

1. Экстракция липидов из биологического материала.
Теоретическое введение. При экстракции липидов принимают во внимание то, что они способны не только к гидрофобным, взаимодействиям, но и к образованию водородных, электростатических и ковалентных связей (сложноэфирных, амидных, гликозидных). Относительно неполярные растворители (хлороформ, бензол, диэтиловый эфир) разрушают комплексы, образованные гидрофобными взаимодействиями в жировой ткани, комплексы альбумина с жирными кислотами. Полярные растворители (этанол, метанол) разрушают водородные и электростатические связи. Их применяют в смеси со слабополярными растворителями при экстракции липидов из плазматических мембран, митохондрий, эндоплазматического ретикулума. Липиды, находящиеся в комплексах, образованных ковалентными связями, растворителями не экстрагируются. Их можно выделить только после гидролиза комплекса слабыми растворами кислот или щелочей в органическом растворителе.
Наиболее распространенным методом экстракции липидов является метод Фолча. Экстракцию проводят смесью хлороформ—метанол (2:1) из расчета 20 частей, экстрагирующей смеси на одну часть ткани. Метод позволяет выделить 90—95% всех клеточных липидов. Смеси растворителей, содержащие спирт, экстрагируют также нелипидные вещества (сахара, аминокислоты, соли и т. д.). Для удаления нелипидных примесей, экстракт липидов промывают водой или слабыми солевыми растворами. Однако это приводит к частичной потере кислых липидов. Очистку экстракта можно провести также гельфильтрацией на сефадексе.
Липиды легко подвергаются окислению и гидролитической деградации. Чтобы затормозить эти процессы, экстракцию липидов проводят при комнатной температуре, применяя растворители, из которых предварительно удален кислород. Извлеченные липиды не упаривают досуха и не оставляют в упаренном виде на долгое время, а сразу растворяют. Экстракты липидов следует хранить в плотно закрытой посуде при -20°С и ниже в присутствии инертных газов. Можно применять антиоксиданты, например 2,6-ди-трет-бутил-n-крезол, который в концентрации 0,005% эффективно предотвращает окислительное расщепление ненасыщенных липидов.
Внимание! Все органические растворители: хлороформ, метанол, гексан, диэтиловый эфир — в той или иной степени ядовиты, поэтому работать с ними нужно под тягой и ни в коем случае не насасывать в пипетку ртом.
Диэтиловый эфир легко воспламеняется, а пары его в воздухе образуют опасные горючие смеси (летуч, кипит при 36°С), поэтому при работе с ним необходимо соблюдать осторожность!
При работе с липидами необходимо пользоваться стеклянной посудой с пришлифованными стеклянными пробками. Смазкой для шлифов пользоваться нельзя! Можно прикрывать колбы и пробирки алюминиевой фольгой.
2. Фракционирование липидов методом адсорбционной хроматографии.
Теоретическое введение. Наиболее эффективным и широко применяемым методом фракционирования сложных смесей липидов является хроматография. Главную роль при аналитическом фракционировании играет адсорбционная хроматография в тонком слое сорбента. Этот метод также применяется в препаративных целях, когда разделению подвергается небольшое количество липидов (50—300 мг). Если масса липидов превышает 300 мг, используют колоночную хроматографию, хотя по разделяющей способности и времени разделения этот метод часто уступает тонкослойной и газовой хроматографии.
Однократного хроматографирования обычно бывает недостаточно для выделения индивидуальных веществ, в связи с этим полученные фракции подвергают препаративной тонкослойной хроматографии или колоночной хроматографии другого типа. При колоночной хроматографии липидов используют не только принцип адсорбции, но и принцип распределения между двумя несмешивающимися жидкостями, гель-фильтрации, ионного обмена.
В основе адсорбционной хроматографии лежит разделение липидов в соответствии со степенью их полярности. Адсорбентом при тонкослойной хроматографии чаще всего служит силикагель. При колоночной хроматографии широкое применение получили три адсорбента: силикагель, окись алюминия, флоризил (силикат магния).
Прочность взаимодействия липида с адсорбентом определяется главным образом водородными и ионными связями, в меньшей степени — силами Ван-дер-Ваальса.
Хроматография на силикагеле.
Силикагель является продуктом полимеризации ортокремниевой кислоты (H4SiО4). Он выпускается рядом фирм в виде зерен различной величины. Адсорбционные свойства силикагеля обусловлены присутствием на поверхности зерен гидроксильных групп, которые за счет водородных связей взаимодействуют друг с другом и водой. Гидратированный силикагель мало активен как адсорбент. При нагревании от 50 до 150°С происходит дегидратация, приводящая к значительному увеличению адсорбционной способности силикагеля.
При хроматографии 1—3 г липидов пользуются колонками диаметром 35 мм и длиной 50—70 см. При разделении больших количеств применяют большие колонки, при этом максимальное весовое отношение липид/адсорбент не должно превышать 1:50 (для фосфолипидов 1:100). Отношение высоты колонки к площади ее сечения должно быть равно 5:1, длина колонки не должна быть больше 1 м.
При аналитической хроматографии толщина слоя силикагеля обычно не превышает 0,25 мм. Для препаративных целей используют слои толщиной 0,75—1,0 мм на пластинках размером 20Х20 см. В некоторых случаях для лучшего разделения используют удлиненные пластинки (34X20 см). Для аналитического разделения липидов можно применять готовые пластинки. Они представляют собой тонкий слой силикагеля, закрепленный на алюминиевой фольге с помощью крахмала. Для подготовки пластинок к работе их необходимо активировать.
Хроматография на окиси алюминия.
При проведении препаративных работ хроматография на окиси алюминия имеет ряд преимуществ по сравнению с хроматографией на силикагеле: требуются меньшие объемы растворителей, скорость тока через колонку выше.
Для хроматографии фосфолипидов используют окись алюминия IV степени активности. Ее получают путем активации коммерческой окиси алюминия при 110°С в течение 12 ч, после чего на каждые 100 г адсорбента добавляют 10 мл воды и встряхивают смесь в закрытом сосуде в течение 2 ч. При выделении больших количеств фосфолипидов используют стеклянные колонки с отношением диаметра к длине, равным 1:2,5, в которые помещают до 1000 г адсорбента. При работе с меньшим количеством адсорбента, до 100 г, применяют колонки с отношением 1:5. Нагрузка колонки не должна превышать 0,5 мг липидного фосфора на 1 г адсорбента.
Однако необходимо отметить, что хроматография многих липидных веществ на окиси алюминия сопровождается значительными потерями в результате гидролиза.
ЛАБОРАТОРНАЯ РАБОТА № 26. СПЕКТРАЛЬНОЕ ОПРЕДЕЛЕНИЕ ФЕРМЕНТОВ
Опыт №1. определение активности пероксидазы по Бояркину
Метод основан на измерении времени, за которое опытный раствор достигает определенной оптической плотности.
В качестве субстрата используется бензидин, в результате окисления которого образуется соединение синего цвета.
Материалы и оборудование: клубни картофеля, 1% раствор гидрохинона, 3% раствор перекиси водорода.
Ножи, терки, марля, воронки, конические колбы на 50 мл, пробирки в штативе, пипетки на 1 и 10 мл.
Растения комнатные, проростки пшеницы, ячменя или других культур, клубни картофеля, ацетатный буферный раствор с рН 5,4; раствор бензидина на ацетатном буфере; 0,03% раствор перекиси водорода.
Мерные колбы на 25 мл, пипетки на 2 мл, фарфоровые ступки с пестиком, секундомеры, центрфуга с пробирками, ФЭК.
