зованным
Повышена чувствительность к
ультрафиолетовым лучам; депигментиро-
ванные участки подвержены солнечным
ожогам. Более чем в 50% случаев болезнь
проявляется до 20 лет. Заболевание может
прогрессировать, а в некоторых случаях
подвергаться обратному развитию.
Повышен риск рака кожи.
Популяционная
частота
— 1 : 100.
Соотношение
полов
— Ml
:
Ж1.
Тип
наследования
— предположительно аутосомно-доминантный
с неполной пенет- рантностью.
Дифференциальный
диагноз: разные
формы альбинизма.
ЛИТЕРАТУРА
Hafer
М
The genetic of vitiligo. - Acta Derm
Ve- nereol, 1983. v. 63, p.
249—251
Влагалища
аплазия
Синоним
синдром Рокитанского- Кюс- тера—Хаушера.
Минимальные
диагностические признаки: аплазия
влагалища, рудиментарная матка, аменорея;
кариотип 46.ХХ.
Клиническая
характеристика
Отмечается недоразвитие производных
мюллеровых протоков: фаллопиевых труб,
тела и шейки матки, верхней части
влагалища. Иногда имеются соединительнотканные
тяжи или рудиментарные фаллопиевы
трубы. Наружные гениталии развиты
по женскому типу. Девственная плева
интактна, влагалище укорочено и
заканчивается слепо. Основная жалоба
— первичная аменорея. Вторичные половые
признаки развиты нормально. Яичники
не изменены. Уровень стероидов и го-
надотропинов в плазме нормальный. Часто
наблюдается односторонняя аплазия
почки, тазовая почка, эктопия почки.
Повышена частота скелетных дефектов,
особенно аномалий позвоночника.
Больные бесплодны.
Тип
насчедования
не установлен. Описаны случаи заболевания
у сестер
Дифференциальный
диагноз:
синдром тес- тикулярной феминизации,
неполная поперечная перегородка
влагалища; неполное слияние мюллеровых
протоков.
ЛИТЕРАТУРА
Jones
Н И J. Wheeless С
R
Salvage of the rep
roductive
potential of women with anomalous development of the Mullenan
ducts: 1868 1968 2068. — Am J. Obstet.
Gynecol., 1969. v 104. p.
348
Wulfsberg
E. A .
Grigbsy
Т.
M
Rokitansky sequence in association with the
facio-auriculo-vertebral sequence- part of a mesodermal malformation
spectrum1' — Am. J Med
Genet. 1990, v 37 p
100 102.
Влагалища
атрезия
Синоним:
аплазия мюллеровых протоков.
Минимальные
диагностические признаки: атрезия
влагалища при нормальном карио- типе
(46.ХХ).
Кчиническая
характеристика.
Различают 4 формы атрезии влагалища:
гименальная, ретрогименальная,
вагинальная и церви- кальная. Нижняя
часть влагалища замешена фиброзной
тканью. Верхние его отделы, шейка и тело
матки, фаллопиевы трубы, яичники и
наружные половые органы сформированы
правильно В пубертатном периоде
появляются вторичные половые признаки,
но менструации отсутствуют: возможен
гид- рометрокольпос. Атрезия влагалища
может сочетаться с атрезией анального
отверстия (полной или свищевой) и
агенезией мочевой системы
Поп
)■. тяционная час mama
от
2 10 000 до 4: 10 000
Тип
наследования
— предположительно аутосомно-доминантный,
ограниченный полом
Дифференциальныи
диагноз: все
формы женского псевдогермафродитизма,
аномалии почек, гениталий и среднего
уха.
ЛИТЕРАТУРА
Shakier
М
Н
Aplasia of the mullenan system: evidence for probable
sex-limited autosomal dominant inheritance. —
Birth Defects. 1978. v. X1V(6 c). p.
157—165.
Волмана
болезнь
Wolman
disease MIM 278000
Синоним:
первичный семейный ксантома- тоз с
кальцификацией надпочечников.
