln a(Mn+),
где E0(Mn+/ M0) – стандартный электродный потенциал, В; R – универсальная газовая постоянная, 8,314 Дж/моль∙К; F – постоянная Фарадея, 96487 (96500) Кл/моль;
Т – температура, К;
n – заряд иона или число электронов; a(Mn+) – активность ионов металла, моль/л
Подставим в формулу значения R, F, T = 298,15 K и, переведя нату-ральный логарифм в десятичный (коэффициент перевода 2,303), получим
8,314 ∙ 2,303∙298,15
EP(Mn+/ M0) = E0(Mn+/ M0) + ———————— ∙ lg a(Mn+) = n ∙96487
0,059
= E0(Mn+/ M0) + −−−−− ∙ lg a(Mn+). n
Если [Mn+] = 1 моль/л, то ЕР = Е0.
Абсолютное значение потенциала измерить невозможно. В связи с этим измеряют разность потенциалов между данным электродом и неко-торым электродом сравнения, потенциал которого условно принимают рав-ным нулю. В качестве стандартного электрода сравнения используют стан-дартный водородный электрод.
Электродные потенциалы, измеренные по отношению к водо-родному электроду в стандартных условиях, называются стандартными электродными потенциалами (Е0). В зависимости от величины и знака стандартного электродного потенциала все металлы можно расположить в ряд, который фактически представляет собой ряд активности .
На основании ряда стандартных электродных потенциалов можно сделать два практически важных заключения:
Металлы, обладающие более отрицательным электродным потенци-алом (более активные металлы), способны вытеснять металлы с менее от-рицательным электродным потенциалом (менее активные металлы) из вод-ных растворов солей.
Все металлы, имеющие отрицательный стандартный электродный потенциал, то есть находящиеся в электрохимическом ряду напряжений ме-
таллов выше водорода, способны вытеснять его из растворов кислот. Например, цинк (Е0 = –0,76 В) более сильный восстановитель, чем
медь (Е0 = +0,337 В) и, следовательно, будет вытеснять её из растворов,
содержащих ионы Cu2+:
Zn (T) + Cu2+(P-Р) = Cu (T) + Zn2+(P-Р).
Для водородного электрода уравнение Нернста имеет вид:
Е(Н+/Н2) = (RT/nF) ∙ ln (а2/р(Н2)),
где р(Н2) – относительное парциальное давление водорода (безразмерная ве-личина). С учётом того, что lg a = − pH, после подстановки постоянных для температуры 298 К, уравнение принимает вид:
Е(Н+/Н2) = − 0,02951 ∙ lg р(Н2) – 0,059 ∙ рН.
Для кислородного электрода (О2 + 4Н+ + 4ē 2Н2О) уравнение Нернста (после подстановки постоянных для температуры 298 К) имеет вид:
E(O2/H2O) = 1,23 + 0,0417 ∙ lg p(O2) – 0,059 ∙ рН.
Равновесие на простом редокси-электроде записывается уравнением:
Ox + nē Red,
где Ох – окисленная форма вещества; Red – восстановленная форма веще-ства. Уравнение Нернста для расчёта потенциала редокси-электрода имеет вид:
RT a(Ox) E(Ox/Red) = E0(Ox/Red) + ——— ∙ ln —‒‒—, nF a(Red)
где E0(Ox/Red) – стандартный потенциал.
Гальванический элемент представляет собой устройство, в котором химическая энергия окислительно-восстановительной реакции непосред-ственно превращается в электрическую энергию.
Непременное условие работы гальванического элемента – разделение единого окислительно-восстановительного процесса на процесс окисления и процесс восстановления, которые протекают на различных пространственно разделённых электродах. Электрод, на котором идёт процесс окисления, называют анодом. Электрод, на котором идёт процесс восстановления, назы-
вается катодом.
Реакция, поддерживающая протекающий во внешней цепи электриче-ский ток, называется токообразующей.