Приготовление бензидина.В мерную колбуна 200 мл, наполненную на 2/3 дистиллированной водой прибавляют 2,3 мл (2,4 г) ледяной уксусной кислоты и 184 мл бензидина. Колбу подогреваютпримерно до 50-60ºС на водяной бане при постоянном взбалтывании. После полного растворения бензидина (10-15 мин) добавляют 5,45 г ацетата натрия, полностью его растворяют, колбу охлаждают и доливают водой до метки. В темном месте раствор можно хранить 7-10 дней.
Ход работы: Растереть в ступке с водой (или с ацетатным буферным раствором рН 5,4) 50-100 мг растительного материала. Перенести его в мерную колбочку на 25 мл и довести до метки. Вытяжку настоять 5-10 мин и отцентрифугировать в течение 10 мин при интенсивности вращения 3000 мин-1.
Далее работают с надосадочной жидкостью (осадок отбрасывают). В надосадочной жидкости определяют активность пероксидазы, о которой судят по времени образования синей окраски окисленного бензидина.
Для измерения активности лучше брать такие разведения вытяжки, при которых окраска изменяется в течение 10-40 с, т.к. активность фермента прямо пропорциональная его концентрации только в начале реакции. Определение ведут на ФЭК с желтым светофильтром.
В две кюветы толщиной 2 см наливают по 2 мл вытяжки, 2 мл буферного раствора, 2 мл бензидина в каждую. Ставят кюветы в кюветодержатели прибора, одну (контроль), другую (опыт), при помощи оптических клиньев уравнивают оба световых потока (стрелка должна быть в нулевом положении). Уравнивают световые потоки сначала при чувствительности «1», затем при чувствительности «2», при которой и ведут определение. В контрольной кювете оптическую плотность сместить на деление 0,250.
В контрольную кювету наливают 2 мл воды, в опытную – 2 мл 0,03% раствора перекиси водорода пипеткой с широким носиком. Сильной струей перемешать содержимое кюветы. Одновременно с падением первой капли включить секундомер. В опытной кювете раствор синеет и стрелка передвигается к нулевому положению. Отмечают время от начала приливания перекиси водорода до достижения раствором заданной оптической плотности (нулевого положения).
Провести три определения и рассчитать среднее значение. Активность ферментоа рассчитывают по формуле
А = D•(α•β•γ)/(t•d),
где D – оптическая плотность, равная 0,250; d – толщина слоя жидкости (толщина кюветы), см; t – время, с; α – отношение количества жидкости, взятой для приготовления вытяжки, мл, к массе навески, г; β – степень дополнительного разведения вытяжки (если это потребуется); γ – степень постоянного разведения вытяжки в реакционной смеси (при данных условиях – 4).
Результаты отражают изменение оптической плотности за 1 с на 1 г сырой массы ткани. Определяют активность пероксидазы в листьях верхнего и нижнего ярусов или в проростках злаков, выращенных в разных условиях. Результаты опыта записывают в таблицу .
Таблица - Определение активности пероксидазы
Растение, ярус |
Навеска, г |
Объем экстракта, мл |
Разведение вытяжки в реакционной смеси, γ |
Толщина слоя жидкости в кювете, см |
Время изменения окраски раствора, с |
Активность пероксидазы, изменение оптической плотности за 1 с на 1 г сырой массы |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Опыт №2. Определение активности НАДФ-зависимойизоцитратдегидрогеназы
НАДФ-зависимая изоцитратдегидрогеназа (НАДФ-ИДГ) (изоцитрат: НАДФ-оксидоредуктаза декарбоксилирующая, КФ 1.1.1.42) катализирует обратимую реакцию окислительного декарбоксилирования изолимонной кислоты, в ходе которой образуется α-кетоглутарат, СО2 и восстановленная форма НАДФ – НАДФН:
изоцитрат + НАДФ ↔ α-кетоглутарат + СО2+ НАДФН + Н+
Константа равновесия реакции, катализируемой НАДФ-ИДГ в среднем равна 1, но в физиологических условиях равновесие в большинстве случаев сдвинуто в сторону образования кетокислоты.
Молекулярная масса мНАДФ ИДГ 58 кДа, фермент состоит из одной полипептидной цепи. Молекулярная масса цитНАДФ ИДГ выше, чем у митохондриальной, она равна, по данным разных авторов, от 75 до 96 кДа. Фермент состоит из 2 субъединиц. Фермент локализован как в митохондриальной, так и цитоплазматической фракциях клеток. У взрослых животных в органах с активно протекающими биосинтетическими процессами (печень, жировая ткань) значительная часть (70 – 80 %) активности НАДФ-ИДГ сосредоточена во внемитохондриальной фракции. В головном мозгу новорожденных животных в период интенсивной миелинизации, которая характеризуется активными биосинтетическими процессами, значительная часть изоцитрата окисляется с помощью НАДФ-ИДГ, локализованной в цитоплазме. В ходе постнатального развития животного активность НАДФ-ИДГ в цитоплазме клеток мозга уменьшается и достигает значений, как у взрослых особей.
В митохондриях печени около 8– 10% от общего содержания изоцитрата подвергается окислительному превращению с помощью НАДФ-зависимой изоцитратдегидрогеназы. Для протекания НАДФ-ИДГ реакции необходимы ионы Mg2+ или Mn2+, поскольку истинным субстратом реакции является комплекс двухвалентного катиона с недиссоциированным изоцитратом (в отличие от НАД-ИДГ, где активной формой является изоцитрат–-). Активность фермента обусловлена наличием сульфгидрильных групп, причем, из 5 SH-групп только 2 связаны с активностью фермента, одна из них находится в активном центре, а другая необходима для поддержания активной конформации фермента.НАДФ-зависимая ИДГ ингибируется НАДН, АТФ и высокими концентрациями НАДФН.
Принцип метода.В процессе окислительного превращения изолимонной кислоты до α-кетоглутаровой происходит восстановление НАДФ; количество восстановленного НАДФ эквимолярно количеству окисленого изоцитрата.
Изменение содержания НАДФН регистрируется на спектрофотометре при длине волны 340 нм.
Реактивы:
1.0,1 Мтрис-НСl-буфер, рН 7,4.
2. 0,02 МMnCl2•4 H2O.
3. 0,003 МНАДФ
4. 0,08 Мраствор D,L-изоцитрат Na.
Оборудование: Спектрофотометр СФ-46 (ЛОМО), автоматические пипетки объемом 10-100 мкл и 200-1000 мкл, пробирки, спектрофотометрические кюветы, весы аналитические
Ход работы:
Навески НАДФ и изоцитрата растворяют перед приготовлением инкубационной среды. В пробу вносят компоненты инкубационной среды, которые заранее смешивают в указанных пропорциях в объеме, зависящем от количества проб.
-
1.трис-НСl
- 1,5 мл
2. Mn2+
- 0,5 мл
3.НАДФ
- 0,5 мл
4.D,L- изоцизоцитрат
- 0,1
5.H2O
- 0,3 мл
В кювету вносят по 2,9 мл готовой инкубационной среды, затем, не вынимая кювету из прибора, добавляют 0,1 мл суспензии митохондрий или цитоплазмы, перемешивают. Реакцию начинают внесением препарата, содержащего фермен (0,1 млцитоплазмы либо суспензии митохондрий).
Изменение оптической плотности раствора в результате восстановления НАДФ регистрируют в течение 3 - 5 мин с интервалом в 1 мин
Активность фермента рассчитывают по формуле:

где ∆ Е – изменение оптической плотности за 1 мин;
3 – объем исследуемой пробы;
1000 – коэффициент перевода микромолей в наномоли;
б – содержание белка в исследуемом образце, мг;
6,22 – коэффициент микромолярной экстинкции НАДФН (мМ-1•см-1).
