

Краткая теория по базисным функциям
.doc
Краткая теория по базисным функциям.
-
Не совсем научное, но более понятное введение.
HΨ=EΨ – уравнение Шредингера, уже знакомое вам. Здесь впервые в курсе квантовой химии вы видите волновую функцию. И с этого момента они будут везде, т.к. волновая функция – это как алфавит, но на языке квантовой механики. Чтобы описать любую химическую систему вы «раскладываете» ее до отдельных электронов.
Каждый электрон «сидит» на какой-либо атомной орбитали, в зависимости от соединения, которые вы изучаете. Между электронами происходит постоянное взаимодействие, т.е. орбитали, на которых находятся электроны, взаимодействуют между собой, иначе говоря – интерферируют(если мы смотрим с точки зрения квантовой механики, где орбитали представляются в виде волновых функций. Взаимодействие волн – это интерференция, подробнее об этом явлении вам рассказывали на физике) или перекрываются. Перекрывание бывает эффективное и неэффективное, т.е. в некоторых случаях интерференция орбиталей приводит к образованию хим связи, а в некоторых нет. От чего это зависит вы мне расскажете на защите или на к/р =)
В ходе взаимодействия двух электронов, находящихся на АО, в ряде случаев образуется МО (связывающая или разрыхляющая – это другой вопрос), которая представляет собой комбинацию из 2-х АО. Если перейти к языку квантовой механики, то МО – ЛКАО. Знакомое сочетание? Каждая молекулярная орбиталь представляет собой линейную комбинацию атомных орбиталей! Вот тут-то и появляются базисы. Базис – это алфавит. Набор волновых функций, с помощью которых вы можете описать ваше химическое соединение. Описываете вы его выстраивая орбитали: АО, из них МО. Таким образом, любая программа «видит» ваше соединение.
-
Перейдем к более формальной теории.
Базисный набор— набор функций, который используется для построения молекулярных орбиталей, которые представляются как линейная комбинация функций этого набора с определенными весами или коэффициентами. Обычно этими функциями являются атомные орбитали, центрированные на атомах, хотя иногда функции центрируют на связях, на половинах p-орбитали и т. п.
Выбор базиса представляет собой компромисс между вычисли- тельными возможностями и желаемой точностью. Существует два типа базисных функций (атомных орбиталей!) – орбитали слэйтеровского типа (STO – Slater Type Orbitals) и гауссовского типа (GTO – Gaussian Type Orbitals)
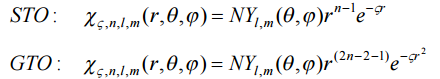
где N – нормировочный множитель, Yl,m – сферическая гармоника
Экспоненциальная зависимость от расстояния между ядром и электроном отражает точный вид орбитали атома водорода. Экспоненциальная зависимость гарантирует хорошую сходимость с увеличением числа функций. Но трёх- и четырёхцентровые интегралы нельзя рас- считать аналитически в базисе этих функций. Поэтому функции STO используются в основном для расчётов атомных или двухатомных сис- тем, где нужна высокая точность, а также в полуэмпирических методах, где трёх- и четырёхцентровые интегралы отбрасываются.
Экспоненциальная зависимость отличает GTO от STO в двух аспектах:
1) функции GTO плохо описывают поведение электрона вблизи ядра;
2) GTO слишком быстро спадают с удалением от ядра по сравнению с STO, вследствие чего "хвост" волновой функции плохо описывается с помощью GTO, что видно из графика

-
Классификация базисных наборов
*Курсивом даны мои комментарии к теории
Одним из важнейших факторов является количество используемых функций. Наименьшее число функций образует минимальный базисный набор. Этот набор соответствует числу функций, достаточных для размещения электронов в нейтральном атоме. Для водорода и гелия – одна s-функция(тут все просто. Одна функция на каждую орбиталь. У водорода 1 электрон сидит на1 орбитали. В базисе SZ будет 1 базисна функция типа 1s). Для атомов 2-го периода – 1s, 2s и три 2p функции. Следующим улучшением базисного набора является удваивание числа функций. Такой базис называется "дабл-зета" (Double Zeta) – DZ. Термин "зета" происходит от греческой буквы ς, которая обо- значает орбитальную экспоненту. Этот базис содержит две s-функции для атомов водорода и гелия, четыре s-функций и шесть p-функций для атомов 2-го периода(просто число функций из минимального базисного набора удваивается). Поэтому этот базис называется также двух- экспоненциальным. Необходим определённый компромисс для значений орбитальных экспонент. Базис DZ имеет двойное количество орбиталей с различными экспонентами. Более компактная функция соответствует большему значению экспоненты, и эта функция вносит больший вклад в образование σ-связи. Более диффузная функция имеет меньшее значение экспоненты и лучше описывает π-связь. Таким образом, двойной набор функций позволяет лучше описывать распределение электронов в разных направлениях
Химическая связь образуется валентными электронами. Удваивание числа функций позволяет также лучше описывать и óстовные 1s- электроны. Но эти орбитали практически не вносят вклад в образование связей. Поэтому почти всегда удваивают количество только валентных орбиталей. Такой базис называется валентно расщеплённым (split valence basis). Обычно термин DZ соответствует валентно расщеплённому базису, который также обозначается как VDZ (valence double zeta). Существуют также трижды, четырежды и более расщеплённые базисы. Из них более часто используется базис TZ (Triple Zeta) (не сложно догадаться, что для расчета числа базисных функций в данном базисном наборе, необходимо минимальное количество функций умножить на 3).