Электродвижущая сила (ЭДС) гальванического элемента (ЕЭ) равна разности потенциалов катода (ЕК) и анода (ЕА) при отсутствии тока в цепи:
ЕЭ = ЕК − ЕА.
Чем дальше в ряду стандартных электродных потенциалов находятся друг от друга два металла, тем больше ЭДС созданного ими гальванического элемента. Но какие бы металлы ни брали для создания электродов гальвани-ческого элемента, электродвижущая сила элемента всегда должна иметь по-ложительное значение.
Для электродов гальванического элемента можно использовать один металл. Возможно создание так называемого концентрационного гальваниче-ского элемента. В этом случае две пластины из одного и того же металла по-гружаются в растворы одного и того же электролита, но с различной актив-ностью потенциалопределяющих ионов. Токообразующим процессом в та-
ком элементе является процесс выравнивания активностей ионов у обоих электродов.
Стандартную ЭДС гальванического элемента можно рассчитать по из-вестным значениям стандартной энергии Гиббса реакции:
E0 = − ∆G0/nF.
Напряжение гальванического элемента меньше ЭДС из-за поляризации электродов и омических потерь:
U = EЭ – I ∙ r − ∆ EЭ,
где I – сила тока; r – омическое сопротивление; ∆EЭ – поляризация элемента, равная сумме поляризаций катода и анода.
С увеличением плотности тока возрастают поляризация и омическое падение напряжения. Таким образом, при увеличении плотности тока напря-жение элемента падает. По мере работы элемента (разряда) уменьшается концентрация исходных реагентов и увеличивается концентрация продуктов реакции, поэтому в соответствии с уравнением Нернста ЭДС элемента уменьшается. Кроме того, возрастает поляризация элемента. Поэтому при разряде элемента напряжение его постепенно падает.
Лекция 14
ЭЛЕКТРОЛИЗ. КОРРОЗИЯ И ЗАЩИТА МЕТАЛЛОВ
(2 ЧАС)
Цель:
Ознакомить студентов с электрохимическими процессами, протекающими на электродах при электролизе растворов и расплавов электролитов, с методами подбора способов защиты металлических конструкций и инженерных сооружений от коррозионных разрушений с использованием электрохимических процессов.
План:
Электролиз расплавов и растворов электролитов.
Законы Фарадея. Выход по току.
Основные виды коррозии.
Химическая и электрохимическая коррозия металлов.
Основные методы защиты от коррозии.
Литература:
Коровин Н.В. Общая химия. – М.: ВШ, 2005, Гл. 9, 10 §§ 9.6-9.8, §§ 10.1-10.4,
С.284-337.
Урядова Л.Ф., Чичирова Н.Д. Химия. – Казань.: КГЭУ, 2002. Ч.1, §§ 11.5-12.5,
С. 102-112.
Будяк Е.В. Общая химия: учебно-методическое пособие [электронный ресурс]. 3-е изд., перераб. и доп.– СПб.: Издательство «Лань», 2011. - 384 с. – Режим доступа: http://e.lanbook.com.
Лекционный материал:
Электролиз – это окислительно-восстановительный процесс, протекающий на электродах при прохождении постоянного электрического тока через раствор или расплав электролитов.
Для осуществления электролиза к отрицательному полюсу внешнего источника тока присоединяют катод, а к положительному полюсу – анод, после чего погружают их в электролизер с раствором или расплавом электролита. На поверхности катода ионы, молекулы или атомы присоединяют электроны, т.е. протекает реакция электрохимического восстановления. На аноде (положительном электроде) происходит отдача электронов, т.е. реакция окисления.
Таким образом, сущность электролиза: Катод (−) : восстановление;
Анод (+) : окисление.
Возможными анодными процессами при электролизе являются:
1. Растворение электрохимически активного металла (растворимый анод)
Ме0 → Меn+ + nē.
2. Разряд анионов из раствора или расплава электролита на инертном (нерастворимом) аноде.