Опыт №3. Определение активности2-оксоглутаратдегидрогеназы
Реакции окислительного превращения 2-оксоглутарата с образованием сукцинил-КоА, углекислого газа, восстановленного НАД катализирует ферментная система, состоящая из 3 основных ферментов: собственно дегидрогеназа 2-оксоглутарата [E1], липоатсукцинилтрансферазы[E2] и липоамиддегидрогеназы[E3]. Эти ферменты при участии КоА, НАД+, ФАД, Лип S2Н, ТДФ и Мg2+ осуществляют последовательность реакций окислительного декарбоксилирования 2-оксоглутарата.
Суммарное уравнение имеет вид:
α -кетоглутарат + НАД++КоА-SH → сукцинил КоА +СО2+ НАДН + Н+.
Локализованная в митохондриях ферментная система представляет собой высокоорганизованный комплекс функционально связанных ферментов. Значение этого комплекса в клеточном метаболизме обусловлено широким участием его субстратов и продуктов в обмене веществ. Важность их изучения не вызывает сомнений: во-первых, реакция окислительного декарбоксилирования 2-оксоглутарата локализирована в месте пересечения нескольких метаболических путей: 2-оксоглутарат является общим интермедиатом обмена углеводов и белков; во-вторых, ключевая роль оксоглутаратдегидрогеназного комплекса обусловлено тем, что он катализирует основной путь биодеградации оксокислоты.
Для изучения скорости окисления 2-оксоглутарата предложен ряд методов. Одни из них основаны на определении суммарной активности 2- оксоглутаратдегидрогеназного комплекса по скорости восстановления НАД или какого-либо искусственного акцептора электронов (2,6-дихлорфенолиндофенола, солей тетразолия, феррицианида и т.д.) Однако контроль активности комплекса по восстановлению НАД носит ограниченный характер, т.к. показано, что 2- оксоглутаратдегидрогеназа почек, печени быстро инактивируется из-за присутствия протеазы, вызывающей частичный протеолиз липоатсукцинилтрансферазы, что приводит к диссоциации ее первого и третьего компонентов. Подвергнутый ограниченному протеолизу комплекс утрачивает способность восстанавливать НАД.
В качестве одного из продуктов реакции окислительного превращения 2-оксоглутарата образуется углекислый газ. Реакция окислительного декарбоксилирования, катализируемая первым компонентом комплекса, является наиболее медленной из трех реакций и, следовательно, ограничивает скорость всего последующего процесса. Поэтому об активности энзима можно судить по количеству продукта декарбоксилирования 2-оксоглутарата – углекислого газа, связанного какой-либо щелочью. На этом принципе основан радиометрический метод тестирования активности 2-оксоглутаратдегидрогеназы.
Широко распространено определение активности 2-оксоглутаратдегидрогеназы методом, описанным Gubler, где в качестве искусственного акцептора электронов используется феррицианид калия. В процессе окисления субстрата происходит восстановление феррицианида, окраска последнего изменяется до бесцветной. Этот метод легко воспроизводим, доступны все реактивы, что также имеет немаловажное значение. Метод может быть использован для определения скорости окисления не только 2-оксоглутарата, но и пирувата.
Однако известен метод определения активности следующего митохондриального фермента цикла Кребса – суцинатдегидрогеназы с феррицианидом в качестве конечного акцептора электронов. Поэтому, чтобы исключить вклад суцинатдегидрогеназы в восстановление феррицианида, определение активности дегидрогеназы 2-оксоглутарата проводят в присутствии малоната в среде инкубации. Как известно, последний является конкурентным ингибитором энзима (СДГ).
Реактивы: 0,15 М Калий-фосфатный буфер рН 7,4; 0,2 М MnSO4; 0,02 М ЭДТА-динатриевая соль (трилон Б); 0,06 М АТФ –динатриевая соль; 0,0067 М K3[Fe (CN)6];0,05 М малонат калия,0,25 М сахароза, рН 7,4; 0,4 М α-кетоглутарат, рН; 7,4,0,25 М сахароза, рН 7,4; суспензия митохондрий
Оборудование: Спектрофотометр СФ-46 (ЛОМО), автоматические пипетки объемом 10–100 мкл и 200–1000 мкл, пробирки, спектрофотометрические кюветы, весы аналитические
Ход работы
№ п/п |
Реактивы |
Инкубационная смесь (объем, мл) |
|
контроль |
опыт |
||
1. |
0,15 МКалий-фосфатный буфер рН 7,4 |
0,5 |
0,5 |
2. |
0,2 МMnSO4 |
0,1 |
0,1 |
3. |
0,02 МЭДТА-динатриевая соль (трилон Б) |
0,1 |
0,1 |
4. |
0,06 МАТФ-динатриевая соль |
0,1 |
0,1 |
5. |
0,0067 МK3[Fe (CN)6] |
0,7 |
0,7 |
6. |
0,05 Ммалонат калия |
0,1 |
0,1 |
7. |
0,25 Мсахароза, рН 7,4 |
1,6 |
1,6 |
8 |
0,4 Мα -кетоглутарат, рН 7,4 |
– |
0,1 |
9 |
0,25 Мсахароза, рН 7,4 |
0,1 |
– |
10 |
Суспензия митохондрий |
0,4 |
0,4 |
Инкубационную смесь разливают по 3,2 мл; 0,4 Мраствор 2-оксоглутарата в количестве 0,1 мл вносят в опытные пробы. Контрольные пробы вместо субстрата содержат 0,1 мл0,25 Мраствора сахарозы.
Суспензию митохондрий готовят, добавляя 0,25 М раствор сахарозы к осадку митомондрий из расчета 4 мл на осадок из 1 гткани. Реакцию начинают прибавлением 0,4 мл суспензии митохондрий, которую вносят в заранее подготовленную среду инкубации. Пробы (контрольную и опытную) инкубируют в водяной бане при 250С 20 минут. Реакцию останавливают добавлением 0,3 мл 50 % ТХУ. Пробы центрифугируют 15 мин при3000 g и фотометрируют при длине волны 417 нм на спектрофотометре.
Разница между экстинкцией контроля (без субстрата) и опыта соответствует количеству восстановленной красной кровяной соли, что эквивалентно количеству окисленного 2-оксоглутарата в пробе. Количество восстановленного феррицианида определяли по калибровочной кривой и активности рассчитывают по формуле:

где С1– содержание феррицианида (мкмоль) в контрольной пробе;
С2– содержание феррицианида (мкмоль) в опытной прбе;
1000 – коэффицент перевода микромолей в наномоли;
б – содержание белка в пробе (в мг);
20 – время инкубации проб в мин.
Активность фермента выражают в наномолях K3[Fe (CN)6] на мг белка за 1 мин.
Опыт № 3. Изучение участия пиридиннуклеотидов в биохимических реакциях окисления-восстановления.
Теоретическое введение. Пиридиннуклеотиды, входящие в качестве коферментов в состав более чем 150 так называемых «пиридинзависимых» дегидрогеназ, играют существенную роль в реакциях окисления-восстановления (главным образом, в процессе дыхания, т. е. в процессе переноса электронов от органических субстратов к молекулярному кислороду). Спектрофотометрическое определение восстановленных пиридиннуклеотидов служит в связи с этим одним из важных методов современной энзимологии.
