64
Геманпюмы
и тромбоцитопении синдром
эия,
мышечная гипотония. Довольно рано на
глазном дне обнаруживается симптом
«вишневой косточки»; к концу 1-го
года жизни наступает слепота, обусловленная
атрофией зрительных нервов, интеллект
снижается до степени идиотии. Постепенно
развивается полная обездвиженность,
появляются судороги, преимущественно
тонические, не поддающиеся
противосудорожной терапии. В конечной
стадии заболевания отмечается де-
церебрационная ригидность. Смерть
обычно наступает в 3—4 года. Вследствие
дефицита гексозаминидазы А в головном
мозге, печени, селезенке и эритроцитах
накапливаются
GM.-ганглиозиды
Популяционная
частота — 1 ;
5000 среди евреев-ашкенази родом из Польши
и Литвы Тип
нас и'Оования
— аутосом но-рецессивный. Ген
локализован на 15q23-q24.
ЛИТЕРАТУРА
Barnes
IX.
Mislra
V.
P
. Young £
P
An adult onset hexosaminidase A deficiency syndrome with sensory
neuropathy and intemuclear ophthalmoplegia J. Neurol.
Neurosurg. Psychiatry. 1991. v 54.
p. 1112— 1113.
Kelh
Т.
E..
Chase С
A
Kaback U M etal
Tay- Sachs disease: high gene frequency in a non-Jewish population.
— Am J. Hum Genet., 1975,
v. 27, p. 287—291.
O
'Brien J S. Okada S.. Gillerup D L el al
Tay- Sachs disease: detection of heterozygotes and homo- zygotes by
serum hexosaminidase assay. — N Engl. J.
Med.. 1970, v 283, p
15 20
Смг-ганглиозидоз,
тип II
Gm
-gangliosidosis, type II
MIM: 268800
Синонимы:
См,-ганглиозидоз с дефици-
том
гексозаминидазы А и В; болезнь Санд-
хоффа.
Минимальные
диагностические признаки:
отставание
в психомоторном развитии; сим-
птом
«вишневой косточки» на глазном
дне;
дефицит гексозаминидазы А и В.
Клиническая
характеристика.
Типичны
отставание в психомоторном
развитии в
первые 6 мес жизни,
гипотония, слепота. На
глазном дне
определяется симптом «вишне-
вой
косточки». Характерны гепатосплено-
мегалия.
кардиомегалия, «кукольное лицо»;
возможна
макроцефалия. Неврологические нарушения
появляются к 8 мес жизни. По мере
прогрессирования появляются клони-
ческие судороги, спастический тетрапарез.
Смерть наступает в 2—3 года. В костном
мозге обнаруживаются «пенистые» клетки.
В основе заболевания лежит дефицит
ферментов гексозаминидазы А и В, что
приводит к накоплению ганглиозида
Gm.
в
нервной системе и внутренних органах.
Тип
наследования
— аутосом но-рецессив- ный.
Дифференциачьный
диагноз:
GM
-ганглио-
зидоз, тип I.
ЛИТЕРАТУРА
Der
Kalouslian I Н
Khoun
V/
Y.
Hallal
R. ei al
SandhofT disease: a prevalent form of infantile Gm
.-gangliosidosis in Lebanon. — Am J. Hum.
Genet. 1981. v. 33. p
85—89
Low
den J. A.. lyes E. J Keene D L el al.
Carrier detection in Sandhoffdisease —Am. J
Hum Genet., 1978. v. 30. p.
138 -145.
Neufeld
E F Natural
history and inherited disorders of a lysosomal ensyme.
beta-hexosaminidase — J Biol. Chem.. 1989.
v. 264, p 1927 1930.
TanakaA..
OhnoK.. Suzuki К
Gm
-gangliosidosis В variant analysis of
beta-hexosaminidase alpha gene abnormalities in seven patients. —
Am J. Hum. Genet . 1990. v. 49.
p. 329—339
Гемангиомы
и тромбоцитопении синдром
Hemangioma
and thrombocytopenia
syndrome
MIM:
141000
Синоним:
синдром Казабаха— Мерита.
Минимальные
диагностические признаки: гемангиома.
экхиматозная и петехиальная сыпь.
Клиническая
характеристика.
У всех больных имеется стремительно
разрастающаяся гемангиома в сочетании
с тромбоци- топенией
(рис. 35). Кроме
тромбоцитопении, обнаруживаются
снижение гемоглобина и числа
эритроцитов (75%), гипофибри- ногенемия,
снижение протромбинового индекса.
Клинически это проявляется множественными
экхимозами и петехиями. Заболевание
может проявиться в период с рождения
до 70 лет (чаще всего в раннем возрасте).
Соотношение
полов
— Ml
:
Ж1.
Тип
насчедования
неизвестен.Дифференциачьный диагноз: цероидлипо- фусциноз; Ом,-ганглиозидоз; лейкодистро- фии; gMi-ганглиозидоз, тип II.
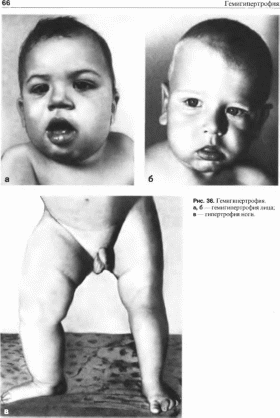
Гемнгнпертрофня
65
Рис.
35.
Синдром гемангиомы и тромбоцитопснии.
Гигантская гемангиома ноги и множественные
подкожные 1смаш помы
ЛИТЕРАТУРА
David
Т.
J..
Evans D J . Stevens R F.
Haemangi- oma with thrombocytopenia (Kasabach Merrill syndrome) —
Arch Dis. Child., 1983, v. 58,
p. 1022 1023.
Гемигипертрофия
H
emihy pert rophy MIM: 235000
Минимальные
диагностические признаки: асимметрия
тела.
Клиническая
характеристика.
Выраженность гемигмпертрофни
значительно варьирует от увеличения
всей половины тела (тотальная
гемигипертрофия) до увеличения частей
тела (частичная или сегментарная
гемигипертрофия)
(рис. 36, а, б, в).
Иногда увеличиваются несмежные участки
тела (перекрестная гемигипертрофия).
Гипертрофируются не только кожа,
мягкие ткани и кост- но-мышечная система,
но также и внутренние органы пораженной
половины. Специфических изменений
лабораторных показа
телен
не выявлено. При рентгенологическом
обследовании находят опережение
костного возраста гипертрофированных
конечностей, увеличение внутренних
органов. Приблизительно в 20 30% случаев
встречаются пиг- менные невусы.
гемангиомы. умственная отсталость.
аномалии мочеполовой системы. Больные
предрасположены к опухолям почек
(опухоль Вильмса), коры надпочечников
и печени (в детском возрасте). Заболевание
проявляется вскоре после рождения; с
возрастом выраженность асимметрии
может нарастать или уменьшаться.
Соотношение
по юв — Ml : ЖI
Тип
наследования
неизвестен.
Дифференциальный
диагноз
гемиатрофия врожденная, синдром Рассела
Сильвера, синдром Беквита Видемана.
ЛИТЕРАТУРА
Fraumeni
J F,
Geiser
С
F.
Mamnumg
M. D Wilms
tumor and congenital hemihypertrophy report of five new cases and
review of literature. Pediatrics, 1967, v.
40. p. 886.
5
173