Примером такого процесса может служить анодное выделение кислорода при электролизе воды:
4ОН− − 4ē → О2 + 2Н2О.
3. Перемена валентности ионов в сторону увеличения положительного заряда:
Fe2+ − 1ē → Fe3+, [Fe+2(CN)6]4− − 1ē → [Fe+3(CN)6]3−.
Из числа возможных анодных процессов в первую очередь идет процесс с наиболее отрицательным потенциалом.
Возможными катодными процессами при электролизе являются:
1. Разряд катионов металла или водорода: Меn+ + nē → Ме0,
2Н+ + 2ē → Н2.
2. Изменение валентности ионов, присутствующих в растворе, в сторону уменьшения их положительного заряда или увеличения отрицательного заряда:
Sn4+ + 2ē → Sn2+,
[Fe+3(CN)6]3− + 1ē → [Fe+2(CN)6]4−.
Из числа возможных катодных процессов, в первую очередь идет процесс с наиболее положительным потенциалом.
случае близости потенциалов катодных процессов, возможно их одновременное протекание.
результате электролиза выделяются соответствующие продукты восстановления и окисления, которые, в зависимости от условий, могут вступать в реакции с растворителем, материалом электрода и т.п., − так называемые вторичные процессы.
Окислительно-восстановительное действие электрического тока может быть во много раз сильнее действия химических окислителей и восстановителей. Меняя напряжение на электродах, можно создать почти любой силы окислители и восстановители, которыми являются электроды электролитической ванны или электролизёра.
Например, известно, что ни один самый сильный химический окислитель не может отнять у фторид-иона F− его электрон. Но это возможно при электролизе,
например, соли NaF (CaF2). В этом случае на катоде выделяется из ионного состояния металлический натрий или кальций:
Na+ + ē = Na0; (Ca2+ + 2ē = Ca0).
На аноде окисляется фторид-ион, переходя в свободное состояние:
F− − ē = F0; 2F0 → F20.
Для составления уравнений электролиза растворов с инертными (угольным, графитовым – С; платиновым − Pt) электродами необходимо знать следующие
правила: |
|
1. |
Если металл расположен в ряду напряжений металлов до Al│ |
включительно, то на катоде разряжаться будет Н+: |
|
Н+ + 1ē → Н0; 2Н0 = Н2↑. |
|
2. |
Если металл расположен в ряду напряжений металлов правее |
Н2│Меn+, то на катоде разряжаться будет сам металл: Меn+ + nē → Ме0.
3. Если металл расположен в ряду напряжений металлов правее Al│→, но левее│← Н2, то на катоде параллельно будут протекать два процесса:
Меn+ + nē → Ме0 и Н+ + 1ē → Н0; 2Н0 = Н2↑.
Li K Ba Sr Ca Na Mg Al |
Mn Zn Cr Fe Cd Co Ni Sn Pb H2Cu Hg Ag Pt Au |
|
→ |
→ |
← → |
Н+ +1ē → Н0, 2Н0 = Н2 |
Меn+ + nē → Ме0 |
Меn++ nē → Ме0 |
Н+ + 1ē → Н0; 2Н0 = Н2↑
4. Если анион расположен в ряду анионов до →│ОН−, то на аноде разряжаться будет сам анион:
Cl− − 1ē = Cl0; 2Cl0 = Cl2↑.
5. Если анион расположен в ряду анионов правее │→ ОН−, то на аноде разряжаться будет ОН−:
ОН− − 1ē = ОН0; 4ОН− = 2Н2О + О2↑.
Ряд анионов: S2− I− Br− Cl− →│ОН− SO42− NO3− CO32− PO43− F−
―――――――> химическая активность уменьшается.
Количественно соотношения при электролизе между выделившимся веществом и прошедшим через электролит электричеством выражаются законами Фарадея.