В основе метода лежит существенное отличие спектров окисленной и восстановленной форм кофермента. Спектр поглощения окисленной формы — NAD+ имеет максимум при 260 нм. Для восстановленной формы — NADH характерен более длинноволновый максимум поглощения — при 340 нм. Измеряя в ходе окислительно-восстановительной реакции оптическую плотность объекта при 340 им, можно установить (зная коэффициент поглощения и длину кюветы) количество нуклеотида в пробе. С другой стороны, учитывая, что окисление-восстановление 1 мкмоля кофермента сопровождается восстановлением-окислением 1 мкмоля субстрата, этим же методом можно определить количество прореагировавшего субстрата.
Исследуемая реакция протекает по следующей схеме:
фермент
субстрат + NAD+ ---------------→ продукт реакции + NADH
Как правило, все компоненты, кроме определяемого, присутствуют в избытке. В расчетах исходят из величины коэффициента молярной экстинкции NADH: при окислении 1 мкмоль кофермента в 1 мл смеси оптическая плотность при 340 нм в кювете толщиной 1 см меняется на 6,22 ед. Количество определяемого вещества в пробе (в мкмолях) М рассчитывается по формуле:

где ∆D340 — изменение оптической плотности смеси при 340 нм;
V — объем пробы (мл).
Энзиматический метод количественного определения
aлкогольдегидрогеназа
O NAD++CH3–CH2–OH
CH3–C+NADH
H


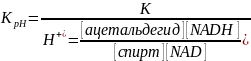

Принцип метода. NAD восстанавливается в присутствии этанола и алкогольдегидрогеназы. Количество образовавшегося NADH равно исходном количеств NAD+ и определяется спектрофотометрически:
При рН = 8,8 и концентрации спирта 0,5 М можно получить практически полное восстановление NAD+.
Реактивы.
1 Этанол 0,5 М, приготовленный на 0,1 М трис-буфере с рН= 10,1 (молекулярная масса этанола 46,07).
2. Алкогольдегидрогеназа: растворить в 1—2 мл дистиллята 1—2 мг белка непосредственно перед использованием. В случае использования суспензии учесть, что одной ферментной единицы (см. обозначения на упаковке белка) достаточно для полного превращения 1 мкмоля субстрата в 1 мин.
3. Раствор NAD+: 1—2 мл концентрации 1- 4 мМ; рН = 6,5 (молекулярная масса NAD+=633).
Процедура определения. В две спектрофотометрические кюветы помещают по 0,39 мл Н2O (дистиллят); 2,5 мл раствора этанола 0,5 М на 0,1 М трис-буфере с рН—10,1 и 0,1 мл раствора NAD. В опытную кювету добавить 0,05 мл раствора алкогольдегидрогеназы, содержащего 1—2 мг белка в 1 мл.
Измеряют по точкам спектры поглощения растворов в опытной кювете и в контрольной для области 220—380 нм с шагом 5 нм. Учитывая поглощение контроля, оценивают ∆D340 в опытной кювете. Рассчитывают количество NADH в растворе и количество прореагировавшего NAD+. Последнюю величину сравнивают с исходной.
Спектры поглощения, снятые по точкам, необходимо представить в виде таблиц и графиков (ордината — D, абсцисса — λ в нм).
Практическое задание
Кофермент А принимает участие в качестве кофактора во многих ферментативных реакциях протекающих в организме человека и животных. Перепишите формулу кофермента А в тетрадь:

Карандашом обведите формулу пантотеновой кислоты в составе кофермента, также укажите в его составе аденин и рибозу.
2. В состав кофермента ФАД входит витамин рибофлавин. Перепишите в тетрадь формулу ФАД:

Карандашом обведите формулу рибофлавина.
3. Витамин никотинамид входит в состав кофермента НАД:

В тетрадь перепишите формулу НАД, карандашомобведите формулу витамина.
ЛАБОРАТОРНАЯ РАБОТА № 27. КОЛИЧЕСТВЕННОЕ ОПРЕДЕОЛЕНИЕ УГЛЕВОДОВ.
Количественное определение глюкозы
Глюкоза при нагревании с ортотолуидиновым реактивом дает зеленую окраску, интенсивность которой пропорциональна концентрации глюкозы.
Реактивы: ортотолуидиновый реактив, стандартный раствор глюкозы 1 мг/мл, раствор глюкозы неизвестной концентрации.
Оборудование: Спектрофотометр, пипетки, водяная баня, штатив, пробирки.
Ход работы
К 0,2 мл стандартного раствора глюкозы и 0,2 мл раствора с неизвестной концентрацией добавить по 2 мл ортотолуидинового реактива. Пробирки со смесью поместить в кипящую водяную баню на 8 минут. Пробирки вынуть, охладить под струей водопроводной воды до комнатной температуры.
Оптическую плотность раствора измеряют при длине волн 590 – 650 нм. Расчет ведут по формуле:

где:
сх – концентрация глюкозы в исследуемом растворе (мг/мл);
сст – концентрация глюкозы в стандартном растворе (мг/мл);
Ах – оптическая плотность исследуемого раствора;
Аст – оптическая плотность стандарта.
Задание 1.
Определите число оптических изомеров для альдопентоз.
Изобразите проекции Фишера всех линейных изомеров альдопентоз.
Определите отношение изомеров к D– или L–ряду по последнему от карбонильной группы хиральному центру.
Найдите среди изображенных изомеров D–рибозу.
Задание 2.
Определите число оптических изомеров у альдогексоз.
Изобразите проекции Фишера всех линейных изомеров альдогексоз.
Определите отношение углеводов к D– или L–ряду.
Найдите среди изображенных изомеров D–глюкозу, D–галактозу, L–глюкозу.
ЛАБОРАТОРНАЯ РАБОТА № 28. СПЕКТРОФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ Β -ЛИПОПРОТЕИНОВ ПЛАЗМЫ КРОВИ.
Теоретическое введение. Большинство липидов находится в крови не в свободном состоянии, а в составе белково-липидных комплексов: хиломикроны, α-липопротеиды и β -липопротеиды. Липопротеиды можно разделить различными методами: электрофореза, тонкослойной хроматографии, ультрацентрифугирования в солевых растворах различной плотности. При ультрацентрифугировании выделяются хиломикроны и липопротеиды разной плотности: высокой (ЛВП—α-липопротеиды), низкой (ЛНП—β -липопротеиды) и очень низкой (ЛОНП—пре- β -липопротеиды) и др.
Фракции липопротеидов отличаются по количеству белка, относительной молекулярной массе липопротеидов и процентному содержанию отдельных липидных компонентов. Так, α-липопротеиды, содержащие большое количество белка (50—60%), имеют и более высокую относительную плотность (1,063—1,21), тогда как β -липопротеиды и пре-β -липопротеиды содержат меньше белка и значительное количество липидов — до 95% от всей относительной молекулярной массы и низкую относительную плотность (1,01 —1,063).
Принцип метода. В основу метода положена способность β -липопротеидов (ЛПНП) осаждаться в присутствии хлорида кальция и гепарина; при этом изменяется мутность раствора. По степени помутнения раствора и судят о концентрации β -липопротеидов в сыворотке крови. Считают, что гепарин способен образовывать с β -липопротеидами комплекс, который под действием хлорида кальция выпадает в осадок.
Реактивы, оборудование и исследуемый материал:
1. Хлорид кальция (СаСI2) 0,27% раствор (готовят из безводного реактива).
2. Гепарин, 1% раствор (готовят ex tempore), 1 мл его должен содержать 1000 ЕД (лучше кристаллический).
3. ФЭК.
4. Микропипетка вместимостью 0,2 мл.
5. Сыворотка крови.