Заболевание
описано в 1961 г. М. Wolman с
соавт.
Минимачьные
диагностические признаки:Vagina, absence of m1m: 227000
Vaginal atresia mim 158300
Рис.
32. Синдром
скрученных волос и глухоты.
а
редкие скрученные волосы;
б микроскопическая
картина волос: неровные контуры.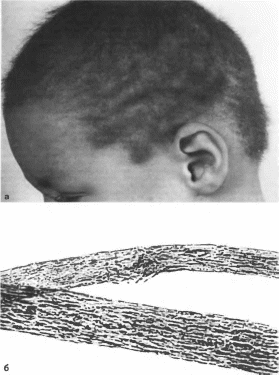
гепатоспленомегалия,
увеличение и калыди- фикация надпочечников,
«пенистые» клетки в костном мозге,
вакуолизация лимфоцитов.
Клиническая
характеристика.
В первые недели жизни появляются рвота,
частый жидкий стул, отставание в весе.
Отмечаются выраженная гепатоспленомегалия.
ксанто- матоз кожи. Рентгенологически
выявляют увеличенные надпочечники с
кальцификата- ми. При гистологическом
исследовании в пе- ченн, селезенке,
надпочечниках, тимусе, костном мозге
обнаруживают «пенистые» клетки с
высоким содержанием триглицери- дов и
эфиров холестерина.
Метаболический
дефект заключается в дефиците кислой
липазы
Тип
наследования
- аутосомно-рецессив- ный. Ген локализован
на 10q24-q25.
Дифференциальный
диагноз:
болезнь Ни- мана Пика: липогрануломатоз
Фарбера.
ЛИТЕРАТУРА
Burton
В.
К . ReedS.
P. Acid lipase
crossreacnng material in Wolman disease and cholesterol ester
storage disease. — Am. J. Hum. Genet..
1981. v. 33, p. 203
208.
Волос
скрученных и глухоты синдром
Pili
torti-deafness syndrome M1M: 262000
Описан
в 1965 г. R. Bjornsiad.
Минимальные
диагностические признаки: скрученные
волосы, глухота.
Клиническая
характеристика.
Наблюдаются аномалии волос и
нейросенсорная глухота. проявляющаяся
на 1-м году жизни Волосы желобоватые,
плоские, скрученные, ломкие
(рис. 32, а, б).
Потеря слуха обусловлена дефектом
улитки. Отмечается связь между
степенью поражения волос и тяжестью
глухоты.
Тип
наследования
— предположительно аутосом но-рецесснвны
й
Дифференциальный
диагноз:
синдром Мен- кеса.
ЛИТЕРАТУРА
Crandall
В
F.
Sumic
L., Sparkes R Wright S A familial
syndrome of deafness, alopecia, and hypogonadism. —
J. Pediatr., 1973, v. 82.
p. 461 465.
Cremers
С
W.
Geerls S. J.
Sensorineuronal hearing loss and pili torti —
'Чпп. Otol. Rhinol. Laryn- gol , 1979. v.
88. p. 100—104.
Волосо-зубо-костный
синдром
Tricho-dento-osseous
syndrome MIM: 190320
Минимальные
диагностические признаки. гипоплазия
эмали зубов: тауродонтизм; курчавые
волосы; кортикальный склеростеоз.
Клиническая
характеристика
Наблюдается долихоцефалия, выступающие
лобные бугры. Волосы плотные, курчавые,
ресницы изогнутые (80%), с возрастом
волосы могут стать прямыми. Эмаль зубов
тонкая, легко стирается. Зубы мелкие,
широко расставленные, в 90% случаев
имеют увеличенную полость: возможна
задержка прорезывания или адонтия.
Аномальное расположение пульпы
иногда приводит к ее инфицированию
и абсцедированию. Отмечается расслоение
ногтей (50'i), иногда только
на стопах. В 30% случаев наблюдаются
склероз кортикального слоя костей,
множественные переломы, а также
склерозирование основания черепа
и сосцевидного отростка.