1 закон Фарадея. Количества веществ выделяющихся на электродах при электролизе, прямо пропорциональны количеству электричества, прошедшего через электролит. Количество вещества, выделяющегося при прохождении через электролит 1 кулона электричества, называется электрохимическим эквивалентом.
2 закон Фарадея. Количества веществ, выделяющихся на электродах при прохождении одинакового количества электричества, прямо пропорциональны их химическим эквивалентам. Следовательно, для восстановления на катоде и окисления на аноде одного грамм-эквивалент любого вещества необходимо затратить одно и то же количество электричества, а именно 96487 кулонов. Эта константа называется числом Фарадея (F). Из законов Фарадея вытекает математическое выражение:
MЭ ∙ Q VЭ∙ Q
m = -----------, V = -----------,
F F
где m – масса окисленного или восстановленного вещества, МЭ – молярная масса эквивалента вещества, г/моль,
VЭ – объём молярной массы эквивалента газа,
Q – количество прошедшего через систему электричества, Кл(А ∙ с); F – постоянная Фарадея, 96 487 (96 500) Кл/моль.
Или:
m = (MЭ ∙ I ∙ t) / F = (MЭ ∙ I ∙ t) / 96 487; V = (VЭ ∙ I ∙ t) / F,
где МЭ – масса моль-эквивалента вещества, г/моль; VЭ – объём молярной массы эквивалента газа;
I – сила тока, А;
t – продолжительность электролиза, с.
При протекании на электроде нескольких реакций на превращение вещества тратится определённая доля количества электричества (В), называемая выходом вещества по току, и определяемая из выражения:
В = Qi / Q,
где Qi – количество электричества, израсходованное на превращение i-го вещества.
Расход энергии W ((Вт∙ч)/кг) на превращение при электролизе 1 кг вещества i, рассчитывают по формуле:
W = (F ∙ U ∙ 103) / MЭ ∙ Bi ,
где U – напряжение на электролизёре, В.
Коррозия – это самопроизвольный процесс разрушения металлов и сплавов в результате химического, электрохимического и биологического взаимодействия их окружающей средой.
Коррозионные процессы являются окислительно-восстановительными и протекают, как правило, на границе раздела фаз твёрдого вещества с газом или жидкостью, т.е. взаимодействие происходит по гетерогенному механизму.
Коррозионные процессы классифицируют по:
механизму процесса:
химическая коррозия: электрохимическая коррозия; биологическая коррозия
условиям протекания:
газовая коррозия; коррозия в жидкостях-неэлектролитах;
коррозия в водных растворах электролитов (солевая, кислотная, щелочная и
т.п.);
атмосферная коррозия; аэрационная коррозия; подземная коррозия;
коррозия под действием блуждающих токов; коррозия под напряжением;
- характеру коррозионных поражений:
равномерная; неравномерная; структурно-избирательная; пятнами; язвами; межкристаллитная; транскристаллитная; подповерхностная.
Химическая коррозия − это самопроизвольное разрушение металлов под действием газов и органических жидкостей-неэлектролитов, не проводящих электрический ток.
Электрохимическая коррозия протекает при контакте металла с электропроводящей средой (водой, водными растворами солей, кислот, щелочей; расплавленными солями и щелочами). Разрушение металла происходит в результате образования на поверхности металла гальванических пар. Причиной образования гальванических пар является наличие в металле примесей других металлов или неметаллов. В гальванических парах анодом является металл, а катодом − примеси. В такой системе анод (основной металл) растворяется (окисляется), а катод восстанавливается.
На анодных участках происходит процесс окисления металла:
M0 – nē → Mn+.
На катодных участках – процесс восстановления окислителя: Ок + nē → продукт восстановления Ок.
Электроны по металлу перемещаются от анодных участков к катодным; внутренняя цепь коррозионного микрогальванического элемента замыкается
ионами, движущимися в растворе электролита (анионы перемещаются в растворе от катода к аноду).
Наиболее распространёнными окислителями при электрохимической коррозии являются молекулы кислорода и ионы водорода, имеющиеся в электролите.