Валентно-расщепленные базисные наборы Попла:
В целом, разновидностей базисов гораздо больше, я останавливаюсь на тех, с которыми вы столкнетесь в курсе квантовой химии.
Недостатком всех базисов, оптимизированных по энергии, является то, что они главным образом зависят от волновой функции внутренних электронных слоёв. Энергия электронов орбитали 1s включает бόльшую часть общей энергии. Поэтому минимизация энергии в большей степени оптимизирует базисный набор для όстовных электронов, чем для валентных. Но с точки зрения химии последние наиболее важны. Кроме того, такие свойства, как поляризуемость, зависят в основном от "хвоста" волновой функции, который не важен для расчёта энергии. Базисные наборы, хорошо описывающие внешнюю часть волновой функции ("хвост"), должны быть очень большими. Это не самый эффективный способ построения базисного набора для описания внешней части волновой функции. Вместо этого в энергетически оптимизированные базисы включают диффузные функции – функции с малыми экспонентами ζ. Диффузные функции необходимы тогда, когда в структуре присутствуют слабо связанные электронные пары, как, например, в анионах или в возбуждённых состояниях. Диффузные функции необходимы также для исследования свойств, зависящих от "хвоста" функции (например, поляризуемость).
Несмотря на то, что электроны внутренних АО практически не участвуют в образовании химических связей, для представления этих АО требуется большее число гауссовых примитив. Причина этого кажущегося противоречия состоит в том, что внутренние АО дают наибольший вклад в полную энергию молекулы, поэтому плохое описание таких АО будет приводить к 7 значительным погрешностям расчета энергетических характеристик. В аббревиатуре валентно-расщепленных базисных наборов M-NPG количество гауссовых функций, описывающих внутренние АО, задается числом M, принимающим значения от 3 до 6. Сжатая часть валентной функции определяется числом N, обычно равным 2 или 3. Наконец, диффузная часть валентной функции часто описыватся одной гауссовой примитивой.
Такое разделение (расщепление) производится только для валентных электронов, поэтому базисные наборы такого типа называются валентно-расщепленными.

Основной недостаток валентно-расщепленных базисов состоит в том, что «центр тяжести» отрицательного заряда данной АО совпадает с ядром атома. Однако, в некоторых соединениях (высокополярных молекулах, малых циклах) активно проявляется тенденция смещения центра заряда. Чтобы учесть этот эффект в базисный набор включают функции более высокого побочного квантового числа: d-типа для тяжелых атомов и p-типа для водорода.
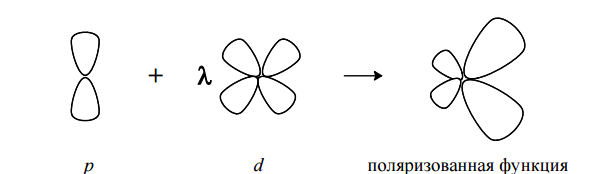
ПРИМЕР:
Один из наиболее популярных базисных наборов 6-31G*. Давайте «расшифруем» его. Это валентно-расщепленный базисный набор, в котором каждая остовная АО представлена 6 гауссовскими примитивами. Каждая валетная орбиталь расщепляется на 2 части, одна из которых описывается 3 функциями, вторая – одной. Также дополнительно для всех неводородных атомов учитываются поляризационные d-функции в количестве 6 штук. Используются орбитали гауссовского типа.
Ниже представлена схема расчета базисных функций для молекулы HF в основных базисах.
Более подробная информация по классификации базисных наборов представлена в учебнике проф. В.Г. Цирельсона, а также в ваших раздаточных материалах «Методы расчета молекул»
КАК РАССЧИТАТЬ ЧИСЛО БАЗИСНЫХ ФУНКЦИЙ
Пример: РАСЧЕТ МОЛЕКУЛЫ HF В РАЗЛИЧНЫХ БАЗИСАХ.
-
Базисные наборы на основе орбиталей слейтеровского типа (STO):
-
SZ – single-:
HF = H (1s) + F (1s + 2s + 2p*3) = 6
-
DZ – double-:
HF = H (1s)*2 + F (1s + 2s + 2p*3)*2 = 12
-
TZ – triple-:
HF = H (1s)*3 + F (1s + 2s + 2p*3)*3 = 18
-
Базисные наборы Попла на основе гауссовых примитивов:
-
Минимальные базисы STO-NG, N = 2, 3, 4, 5, 6:
HF = H (1s) + F (1s + 2s + 2p*3) = 6
Контрактация для
любой АО. Пример, 1s:
(1s)
=![]() aiGi,
aiGi,
при N=3
STO-3G:
1s
=
![]() aiGi
, ai
– коэффициент контрактации.
aiGi
, ai
– коэффициент контрактации.
