

Как нарисовать молекулу
.docМеню программы:

Основные кнопки управления:
![]()
Слева направо: построить молекулу, выделить, вращение молекулы, вращение по кругу, перемещение, выбор, увеличение и уменьшение, еще вариант перемещения.
Как нарисовать молекулу?
В левом верхнем
углу выбираем первый курсор
![]() ,
в меню
,
в меню
![]() выбираем углерод и рисуем «основу»(без
водородов) нашего соединения. Щелчком
мыши «ставим» атом, нажатием на него
«протягиваем» связь. Двойной щелчок по
связи – двойная связь. Убрать атом или
понизить порядок связи – правая кнопка
мыши.
выбираем углерод и рисуем «основу»(без
водородов) нашего соединения. Щелчком
мыши «ставим» атом, нажатием на него
«протягиваем» связь. Двойной щелчок по
связи – двойная связь. Убрать атом или
понизить порядок связи – правая кнопка
мыши.
Если необходимо добавить еще какой-либо атом, которого нет верхнем меню, заходим в BUILT—DEFAULT ELEMENT и выбираем необходимый элемент в таблице.


К огда
нарисовали все, КРОМЕ водородов, заходим
в меню BUILT –ADD HYDROGEN.
Также можно нажать кнопку
огда
нарисовали все, КРОМЕ водородов, заходим
в меню BUILT –ADD HYDROGEN.
Также можно нажать кнопку
![]() (для более новых версий).
Не
забудьте проверить, правильно ли
добавились водороды! При необходимости
– скорректируйте результат.
(для более новых версий).
Не
забудьте проверить, правильно ли
добавились водороды! При необходимости
– скорректируйте результат.
Т еперь
необходимо получить изображение молекулы
для того, чтобы представить ее в отчете.
Ниже описаны опции, которые помогут
настроить его под ваши запросы.
еперь
необходимо получить изображение молекулы
для того, чтобы представить ее в отчете.
Ниже описаны опции, которые помогут
настроить его под ваши запросы.
Чтобы отобразить символы атомом, заходим в меню DISPLAY—LABELS
Также в меню DISPLAY ---RENDERINGS Можно выбрать модель атома(линии, шарики), поварьировать параметры отображения и т.д. Фон экрана меняется в меню FILE—PREFERENCES
Теперь можно переходить к самому расчету. Первое, что необходиом сделать – провести «двойную» оптимизацию геометрии. Первый этап – оптимизация методом молекулярной механики, он служит как вычислительная модель для оценки потенциальной энергии молекулы с учетом всех степеней свободы. Также помогает избежать возможных ошибок, при дальнейших расчетах.
!!!В меню file –startlog начинаем запись файла *.log, в котором будет записана вся информация о расчете!!!
-
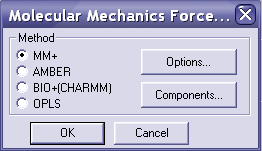
В меню setup выбираете molecular mechanics и MM+, нажимаете ОК.

Затем compute-geometry optimization-OK. При сравнительно небольших молекулах число циклов можно не менять, оставив значение по умолчанию, которое предлагает программа.


При окончании расчета внизу должна появиться подобная запись
![]()
Если написано NO, это значит, что сходимость в энергии не достигнута, т.е. необходимо запустить оптимизацию еше раз, пока не будет достигнута сходимость(converged=YES).
В меню display-labels можно длину связи (bond length), после выбора закрываете окно и у вас на соединении отображаются эти величины. Их нужно переписать или сделать скрин картинки, чтобы потом вставить в отчет!
-
Повторяем оптимизацию, только выбираем semi emperical в меню setup(метод AM1, MNDO и др.) и также compute – geometry optimization, при необходимости увеличивая число циклов для расчета.
Вот что получается по окончанию(успешному) оптимизации:
![]()
В меню DISPLAY-LABELS по очереди выбираете заряд(charge) и длину связи (bond length), после выбора закрываете окно и у вас на соединении отображаются эти величины. Их нужно переписать или сделать скрин картинки.
-
Теперь переходим к построению МО. В меню compute—orbitals появляется следующее окошко. Вводя номер ВЗМО и НСМО(по очереди) можно посмотреть их энергию, а нажав кнопку plot ---ОК можно построить изображение МО на вашей молекуле. Если видно плохо, поменяйте RENDERINGS или нажмите F4 и выбирете парметры для 3d-графика(wire mesh и contour value).