Коррозия с участием кислорода в катодном процессе называется коррозией с поглощением кислорода или коррозией с кислородной деполяризацией. Реакция на катодных участках микрогальванических элементов можно представить следующими уравнениями:
в нейтральной и щелочной средах
O2 + 2H2O + 4ē → 4OH−,
в кислой среде
O2 + 4H+ + 4ē → 2H2O.
Схему работы микрогальванических элементов в данном случае можно записать следующим образом:
ē → ┌──────────┐┌─────┐
A (−) M0/Mn+ | (катодные участки) | O2/OH− (+) K.
![]() анионы
Если электроны с катодных участков
снимаются ионами водорода, то
анионы
Если электроны с катодных участков
снимаются ионами водорода, то
процесс называют коррозией с выделением водорода или коррозией с водородной деполяризацией:
2H+ + 2ē → H2.
Схема работы микрогальванических элементов: ē →
┌──────────┐┌─────┐
A (−) M0/Mn+ | (катодные участки) | H+/H2 (+) K. анионы
Электрохимическая коррозия протекает в местах:
контакта двух разных металлов;
контакта металла с продуктом коррозии;
разной концентрации окислителя на поверхности металла;
механических напряжений, где происходит изменение потенциала металлов.
Защита металлов от коррозии осуществляется различными методами. Выбор того или иного метода определяется его эффективностью в данном конкретном случае, экономической целесообразностью, а также назначением изделия. Все многочисленные методы защиты металлов от коррозии можно объединить в три условные группы: изменение свойств коррозионной среды; изменение свойств корродирующего металла; защитные покрытия.
Основные методы защиты металлов от коррозии
Защитные |
Приготовление |
Электрохимические |
Добавление |
покрытия |
сплавов, |
методы защиты |
ингибиторов |
|
стойких |
|
|
|
к коррозии |
|
|
![]()
![]()
![]()
Неметаллические |
Части машин, |
Протекторная |
Анодные |
покрытия: |
инструменты и |
защита: |
замедлители: |
оксидирование, |
предметы быта |
используют анод – |
карбонат |
фосфатирование, |
изготавливают |
протектор (старые |
натрия, |
азотирование; |
из |
железные детали, |
фосфаты, |
плёнки из |
нержавеющей |
магниевые сплавы |
силикаты, |
высокополимерных |
стали и других |
и т.д.) с более |
хроматы, |
веществ (каучук, |
сплавов, |
отрицательным |
нитраты и др. |
пластмассы), лаки, |
стойких к |
потенциалом, чем |
Катодные |
краски, эмали. |
коррозии |
потенциал металла |
замедлители: |
Металлические |
|
защищаемой |
соли магния, |
покрытия |
|
конструкции. |
цинка, никеля и |
(катодные, |
|
Электрозащита: |
др. |
анодные): |
|
защищаемая |
Ингибиторы |
никелирование, |
|
конструкция |
работающие в |
хромирование, |
|
присоединяется к |
кислотах: |
меднение и т.д. |
|
катоду внешнего |
тиомочевина, |
|
|
источника |
уротропин, |
|
|
электричества. |
производные |
|
|
|
аминов и др. |
Лекция № 15
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
(2 час)
Цель: Ознакомление с явлением комплексообразования, с основными по-ложениями координационной теории, с применением комплексных соединений в электро- и теплоэнергетике для решения экологических и технических проблем.
План:
Комплексные соединения. Координационная теория Вернера.
Классификация и номенклатура комплексных соединений.
Поведение комплексных соединений в растворах.
Значение комплексных соединений в электро- и теплоэнергетике, биоло-гическая роль.
Литература:
1. Коровин Н.В. Общая химия. – М.: ВШ, 2005, В, §§ 3.3-3.4, С.71-80.