Ход работы. В пробирку вносят 2 мл 0,27% раствора СаС12 и 0,2 мл сыворотки крови, перемешивают. Определяют оптическую плотность раствора (Е1) против 0,27% раствора СаС12 по левому барабану в кюветах на 5 мм, при красном светофильтре (630 нм). Раствор из кюветы переливают в пробирку, добавляют микропипеткой 0,04 мл 1 % раствора гепарина, перемешивают и точно через 4 мин снова определяют оптическую плотность раствора (Е2) в тех же условиях:

Расчет. Вычисляют разность оптической плотности и умножают ее на 1000 — эмпирический коэффициент, так как построение калибровочной кривой сопряжено с рядом трудностей. Ответ выражают в г/л. В норме содержание β -липопротеидов составляет 3—4,5 г/л (300—450 мг %). Содержание β -липопротеидов колеблется в зависимости от возраста и пола.
ЛАБОРАТОРНАЯ РАБОТА № 29. ФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ МАССОВОЙ ДОЛИ ПРОТЕИНОВ В БИОЛОГИЧЕСКИХ ОБЪЕКТОВ.
Реактивы и растворы
Раствор A. Готовят путем растворения 2 г Na2CO3 в 100 см3 0,1 М раствора NaOH.
Раствор B. Навеску 10 г винно-красного натрия-калия растворяют в 300 см3 дистиллированной воды, добавляют 5 г CuSO4*5 H2O и, после растворения соли, доводят объем водой до 1 дм3.
Раствор С. Смешивают 49 см3 раствора А с 1 см3 раствора В. Раствор хранится не более суток.
Раствор Ф (реактив Фолина). Методика приготовления и хранения раствора реактива Фолина. Его готовят следующим образом: смешивают 100г Na2WO4*2 H2O, 25 г Na2MoO4, 700 см3 воды, 50 мл 85 %-ной H3PO4 и 100 см3 концентрированной HCl и кипятят с обратным холодильником в течение 10 ч. К смеси добавляют 150 г Li2SO4, 50 см3 воды и несколько капель бромной воды. Избыток брома удаляют кипячением и объем раствора доводят до 1 дм3. Реактив Фолина хранят в темной закрытой склянке до 1 года. Перед использованием устанавливают его нормальность путем титрования стандартизованным раствором NaOH, а затем разбавляют его дистиллированной водой до нормальности, равной единице.
Непосредственно перед анализом готовят 12 %-ный раствор CuSО4*5H2O, 2,5 %-ный и 1 М растворы NaOH и 0,25 М раствор комплексона ІІІ.
Все указанные выше рабочие растворы применяют при фотометрическом определении белка как в исследуемом растворе, так и в стандартных растворах альбумина бычьего сывороточного.
Последние используются для построения градуировочного графика, представляющего зависимость оптической плотности окрашенного раствора от содержания альбумина.
Ход анализа. Выделяют белок из навески средней пробы кормовых дрожжей. В стакан вместимостью 100 – 150 см3 помещают точную навеску высушенных дрожжей массой 0,3 – 0,4 г, добавляют 50 см3 дистиллированной воды и после двухминутного кипячения осаждают белок путем добавления избытка (10 см3) 12 %-ного раствора сульфата меди и 10 см3 2,5 %-ного раствора NaOH. Осадок медно-белкового комплекса отделяют фильтрованием через бумажный фильтр (с синей лентой) и промывают двумя порциями по 10 см3 воды. Осадок вместе с фильтром возвращают в исходный стакан, где его растворяют путем добавления 50 см3 1 M раствора NaOH и 20 см3 0,25 М раствора комплексона ІІІ. Смесь кипятят на песчаной бане в течение 30 мин и постоянном перемешивании. Раствор, содержащий растворенный белок, осторожно переливают в мерную колбу вместимостью 500 см3, оставляя фильтр в стакане. Стенки стакана и нерастворимый осадок промывают тремя порциями по 5 – 10 см3 горячей дистиллированной воды (декантация). Промывные воды собирают в ту же мерную колбу и после охлаждения ее под струей водопроводной воды доводят раствор до метки дистиллированной водой и перемешивают. Отбирают аликвоту (100 см3) полученного раствора, помещают ее в мерную колбу вместимостью 250 см3. Раствор в колбе доводят водой до метки и перемешивают.
С целью отделения взвешенных частиц около 20 см3 раствора фильтруют в сухой стакан через бумажный фильтр с синей лентой, отбрасывая первую порцию (около 10 см3) фильтрата. Отбирают пипеткой 5 см3 очищенного раствора и помещают его в мерную колбу вместимостью 25 см3. Добавляют туда 15 см3 раствора С и осторожно перемешивают содержимое в течение 10 мин.
Затем добавляют 1,5 см3 раствора Ф (реактива Фолина), доводят содержимое до метки водой и тщательно перемешивают. Полученный окрашенный раствор синего цвета фотометрируют в кювете с толщиной поглощающего свет слоя l = 1 см при λ = 690 нм (красный светофильтр). В качестве “нулевого” используют раствор “холостого” опыта, который одновременно получают по указанной методике, заменив в ней 5 см3 анализируемого раствора белка на соответствующее количество воды.
Оптическую плотность измеряют не менее трех раз, принимая за истинный результат измерений среднее арифметическое из двух близких.
По величине оптической плотности раствора, используя градуировочный график, вычисляют массовую долю истинного белка в кормовых дрожжах по формуле
ωбелка = γ 0,025/m(1 – W/100),
где — количество белка, определенное по градуировочному графику, мкг; 0,025 — коэффициент разбавления и соотношения размерностей;
m —навеска дрожжей, г;
W — массовая доля влаги в кормовых дрожжах, %.
ЛАБОРАТОРНАЯ РАБОТА № 30. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ДНК
Реактивы: 5% –й раствор хлорида натрия, дифениламиновый реактив (1г дифениламина в 100 мл ледяной уксусной кислоты,+ 2,75мл H2SO4 конц.), печень, дистиллированная вода.
Оборудование: гомогенизатор, центрифуга, центрифужные весы, центрифужные пробирки, стакан химический на 100 мл, пипетка на 10 мл, цилиндры на 25 и 50 мл, стеклянные палочки, штатив с пробирками, водяная баня
Ход работы
Навеску ткани печени 500 – 100 мг гомогенизируют в 5—10 мл 5% –го раствора хлорида натрия. Полученный гомогенат переносят в центрифужные пробирки, уравновешивают их на центрифужных весах и центрифугируют 10 – 15 минут при 1500 – 3000 об/мин
Надосадочную жидкость делят на 2 части. В одной из пробирок проводят качественную реакцию на ДНК. Вторую пробирку помещают в кипящую водяную баню, предварительно добавив 3 мл 2 % – го раствора серной кислоты для проведения гидролиза (гидролиз проводят в течении часа). Гидролизат охлаждают, центрифугируют. В надосадочной жидкости содержатся составные компоненты нуклеопротеидов
Метод основан на свойстве дезоксирибозы, входящей в состав ДНК, образовывать синее окрашивание прямо пропорциональное концентрации ДНК.
Реактивы: дифениламиновый реактив (1 г дифениламина в 100 мл ледяной уксусной кислоты, + 2,75 мл H2SO4конц.), дистиллированная вода, водный раствор ДНК.
Оборудование: термостат, спектрофотометр, автоматические пипетки, пробирки, штатив.
Ход работы
Вначале необходимо построить калибровочный график. Для этого в 3 пробирки наливают по 1мл раствора ДНК с различной концентрацией (50, 100, 200 мкг/мл) и по 2 мл дифениламинового реактива. Помещают пробирки на 10 – 15 минут в кипящую водяную баню для развития окраски, затем определяют оптическую плотность каждого из растворов при длине волны 595 нм. Калибровочный график строят, откладывая по оси абсцисс концентрацию использованных растворов ДНК, по оси ординат – соответствующие им значения оптической плотности.