Последовательно сохраняем диаграмму МО, а также изображения МО(ВЗМО И НСМО) на флешку/диск для последующего отчета.
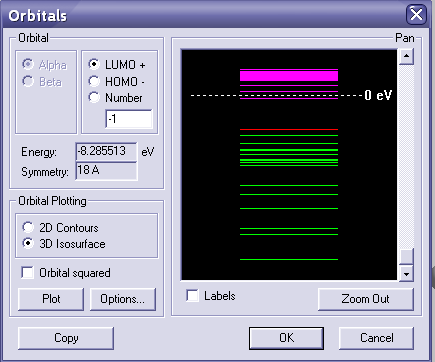

*При расчете номера ВЗМО и НСМО нужно помнить, что полуэмперические методы учитывают ТОЛЬКО валентные электроны!
Теперь можно остановить запись log-файла(file-stop log)
Например, получается вот так:

В вашем log-файле, как и в работе 1 есть информация об энергиях всех орбиталей!
-
Последнее, что необходимо сделать – это построить электростатический потенциал соединения для оценки его реакционной способности. Идем в compute-plot molecular graphs и выбираем следующие параметры:

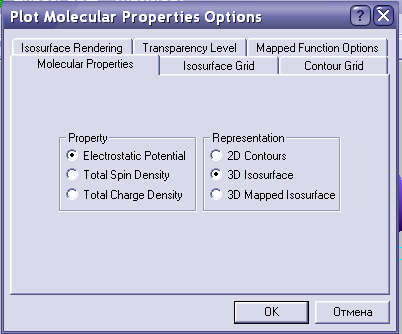
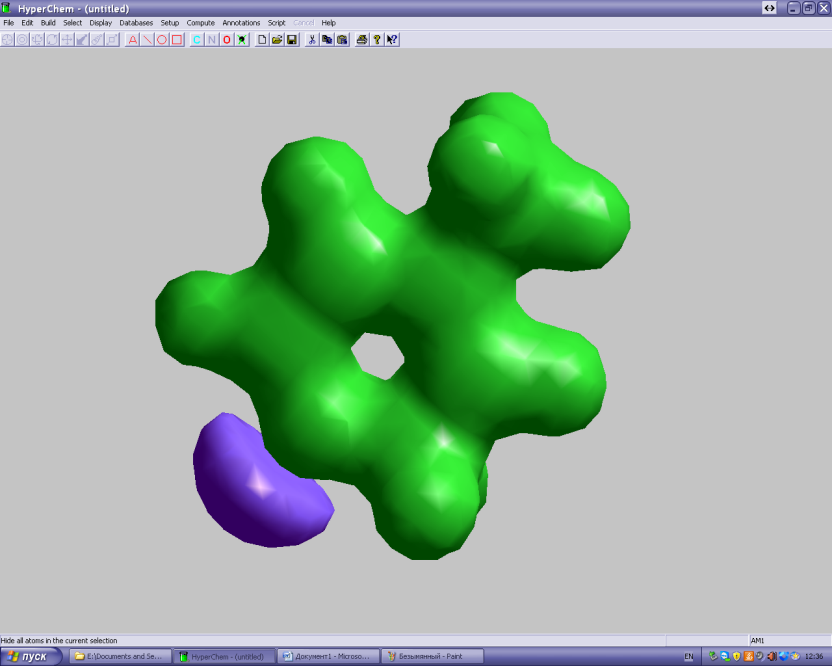
Помните, что нажав F4 можно отрегулировать изображение: выбрать тип поверхности, шаг и т.д. Если у вас есть сильный ЭО заместитель, а потенциал весь положительный, это значит, что значение контура поверхности слишком большое. Можно попробовать его уменьшить и посмотреть, что получается. Например:
Что должно быть в отчете:
-
Сама молекула
-
Заряды и длины связей ПОСЛЕ оптимизации по методу ММ
-
Заряды и длины связей после оптимизации по методу AM1
-
Полные энергии после оптимизации по методу MM и AM1
-
Орбитали(ВЗМО и НСМО), их энергии и изображение
-
Графическое распределение электростатического потенциала
-
Не забудьте сохранить себе log-файл