2. Будяк Е.В. Общая химия: учебно-методическое пособие [электронный ресурс]. 3-е изд., перераб. и доп.– СПб.: Издательство «Лань», 2011. - 384 с. – Режим до-ступа: http://e.lanbook.com.
Гибадуллина Х.В. Химия. Комплексные соединения: Метод. указания к прак-тическим и лабораторным занятиям. - Казань: КГЭУ, 2010.46 с.
Заббарова Р.С. Общая и неорганическая химия. Ч.2. - Казань: КГЭУ, 2010. С.93-
Лекционный материал:
Все многообразие неорганических соединений условно можно разделить на две категории: соединения первого порядка и соединения высшего порядка. К со-единениям первого порядка относятся оксиды, кислоты, соли (СuО, НВr, NaCl и др.). Соединения высшего порядка, или как их называют «комплексные соедине-ния», являются продуктами взаимодействия соединений первого порядка. Напри-мер:
HgI2 + 2KI → K2[HgI4],
Zn(OH)2 + 4NH3 → [Zn(NH3)4](OH)2.
Комплексные соединения были получены еще в середине XIX в. Как оказа-лось, простейшие валентно насыщенные бинарные вещества способны соединять-ся и давать более сложные вещества.
Основным критерием, определяющим принадлежность вещества к коорди-национным (комплексным) соединениям, является то, что их строение можно описать на основании представлений о почти неизменном катионном централь-ном образовании М, вокруг которого несколько различных лигандов L могут рас-полагаться в практически неограниченном числе сочетаний. В общем виде обра-зование координационного комплекса можно изобразить уравнением:
M + x (:L) → [ M(:L)x].
Суммарный заряд образовавшегося комплекса определяется зарядом катио-на М и суммой зарядов лигандов, что видно на примере образования
комплексов Pt2+: [Pt(NH3)4]2+, [Pt(NH3)3Cl]+, [Pt(NH3)2Cl2] , [Pt(NH3)Cl3]−, [PtCl4]2−.
Основные положения координационной теории:
В молекуле любого комплексного соединения один из ионов или атомов металла занимает центральное место и называется комплексообразователем (М).
Вокруг центрального иона или атома располагается, или координируется, некоторое число (х) отрицательно заряженных ионов-анионов или нейтральных молекул (:L), которые называются лигандами.
Комплексообразо-

ватель
-
Лиганды
[Cr(NH3)6] Cl3
Внешняя сфера
Координационное число
Внутренняя сфера
![]()
Центральный ион с расположенными вокруг него лигандами образует так называемую внутреннюю координационную сферу соединения.
Ионы, находящиеся за пределами координационной сферы, составляют внешнюю сферу комплекса.
Число, определяющее количество лигандов, окружающих центральный ион или комплексообразователь, называется координационным числом (КЧ).
Классифицировать комплексные соединения можно по нескольким призна-
кам.
По природе лигандов. Сюда относятся комплексные соединения, в со-став комплексного иона которых входят молекулы, содержащие атомы с донор-ной функцией. Если лиганд − вода, комплекс называется аквакомплексом:
[Co(H2O)6]SO4.
Если лигандом является аммиак – аммиакатом: [Cu(NH3)4]SO4.
Если в состав комплекса в качестве лигандов входят анионы различных кислот, то они называются ацидокомплексами: K4[Fe(CN)6].
Соединения, содержащие в качестве лигандов ОН-группы, называются гид-роксокомплексами: K3[Al(OH)6].
По принадлежности к кислотам, основаниям или солям. По этой клас-сификации различают комплексные основания [Ag(NH3)2]OH; комплексные кис-
лоты Н2[SiF6]; комплексные соли K2[HgI4], K4[Fe(CN)6].
По заряду комплексного иона. В зависимости от заряда внутренней сферы все комплексные соединения можно разделить на три категории:
1. Комплексы катионного характера – заряд комплексного иона положи-тельный, так как лигандами являются нейтральные молекулы. Например,
[Zn(NH3)4]Cl2 − [Zn(NH3)4]2+,
[Cr(H2O)6]Cl3 − [Cr(H2O)6]3+.