Затем берут две пробирки: в контрольную пробирку наливают 1 мл воды, в опытную пробирку – 1 мл водного раствора ДНК (неизвестной концентрации). В каждую пробирку добавляют по 2 мл дифениламинового реактива. Обе пробирки помещают на 10 – 15 минут в кипящую водяную баню. Затем пробы охлаждают и измеряют насыщенность окраски против контроля при λ = 595 нм. Зная оптическую плотность пробы, по калибровочному графику находят количество ДНК в ней.
Состав проб
Про– бирки |
ДНК (мл) |
Дифенил– аминовый реактив (мл) |
H2O (мл) |
|||||
50 (мкг/мл) |
100 (мкг/мл) |
200 (мкг/мл) |
Х (мкг/мл) |
|||||
1 |
1 |
– |
– |
– |
2 |
– |
||
2 |
– |
1 |
– |
– |
2 |
– |
||
3 |
– |
– |
1 |
– |
2 |
– |
||
Х |
– |
– |
– |
1 |
2 |
– |
||
К |
– |
– |
– |
– |
2 |
1 |
||
10 – 15 мин кипящ. водян. баня |
||||||||
охладить, измерить оптич. плотность при λ=595 нм |
||||||||
ЛАБОРАТОРНАЯ РАБОТА № 31. ЭЛЕКТРОФОРЕТИЧЕСКОЕ РАЗДЕЛЕНИЕ БЕЛКОВ
Метод основан на том, что молекулы белка обладают электрическим зарядом, величина и знак которого определяются аминокислотным составом белка, pH и ионной силой окружающей среды. Под влиянием внешнего электрического поля заряженные молекулы передвигаются в растворе к противоположно заряженному полюсу. Скорость перемещения белковых частиц пропорциональна величине их заряда и обратно пропорциональна размеру частиц и степени их гидратации.
Широкое распространение в настоящее время получил так называемый «зональный электрофорез» — электрофорез на твердом носителе (на бумажных полосах, агаре, крахмале, акриламиде), пропитанном буферным раствором с нужным значением pH. Положение белков на бумаге или геле определяют путем фиксации и последующего окрашивания их тем или иным красителем (обычно бромфеноловым синим, амидовым черным или кумасси синим). Количество белка в каждой фракции можно ориентировочно определять по интенсивности окраски связанного красителя. Такое определение не дает строго количественного соотношения белковых фракций, так как количество красителя, связываемого различными белками, неодинаково.
ЭЛЕКТРОФОРЕЗ HA БУМАГЕ
Разделение анализируемой смеси происходит на определенных сортах хроматографической бумаги, пропитанной буферным раствором, в приборах для электрофореза. Белки разделяют при напряжении до 500 B.
Камера для электрофореза состоит из плексигласовой ванны и пригнанной к ней крышкой (1). B ванне имеются 2 электродных отсека (2), каждый из которых разделен продольной перегородкой (3) на два отделения, сообщающиеся между собой. Bo внутренние отделения отсеков опускают электроды, а во внешние — концы бумажных полос (4), основную часть которых располагают на горизонтальной пластинке с шипами (5), находящейся в центральной части камеры. Между горизонтальной пластинкой и наружным отделением электродных отсеков имеются палочки (6), через которые перекидываются бумажные полоски и которые служат для их поддерживания. Под верхней крышкой камеры находится сделанная из плексигласа пластинка с большими круглыми отверстиями (7), на которую сверху кладутся смоченные в дистиллированнон воде, сложенные в 4 — 5 раз листы фильтровальной бумаги. Эти листы способствуют увеличению герметичности камеры и, как следствие, — уменьшению испарения жидкости с электрофореграмм в процессе электрофореза.

Рисунок - Схема прибора для низковольтного электрофореза
Электрофорезом на бумаге студентам предлагается провести разделение белков сыворотки крови. Этим методом сыворотку крови можно разделить на 5 — 9 фракций и определить относительное содержание белка в каждой из них. Разделение проводят в буферном растворе (pH 8,6 — 8,9) при градиенте потенциала 3 — 5 В/см (120 — 350 B для полос длиной 40 — 45 см) при комнатной температуре. Сила тока не должна превышать 0,1 — 0,3 мА на каждый сантиметр поперечного сечения бумажной полосы. Увеличение силы тока более чем в 2 раза недопустимо, так как при этом происходит чрезмерное нагревание, значительное увеличение испарения и в конечном итоге — прогорание бумаги
Реактивы:
1. Буферный раствор. Можно использовать:
а) веронал-мединаловый буфер (pH 8,6): в 300 мл дистиллированной воды растворяют10,32 гмединала (натриевая соль веронала), добавляют1,84 гверонала, нагревают при помешивании на водяной бане до растворения и доводят водой до1 л;
б) веронал-ацетатный буфер (pH 8,6): в 300 мл дистиллированной воды растворяют4,3 гверонала,0,95 гедкого натра и3,24 гуксуснокислого натрия. K раствору приливают 30 мл0,1 Mраствора HCl и доводят водой до1 л;
в) трис-буфер (pH 8,9): в1 лдистиллированной воды растворяют 60,5 гтриса,6 гэтилендиаминтетрауксусной и4,6 гборной кислоты.
2. Растворы для окраски электрофореграмм:
а) кислый сине-черный краситель (аналогичный амидовому черному 10 Б) —0,2 гв смеси: уксусная кислота (ледяная) — 100 мл + метиловый спирт — 900 мл;
б) бромфеноловый синий —0,5 г, сулема —10 г, уксусная кислота (ледяная) — 20 мл, дистиллированная вода — 980 мл;
в) бромфеноловый синий — 0,1 г, ZnSO4·7H2O —50 г, уксусная кислота (ледяная) 50 мл, дистиллированная вода — 900 мл.
3. Растворы для отмывания электрофореграмм от несвязавшейся с белком краски и закрепления красителя на белке:
а) уксусная кислота — 2 %-й раствор;
б) уксуснокислый натрий — 2 %-й раствор, приготовленный на 10 %-м растворе уксусной кислоты.
4. Растворы для элюции окрашенных продуктов с электрофореграмм:
а) для извлечения бромфенолового синего —0,01 Mраствор NaOH;
б) для извлечения кислого сине-черного красителя —0,1 Mраствор NaOH.
Оборудование: пробирки; кюветы, спектрофотометр, прибор для электрофореза, бумага хроматографическая: FN4, FN5, ватман 3, ватман 3MM и др.
Получение сыворотки крови. 2 — 3 мл крови набирают в сухую центрифужную пробирку и оставляют на 1/2 — 1 ч. Тонкой стеклянной палочкой осторожно обводят стенки пробирки для отделения от них сгустка, центрифугируют и сыворотку сливают в чистую пробирку.
Подготовка камеры. Отсеки для электродов наполняют буферным раствором до одинакового уровня (во избежание перетекания буфера), примерно по 800 мл в каждый отсек. Bo внутренние части электродных отсеков погружают электроды. Ha листе хроматографической бумаги (18x45 см) (при использовании тонких сортов бумаги образцы лучше наносить на отдельные полоски шириной 4 —5 см) на расстоянии15 см от одного из его узких сторон простым мягким карандашом (графит препятствует растеканию жидкости) очерчивают места для нанесения проб. Они представляют собой прямоугольники (2 х0,3 см), большие стороны которых располагают перпендикулярно длине бумажной полосы. Расстояние между стартовыми зонами и краями электрофореграммы —2 см. Электрофореграмму пропитывают буфером, в котором будет проходить электрофорез. Для этого ее протягивают через кювету с буферным раствором. Концы бумажных полос (6 —8 см) не смачивают. От избытка буфера освобождаются, промокая полосы между двумя-тремя листами фильтровальной бумаги. Влажную электрофореграмму помещают в камеру на центральную горизонтальную пластинку (5), а концы опускают в наружные отделения электродных отсеков Прибор плотно закрывают крышкой, под которой находятся смоченные водой листы фильтровальной бумаги.