-
Расширенные валентно расщепленные (SV) базисы:
2.1 n-ijG (например, 3-21G, 6-21G, 4-31G, 5-31G, 6-31G):
HF = H (1s*2) + F (1s + (2s + 2p*3)*2) = 11
Контрактация для остовной АО, например, 1sF:
1sF
=
![]() aiGi,
при N=6,
aiGi,
при N=6,
6-31 G: 1sF
=
![]() aiGi
aiGi
Контрактация для любой валентной АО, например, 1sН:
1sН
= c1![]() aiGi
+ c2
aiGi
+ c2![]() ajGj,
при i=3, i=1,
ajGj,
при i=3, i=1,
6-31 G: 1sН = c1aiGi+ c2Gj
ci – варьируемый коэффициент разложения МО по базисным функциям.
2.2 n-ijkG (6-311G):
HF = H (1s*3) + F (1s + (2s + 2p*3)*3) = 16
Контрактация для
остовной АО, например, 1sF:
1sF
=
![]() aiGi,
при N=6,
aiGi,
при N=6,
6-311 G:
1sF
=
![]() aiGi
aiGi
Контрактация для любой валентной АО, например, 1sН:
1sН
= c1![]() aiGi
+ c2
aiGi
+ c2![]() ajGj+
c3
ajGj+
c3![]() akGk
; i=3, j=1, k=1,
akGk
; i=3, j=1, k=1,
6-311 G:
1sН
= c1![]() aiGi+
c2Gj+
c3Gk
aiGi+
c2Gj+
c3Gk
-
Широкие базисы
3.1 Поляризационные функции:
Базис*: 3-21 G*,6-21 G*,4-31 G*, 5-31 G*, 6-31 G*, 6-311 G*:
для каждого неводородного атома (исключения - Н и Не) добавлен набор из 6-ти поляризационных d-функций.
Пример: 6-31 G*:
HF = H (1s*2) + F (1s + (2s + 2p*3)*2 + 3d*6) = 17
Базис**, 3-21 G**, 6-21 G**, 4-31 G**, 5-31 G**, 6-31 G**, 6-311 G**: для каждого неводородного атома добавлен набор из 6-ти поляризационных d-функций. а для атомов Н и Не- 3-и p функции.
6-31 G**:
HF = H (1s*2 + 2р*3) + F (1s + (2s + 2p*3)*2 + 3d*6) = 20
3.2 Диффузные функции:
-
Базис+: 3-21+ G**,6-21+ G**,4-31+ G**, 5-31+ G**, 6-31+ G**,
6-311+ G**: для каждого неводородного атома (за исключением Не) добавлены 4 диффузные функции: 1 s-типа и 3 p-типа.
6-31+ G**:
HF = H (1s*2 + 2p*3) + F (1s + (2s + 2p*3)*2 + 3d*6 + 3s +3p*3) = 24
-
Базис++, 3-21++ G**,6-21++ G**,4-31++ G**, 5-31++ G**, 6-31++ G**,
6-311++ G**: для каждого неводородного атома добавлены диффузные 4 диффузные функции: 1 s-типа и 3 p-типа а для Н и Не- по 1-й s-функции.
6-31++ G**:
HF = H (1s*2 + 2р*3 + 2s) + F (1s + (2s + 2p*3)*2 + 3d*6 + 3s + 3p*3) = 25
! Важное пояснение: логичный вопрос, который может последовать после теории: зачем мы учитываем «пустые» орбитали в молекулах? Если из 3-х p-орбиталей занято 2, то почему кол-во базисных функций не меняется?
-
В прогамме, помимо базиса, вы, что логично, задаете соединение: его состав/формулу, возможно, стркутуру. Каждый атом имеет свое кол-во электронов и программа, естественно, это учитывает. Вы же не можете «вытроить» у углерода 2-й электронный подуровень, исключив некоторые p-орбитали, даже если они пустые? Они же есть, более, того, отсюда вытекает п.2
-
Интерференция даже «пустых» орбиталей вносит вклад в общую энергию системы. Насколько этот вклад большой – зависит от рассматриваемого соединения, от его природы в том числе. Выбор базиса, отчасти, зависит от типа вашего соединения, от состояния, в котором оно находится (это мб промежуточный продукт, цвиттер-ион, комплекс переходного металла и др). Иногда, эффекты от интерференции «пустых» орбиталей достаточно сильны.
-
В рамках «ручного» расчета базисных функций, вы считает кол-во функций для конкретного соединения, которые описывают ОРБИТАЛИ. Есть ли на них электроны, сколько их и тд – это уже другой вопрос