2. Комплексы анионного характера – комплексный ион имеет отрицатель-ный заряд вследствие координации вокруг комплексообразователя отрицатель-ных лигандов, суммарный заряд которых по абсолютной величине превышает за-ряд комплексообразователя. Например,
K2[HgI4] − [HgI4]2−,
K2[Zn(CN)4] − [Zn(CN)4]2−.
3. Комплексные соединения, у которых абсолютные величины зарядов ком-плексообразователя и лигандов равны. Такие комплексные ионы не имеют заряда и называются нейтральными. Например, [Pt(NH3)2Cl2], [Co(H2O)3Cl3], [Fe(CO)5].
Номенклатура комплексных соединений
Катионные комплексные соединения называют следующим образом:
Указывают анион внешней сферы.
Называют число лигандов на греческом языке: 2-ди, 3-три, 4-тетра, 5-пента, 6-гекса, 7-гепта, 8-окта.
Называют лиганд, причем аммиак называют аммин, воду – аква.
Если лигандов несколько, то в первую очередь называют аммиак, затем – воду.
Следующими называют лиганды-ионы, добавляя к ним окончание «о». Например, хлоро (Cl−), гидроксо (ОН−), оксо (О2−), родано (CNS−).
5. Называют комплексообразователь, используя его русское название, в скобках указывается его степень окисления. Например,
[Cr(H2O)6]Cl3 – хлорид гексааквахрома (III),
[Ag(NH3)2]NO3 – нитрат диамминсеребра, [Ag(NH3)2]OH – гидроксид диамминсеребра, [Co(En)3]2(SO4)3 – сульфат трис(этилендиамин) кобальта (III).
Анионные комплексные соединения называют следующим образом:
Указывают число лигандов на греческом языке.
Называют лиганд.
Называют комплексообразователь, используя латинское название элемен-та с добавлением окончания -ат.
Называют катион внешней сферы в родительном падеже. Например,
K2[PtCl4] – тетрахлороплатинат (II) калия,
K3[Fe(CN)6] – гексацианоферрат (III) калия.
Название нейтральных комплексов составляется из названий лигандов с указанием их количества и русского названия комплексообразователя в имени-тельном падеже. Все название комплексного соединения пишется слитно и в име-нительном падеже. Например,
[Pt(NH3)2Cl2] – дихлородиамминплатина, [Co(NH3)3Br3] – трибромотриамминкобальт, [Fe(CO)5] – пентакарбонилжелезо,
Соединения с комплексным катионом и анионом. Названия этих соедине-ний составляются по правилам построения названий комплексных катионов и анионов. Вначале в именительном падеже комплексный анион, затем в родитель-ном падеже – комплексный катион; названия аниона и катиона пишут отдельно. В формуле аниона указывают степень окисления центрального атома.
Например:
[Cu(En)2][PtIICl4] – тетрахлороплатинат(II) бис(этилендиамин)меди(II).
Состояние комплексных соединений в растворе
Комплексные соединения в большинстве случаев являются электролитами и в водных растворах подвергаются диссоциации.
Диссоциация комплексных ионов обычно происходит, подобно слабым электролитам, в незначительной степени и подчиняется закону действия масс.
Количественно охарактеризовать этот процесс можно с помощью константы диссоциации комплексного иона. Для равновесия:
[Zn(NH3)4]2+![]()
![]() Zn2+
+ 4NH3,
Zn2+
+ 4NH3,
КД = [Zn2+] [NH3]4 / [[Zn(NH3)]2+] = 4 ∙10−10.