Тип
наследования
— аутосомно-доми- нантный.
Дифференциальный
диагноз ■
несовершенный амелогенез: гипоплазия
эмали; тауродонтизм; онихолизис;
амелооннхогипогид- ротический синдром
ЛИТЕРАТУРА
Lichtenslein
J. R.
War
son R Jorgenson R. J Mckusick V. A.
The tricho-dento-osseus syndrome. — Am.
J Hum. Genet.. 1972. v. 24.
p. 568 582.
Melnick
A/..
Shields E D . EI-Ka/rawv A
.1/ Tricho-dento-osseous syndrome: a
scanning electron microscopic analysis. Clin Genet., 1977,
v. 12, p 17—27.
Quailroniani
F Shapiro S D.
Young
R. S. et
al Clinical
heterogeneity in the triheo-dento-osseous syndrome. Hum. Genet..
1983. v. 64. p. 116-
121.
Вольфа—Паркинсона—Уайта
синдром
Wolff
Parkinson White syndrome MIM: 194200
Синонимы:
аномалия атриовентрикуляр- ной
проводимости, преждевременное
возбуждение желудочков.
Минимальные
диагностические признаки. укорочение
интервала PQ, расширение
начальной части комплекса QRS.
Кзиническая
характеристика.
Отмечаются пароксизмальная тахикардия,
мягкий сис
толический
шум, усиление I тона, расщепление I и
II тона. Синдром обнаруживают у 13% больных
с врожденной наджелудочко- вой
тахикардией, пароксизмальной наджел-
удочковон тахикардией, трепетанием
предсердий или мерцательной аритмией.
Парок- сизмальная тахикардия может
вызывать обмороки, нарушения мозгового
кровообращения и даже приводить к
смерти, особенно в детском возрасте. В
30% случаев синдром сочетается с врожденной
патологией сердца (аномалия Эбштейна.
транспозиция магистральных артерий,
первичные поражения миокарда, гипоплазия
левых отделов сердца, субаортальный
стеноз, эндокардиаль- ный фиброэластоз,
дефект межжелудочковой перегородки,
открытый артериальный проток, коарктация
аорты, тетрада Фалло, идиопатический
гипертрофический субаортальный
стеноз). Возможна умственная отсталость
На ЭКГ выявляется укорочение
интервала
PQ и расширение комплекса
QRS. Выделяют тнп А. когда
зубец R является единственным
или преобладающим в комплексе QRS
в отведении V|, и тип
В, когда зубец R отрицательный
илн двухфазный.
Попу
гяционния частота
—1:15 ООО
Соотношение
полов
— М1,8 : Ж1
Тип
наследования
— предположительно аутосомно-доминантный.
Днфференциачьный
диагноз:
врожденная наджелудочковая тахикардия
ЛИТЕРАТУРА
CookeR
И /..
\1 спаи J
М..
Van
CappeUe А
И'. De
V'iUenuve
V
Н
Familial congcnilal heart block and hydrops fetus —
Arch. Dis Child , 1980. v 55.
p. 479-480.
Gillelle
P. С
Freed
D , McSuniara D G
A proposed autosomal dominant method of inheritance of the
WolfT Parkinson White syndrome and supraventricular tachycardia
J Pediatr., 1978. v. 93, p.
257 258.
г
Галактоземия
Galactosemia
MIM 230400
Минимальные
диагностичес кие признаки: отставание
психомоторно! о развития; катаракта:
гепатомегалия; отсутствие активности
галактозо-1 -фосфат-у ридилтрансфера-
зы в эритроцитах.
Клиническая
характеристика.
Симптомы заболевания появляются у
новорожденных после приема молока.
Встречаются гепатомегалия (90%).