Проведение электрофореза. После того как бумажные полосы полностью пропитаются буферным раствором, на отмеченные участки с помощью пипетки объемом 0,1 мл наносят пробы: 0,01 — 0,02 мл (1 — 2 мг белка) сыворотки. Камеру закрывают крышкой и включают ток. Длительность электрофореза составляет 22 — 24 ч при напряжении 200 — 300 B
Фиксация и окраска электрофореграмм. По окончании электрофореза выключают ток и тотчас вынимают электрофореграммы из прибора. Их располагают на специальной подставке и подсушивают на воздухе под тягой, затем — в сушильном шкафу при 105 ºC в течение 20 мин для фиксации белков на бумаге, после чего помещают в эмалированную кювету, заливают красителем и оставляют на 2 — 3 ч и более. Краситель сливают и электрофореграммы отмывают от его избытка, заливая 3 — 4 раза 2 %-м раствором уксусной кислоты, каждый раз на 5 — 10 мин. Участки бумаги, не содержащие белка, должны быть полностью освобождены от красителя. Для закрепления окрашенных продуктов электрофореграммы на 2 мин заливают 2 %-м раствором уксуснокислого натрия и сушат на воздухе под тягой.
Определение соотношения отдельных фракций белка. При pH 8,6 белки сыворотки крови заряжены отрицательно и перемещаются в электрическом поле к аноду. Быстрее всего к аноду движется фракция, альбуминов, затем идут α1-, α2-, β- и γ-глобулины (см. Рис. 3).Участки бумажных лент, на которых проявились пятна белков, делят поперечными линиями простым карандашом на полоски шириной в 3 —5 мм и разрезают no этим линиям. Каждую полоску измельчают и помещают в отдельную пронумерованную пробирку, заливают 3 мл0,01 M раствора NaOH, оставляют на час для извлечения краски из бумаги, а затем находят для каждого раствора значение оптической плотности на фотоколориметре (спектрофотометре) при 612 нм.

Рис - Электрофореграмма сыворотки крови человека и кривая распределения белковых фракций
Параллельно обрабатывают контрольную пробу. Для нее вырезают полоску из неокрашенных участков электрофореграммы.
Ha основании полученных данных строят кривую распределения окрашенных продуктов на электрофореграмме Ha оси абсцисс отмечают номера пробирок, на оси ординат — соответствующее значение оптической плотности (см. Рис.3). Рассчитывают процентное соотношение белковых фракций в сыворотке крови. Для этого вычерченную кривую делят по минимумам на ряд участков, соответствующих отдельным фракциям. Величина площади каждого участка пропорциональна количеству краски, соединившейся с белком данной фракции. Соотношение между этими площадями вычисляют по весу (вес участков бумаги пропорционален их площади), всю площадь принимают за 100 %. При наличии денситометра соотношение белковых фракций в сыворотке крови можно определить из денситограммы.
Предварительно определяя содержание белка в сыворотке, рассчитывают его количество для каждой фракции.
ЛАБОРАТОРНАЯ РАБОТА № 32. ОПРЕДЕЛЕНИЕ АМИННОГО АЗОТА ФОРМОЛЬНЫМ ТИТРОВАНИЕМ ПО УПРОЩЕННОМУ МЕТОДУ ЗЕРЕНСЕНА-ГАВРИЛОВА.
Белковые гидролизаты представляют собой смесь аминокислот и простейших пептидов. Сырьем для получения гидролизатов могут служить любые полноценные по аминокислотному составу природные белки. В зависимости от сырья все белковые гидролизаты подразделяются на три группы: гидролизаты из крови и ее составных частей, из казеина, из растительных белков.
Гидролиз белка можно осуществить тремя путями: действием протеолитических ферментов (трипсин, пепсин, папаин), кислот и щелочей. В зависимости от условий гидролиз доводят до полного расщепления белка на аминокислоты или часть его сохраняют на пептиды.
Как известно, расщепление белков в организме до простейших пептидов и аминокислот происходит под действием ферментов пищеварительных соков. Аналогичное расщепление белка возможно и вне организма. Для этого к белковому субстрату (кровь, плазма, сыворотка крови и др.) добавляют ткани поджелудочной железы и слизистую оболочку кишечника, желудочный сок или чистые ферменты – пепсин, трипсин, папаин. Затем смесь помещают в термостат на несколько дней, и белки расщепляются до пептидов и аминокислот. Этот способ расщепления белка называется ферментативным, а полученный при этом раствор - ферментативным гидролизатом. Преимущество ферментативного гидролиза заключается в том, что аминокислоты не разрушаются и не происходит их рацемизация. Недостатками метода является сравнительно медленное течение гидролиза, возможность попадания в гидролизат продуктов расщепления самих протеолитических энзимов и возможность загрязнения препарата бактериями.
Гидролиз может происходить при кипячении белков в растворах кислот – кислотный гидролиз. Кислотный метод гидролиза прост и исключает возможность бактериального загрязнения среды. Его недостатки: в процессе гидролиза может разрушиться одна из аминокислот – триптофан, в более слабой степени окисляются треонин и серин. Кроме того, при кислотном гидролизе белков крови, содержащих углеводы, образуются побочные продукты – гумины. Некоторые из них оказывают неблагоприятный биологический эффект.
Химический гидролиз белка является более жестким процессом, как правило, требующим высоких температур и, сравнительно непродолжительного времени. Он осуществляется с помощью кислот и щелочей. Однако расщепление белка с помощью щелочей мало приемлемо в связи с тем, что при таком виде гидролиза разрушается большая часть диаминокислот, интенсивно разлагаются, взаимопревращаются и рацемизируются другие аминокислоты, например, оксиаминокислоты, цистеин.
При кислотном гидролизе тоже имеет место частичное разрушение некоторых аминокислот, но весьма незначительное, позволяющее в основном иметь представление о составе и характере промежуточных продуктов молекулы белка. В первую очередь, при кислотном расщеплении белка подвергается разрушению триптофан, причем интенсивность разрушения этой аминокислоты связана с наличием примеси углеводов и металлов в гидролизуемом белке. Считают, что триптофан превращается в дикарбоновую аминокислоту, а продукты разрушения триптофана – индоловые ядра, конденсируясь с альдегидами, образуют гуминовые вещества.
При кислотном гидролизе также подвергаются частичному разрушению серин, треонин, цистин, тирозин, фенилаланин. Однако процент разрушения этих аминокислот невелик.
В зависимости от избранной концентрации кислоты и длительности ее воздействия на белок интенсивность гидролиза будет протекать различно.
Помимо пептидов в процессе расщепления белка образуется множество других веществ. В состав белкового материала почти всегда входят углеводы, которые также как и белок подвергаются гидролизу и образуют продукты распада; свободные аминокислоты и продукты их окисления, а также продукты вторичной конденсации аминокислот и сахаридов еще более усложняют состав гидролизата.
Соблюдение определенного температурного режима при гидролизе белка позволяет уменьшить дезаминирование оксиаминокислот, хотя в общем-то распад этих аминокислот невелик.
Разрыв пептидных связей при гидролизе белка не является беспорядочным процессом, а происходит в определенных местах и носит закономерный характер. Пептидные связи, образованные оксиаминокислотами при кислотном гидролизе, менее стойки и раскрываются раньше, чем пептидные связи, образованные другимим аминокислотами.