Величина константы диссоциации характеризует прочность комплексного иона, поэтому ее иначе называют константой нестойкости (КН) комплексного иона. Чем бо-лее устойчив комплексный ион, тем меньшее значение имеет КН. Сравним константу нестойкости [Zn(NH3)4]2+ c константой нестойкости [Zn(CN)4]2−. Диссоциация [Zn(CN)4]2− протекает по схеме:
[Zn(CN)4]2− Zn2+ + 4CN−;
КН = [Zn2+] [СN−]4 / [[Zn(СN)4]2−] = 1,3 ∙10−17.
Из сопоставления констант нестойкости обоих комплексных ионов видно, что цианидный комплекс более прочный, чем аммиачный.
В некоторых справочниках вместо значений констант нестойкости приведе-ны их обратные величины, которые называют константой устойчивости КУ =
1/КН.
Значение комплексных соединений
Комплексные соединения широко используются при получении гальваниче-ских покрытий. При использовании растворов простых солей гальванические по-крытия получаются в крупнокристаллическом виде, имеют плохое сцепление и осыпаются. К тому же при осаждении некоторых металлов, например при покры-тии железа медью, частично протекает химическая реакция замещения
Fe0 + Cu2+ = Fe2+ + Cu0,
ухудшающая качество покрытия. При использовании растворов комплексов по-лучаются плотные мелкокристаллические гальванические покрытия.
Особенно часто для получения гальванических покрытий применяются ци-анидные электролиты, содержащие в растворе наряду с цианидами щелочных ме-таллов такие комплексные соли меди, цинка, золота и других металлов, как
K2[Cu(CN)3], Na2[Zn(CN)4], K[Au(CN)4].
Для омеднения применяется этаноламиновый электролит, содержащий ком-
плексные ионы [Cu(H2NCH2CH2OH)4]2+.
Большое значение имеет комплексообразование при защите металлов от коррозии. Наличие в растворе веществ, дающих с продуктами коррозии раство-римые или нерастворимые вещества, оказывает большое влияние на коррозион-ный процесс, ускоряя или замедляя его.
Проблема отмывки и пассивации теплоэнергетического оборудования, ра-ботающего как на органическом, так и ядерном топливе, является одной из акту-альных задач теплоэнергетики.
Основное назначение химической промывки теплоэнергетических устано-вок состоит в удалении отложений, представляющих собой водонерастворимые неорганические соединения, которые образуются в результате нагревания и выпа-ривания питательной воды, коррозии металла котла и вспомогательного оборудо-вания. По количеству преобладающих в них соединений отложения могут быть железоокисными, кальциевыми, магниевыми, медноокисными, силикатными и т.д.
Самостоятельной задачей теплоэнергетики является пассивация внутренних поверхностей теплоэнергетических установок, основная цель которой – предот-вратить или уменьшить коррозию конструкционных материалов оборудования.
Комплексное решение таких важнейших проблем, как отмывка активных и неактивных отложений, а также пассивация поверхностей теплоэнергетического оборудования, оказывается возможным с использованием комплексонов. Впервые комплексоны были применены в теплоэнергетике для умягчения воды, а затем их стали успешно использовать для отмывки различных по составу отложений, обра-зующихся на внутренних поверхностях нагрева оборудования, изготовленного как из углеродистой, так и аустенитной нержавеющей стали.
Загрязнение окружающей среды соединениями тяжелых металлов – рту-тью, свинцом, кадмием, хромом – приводит к тяжелым отравлениям. Ядовитость или токсичность таких соединений во многих случаях объясняется взаимодей-ствием катионов тяжелых металлов с бионеорганическими комплексами. В ре-зультате этого взаимодействия вытесняются биогенные металлы: железо, цинк, медь, что приводит к нарушению нормальной жизнедеятельности, начинается токсикоз.
Для лечения токсикозов и отравлений часто используются соединения, об-разующие прочные связи с ионами металлов. Такие лекарства называются анти-дотами. При отравлении цинком, кадмием, ртутью применяют трилон Б, называе-мый в медицине тетацином. Комплексонаты этих металлов с тетацином малоток-сичны, имеют большие константы устойчивости, в организме не разрушаются, легко выводятся через почки.