желтуха (78%), анорексия и снижение массы
тела (54%), катаракта (42%), рвота (37"и),
вздутие живота (20%), летаргия (16%), асцит
(14%), спленомегалия. темная моча, бледность
(7%), диарея (7%), геморрагический синдром
(5%). отеки, холелитиаз, ахоличный сул,
дизурия (2' ■) При некоторых формах
синдрома галактоземию можно заподозрить
только на основании гепатоме- галии,
катаракты и задержки психомоторного
развития. Лабораторные исследования
выявляют галактозурию. протеинурию,
патологическую кривую при нагрузке
галактозой. снижение активности
галактозо-1-фос- фат-уридилтрансферазы
в эритроциах. Без лечения больные
погибают в первые месяцы жизни от
инфекций или печеночной недостаточности,
у выживших развиваются ядерная катаракта
и умственная отсталость. При раннем
назначении диеты дети могут развиваться
нормально.
Тип
нааедования
— аутосомно-рецессив- ный. Ген локализован
на 9р13
Дифференциазьный
диагноз:
дефицит га- лактокиназы.
ЛИТЕРАТУРА
Hammersen
G.
Houghton
S., Lew H
Rennes-hke
variant of
galactosemia: clinical and biochemical studies.- J Pediatr..
1975, v. 87, p 50-57.
Tedesco
T A Miller K.
Galactosemia: alterations in sulfate metabolism secondary to
galactose-l-phos- phate uridyltransferase deficiency. —
Science, 1979, v. 205.
p. 1395—1397.
Гамартом
множественных синдром
Multiple
hamartoma syndrome MIM: 158350
Синоним:
синдром Коудена.
Минимальные
диагностические признаки: фибромы
и папилломы на коже и слизистых оболочках,
фиброаденомы молочных желез.
Клиническая
характеристика
Заболевание проявляется множественными
гамарто- мами кожи, слизистых, щитовидной
железы, молочных желез. Наиболее
постоянные признаки — фиброматоз
десен (появляется на 1-м году жизни),
гипертрихоз (развивается в течение
первых 5 лет жизни) и фиброматоз молочных
желез (развивается после периода
полового созревания). Фиброаденомы
молочных желез подвержены раннему
злокачественному перерождению
Гипертрихоз отмечается на туловище,
лице, руках и ногах и может усиливаться
после менархе. Гиперплазия десен
иногда сопровождается гиперкератозом
или паракератозом поверхностного
эпителия, лнбо генерализованным ги-
перкератозным папилломатозом губ,
слизистых рта и глотки Бородавчатые
изменения могут появляться в ротовой
полости, вокруг рта, на разгибательных
поверхностях конечностей; на ладонной
и тыльной поверхности кисти возможна
точечная кератодермия. Гипотиреоз,
тнреоидит, аденома и рак щитовидной
железы чаще встречаются у жешин.
Гинекологическая патология включает
нерегулярные менструации, невынашивание,
фиброму матки и кисты яичников. Со
стороны желудочно-кишечного тракта
выявляют полипоз желудка, толстой и
прямой кишки, подслизистую неврому,
нейромиому, дивертикулы и параректальный
абсцесс. К аномалиям скелета относятся
гипоплазия нижней челюсти, готическое
небо, воронкообразная грудная клетка,
сколиоз и кисты костей. В 5Ci%
случаев отмечается небольшое
снижение интеллекта.
Тип
наследования
— аутосомно-доми- нантный.
Дифференциальный
диагноз,
фиброматоз десен, фиброматоз десен с
аномалиями пальцев; фиброматоз десен
с множественными гиалиновыми фибромами:
фиброматоз десен с дистрофией
роговицы, окуло-цереб- ральный синдром
с гипоиш ментацией.
ЛИТЕРАТУРА
Slarink
Т.
М. Vunder
Venn J P . Arweri F. el at. The
Cowden syndrome: a clinical and genetic study in 21
patients —Clin. Genet. 1986, v. 29.
p. 222-233.
H'ilkop
C.