Выбор метода гидролиза для получения лечебного препарата является существенным моментом.
На первый взгляд расщепление белка с помощью ферментов является более приемлемым, поскольку оно соответствует условиям пищеварения в желудочно-кишечном тракте. При расщеплении белка ферментативным путем нет опасности разрушения аминокислот. Вместе с тем, ферментативный гидролиз таит в себе возможность бактериального загрязнения. Температура, оптимальная для действия ферментов (37-78*C), является наиболее благоприятной и для жизнедеятельности микроорганизмов, особенно если учесть, что в некоторых случаях вместо ферментов пользуются поджелудочной железой и слизистой тонкой кишки, взятыми у животных в момент убоя.
Кроме того, конечный продукт ферментативного гидролиза может быть загрязнен примесями чужеродного белка, каковым является сам фермент.
Расщепление белка с помощью минеральных кислот, с этой точки зрения более приемлемо. Кислая среда в сочетании с высокой температурой полностью устраняет возможность бактериального загрязнения гидролизуемой смеси. Это обстоятельство является особенно существенным в том случае, когда дело идет о массовой заготовке белковых гидролизатов в производственных условиях.
Вместе с тем, кислотный гидролизат может быть получен таким образом, что он будет полноценным по аминокислотному составу. Разрушение некоторых аминокислот (триптофан), имеющее место при полном гидролизе белка, может быть сведено к минимуму, если избрать более мягкие условия гидролиза, вызывающие только частичное расщепление белка.
Белковые гидролизаты применяют при заболеваниях, протекающих с белковой недостаточностью, и при необходимости усиленного белкового питания, например, при заболеваниях желудочно-кишечного тракта с нарушениями всасывания белков, интоксикациях, ожоговой болезни, лучевой болезни и др., а также при невозможности питания через рот (например, после операций на пищеводе, желудке и т. п.) и для улучшения репаративных процессов в послеоперационном периоде.
Принцип метода.
Основан на блокировании формальдегидом при рН=7,0 свободных аминогрупп и титровании щелочью эквивалентного количества карбоксильных групп. Начало и конец титрования определяют потенциометрически.
Реактивы: 1. Раствор 0,1 NNaOH;
2. Раствор 0,1 NHCl;
3. Формалин (40% раствор формальдегида). Перед каждым определением рН формалина доводят до 7,0.
Ход определения.
В химических стаканчик вносят 2 мл испытуемого раствора, прибавляют 18 мл дистиллированной воды и нейтрализуют в зависимости от рН раствора 0,1 NNaOH или 0,1 NHCl до рН=7,0. Вносят 2 мл раствора формольной смеси и титруют 0,1 NNaOH до рН=9,2.
По количеству миллилитра 0,1 NNaOH, пошедшему на титрование, определяется аминный азот в миллиграмм-процентах.
Расчет

где: А – количество миллилитров 0,1 NNaOH, пошедшим на титрование испытуемого раствора;
V – количество испытуемого раствора, взятого на титрование;
Х – количественное содержание аминного азота в миллиграмм-процентах;
1,4 – эквивалент азота, соответвущий 1 мл 0,1 NNaOH.
ОСНОВНЫЕ ПОНЯТИЯ И ТЕРМИНЫ
Ароматичность – особый вид резонансного взаимодействия, при котором сопряжение реализуется в замкнутом цикле.
Биоорганическая химия– наука, изучающая строение, свойства и биологические функции органических веществ, участвующих в процессах жизнедеятельности.
Биополимеры– высокомолекулярные природные соединения, являющиеся структурной основой всех живых организмов и играющие определённую роль в процессах жизнедеятельности.
Биорегуляторы– соединения, которые химически регулируют обмен веществ.
Водородная связь– электростатическое взаимодействие атома водорода связанного с сильно электроотрицательным элементом, с с неподеленной парой электронов другого достаточно электроотрицательноготатома этой же или другой молекулы.
Гибридизация– смешение различных по форме орбиталей (s,p) при образовании связи.
Гомологи– это группа родственных органических соединений с однотипной структурой, каждый последующий член которой отличается от предыдущего на гомологическую разность (СН2)n.
Изомеры– вещества с одинаковым качественным и количественным составом, но различным строением и свойствами.
Индуктивный эффект– способность заместителя поляризовать σ-связь.
Ионная связь– ион-ионные взаимодействия, возникающие в результате полного перехода электронов от одних атомов к другим.
Ковалентная связь– химическая связь, образованная за счет обобществления электронной пары.
Конформеры– молекулы в определенной пространственной ориентации (конформации), в которую её атомы самопроизвольно возвращаются после небольших сдвигов при повороте по σ-связи.
Мезомерный эффект– влияние заместителя, обладающего р- или π- электронами, в результате которого происходит перераспределение π-электронной плотности связи.
Молекулярная спектроскопия– группа физических методов анализа основанных на взаимодействии вещества или смеси веществ, их растворов с различного вида излучениями.
Органические соединения– углеводороды и их производные.
π-связь– связь, образованная при боковом перекрывании негибридизованныхpz-атомных орбиталей с максимумом перекрывания по обе стороны от прямой, соединяющих ядра атомов.
σ-связь– одинарная ковалентная связь, образованная при перекрывании атомных орбиталей по прямой (оси), соединяющей ядра двух связанных атомов с максимумом перекрывания на этой прямой.
Сопряжение– взаимодействие групп атомов, содержащих π- и (или)р-орбитали, приводящее к широкой делокализации электронной плотности в молекуле.
Стереоизомеры– пространственные изомеры, характеризующиеся различием положения заместителей в молекулах относительно друг друга в пространстве.
Таутомерия– явление динамического обратимого превращения изомеров, протекающее с разрывом и образованием связей и сопровождающееся перемещением атомов (чаще всего протона) и реже групп атомов.
Функциональная группа– атом или группа атомов, определяющие принадлежность к определенному классу и ответственные за его химические свойства.
Энантиомеры– оптические изомеры, вращающие плоскость поляризации в разные стороны, но на один и тот же угол.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
Тюкавкина Н А., Бауков Ю И. Биоорганическая химия. М.: Медицина, 2005 - 542с
Овчинников Ю.А. Биоорганическая химия М.: Просвещение, 1987.- 815с.
Биохимия. Лабораторный практикум. Э 45 Учеб. пособие / Сост.
Сенчук В.В., Мохорева С.И., Н.М. Орел и др. – Мн.: БГУ, 2004. – 77 с
Биоорганическая химия: методическое руководство для самостоятельной подготовки студентов к практическим занятиям и экзамену/ сост. О.Ю. Орлова, Л.В. Власова, С.А. Куклина.– Киров: Кировская ГМА, 2009–60 с.
.Райлс А., Смит К., Уорд Р. Основы органической химии (для студентов биологических, медицинских и сельскохозяйственных специальностей). М.: Мир. 1983 - 352с.
Руководство к лабораторным занятиям по биоорганической химии" под редакцией Тюкавкиной Н.А., М.: Медицина, 1985 – 254с.
Николаев А..Я. Биологическая химия. М.: Высшая школа, 1989 - 495с.
Кутыга, О. Н. Лабораторный практикум по биохимии : учебное пособие/ О. Н. Кутыга, О. В. Вострикова – 2-е изд. испр. и доп. – Волгоград : ИУНЛ ВолгГТУ, 2011. – 64 с. Биохимия. Лабораторный практикум. Э 45 Учеб. пособие / Сост. Сенчук В.В.,
Мохорева С.И., Н.М. Орел и др. – Мн.: БГУ, 2004. – 77 с.
trotted.narod.ru›organic/lec-47/47.html