Heterogeneity in gingival fibromatosis. The clinical delineation of
birth defects. XI Orofacial structures. — Birth
Defects, 1971, v. V1I(7), p. 210
- 221.
GMi-ганглиозидоз,
тип I
GMrgangliosidosis,
type I MIM: 230500
Синонимы:
семейный нейровисцеральный липидоз;
недостаточность р-галактозилазы:
болезнь Моркио.
Минимальные
диагностические признаки: висцеромегалия,
характерные изменения лица и костей;
вакуолизация клеток костного мозга и
лимфоцитов, накопление См,-ганг- лиозида
в клетках головного мозга, накопление
ганглиозидов и мукополисахаридов во
всех органах и тканях; дефицит р-галактози-
дазы в мозге, печени, культуре фибробла-
стов, лейкоцитах и моче
Клиническая
характеристика
Дети с раннего возраста резко отстают
в психомоторном развитии. Отмечаются
плоская переносица, гипертелоризм,
выступающий лоб, низко расположенные
уши
(рис. 33).
гипертрофия десен. После 6 мес
выявляется гепа- томегалия. спленомегалия
выражена меньше. Среди аномалий
скелета отмечаются брахидактилия,
кнфосколиоз. множественные сгибательные
контрактуры суставов; рентгенологически
выявляются признаки дизостоза При
офтальмологическом исследовании у
50% больных на глазном дне выявляют
симптом «вишневой косточкн». В начале
2-го года жизни появляются затруднения
при глотании, тонико-клонические
судороги, амавроз; исчезает реакция
на окружающее Дети погибают в возрасте
2—3 лет от сопутствующих бронхолегочных
инфек
ций.
При гистологическом исследовании
находят вакуолизацию лимфоцитов, клеток
костного мозга, гепатоцитов. клеток
почечного эпителия, кожных фибробластов
Гис- тохимически в этих клетках
обнаруживают кислые мукополисахариды.
ганглиозиды и липиды.
Основной
биохимический дефект — недостаточность
лизосомального фермента р- галактозидазы.
Тип
наследования
— аутосомно-рецессив- ный. Ген локализован
на Зр21-р14.2.
Дифференциальный
диагноз:
мукополиса- харидозы; болезнь Нимана
Пика:
Gm--
ганглиозидоз. тнпы I и 11. фукозидоз;
муко- липидозы.
ЛИТЕРАТУРА
Cingliam
R.
Dulra
J С
Perciru
\f L
ei al. GMi-gangliosidosis
clinical and laboratory findings
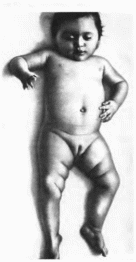
Рис.
33.
GMi-гаиглиозидоз.
тип 1 Запавшая
переносица, гипертелоризм. низко
расположенные ушные раковины,
гепатоспленомегалия.
in
eight families. ■ Hum. Genet.. 1985.
v. 70.
p. 347
354.
Goldman
J. E.. Kalz IX.
Rapin J. el al.
Chronic
GM,-gangliosidosis
presenting as dystonia: I. Clinical
and pathological features.
- Am. Neurol., 1981, v.
9.
p. 465—475.
Singer
H. S.. Schafer /.
A.
Clini al and enzymatic
varinants in
Gmi
generalized gangliosidosis.
Am. J. Hum. Genet.. 1972,
v. 24. p. 454-463.
GMi-ГЭНГЛИОЗИДОЗ,
ТИП II
Gm
-gangliosidosis, type
II
M1M:
230600
Синоним:
ювенильный системный липи-
доз.
Минимальные
диагностические признаки.
спастический
тетрапарез, атаксия, постепен-
ное
снижение реакции на окружающее;
«пе-
нистые» клетки в костном мозге,
накопление
GMi-ганглиозида
в головном мозге, дефицит
Р-галактозидазы
в тканях, лейкоцитах и мо-
че.
Клиническая
характеристика
Типичны
отставание в психомоторном
развитии (с 6 —
12 мес), прогрессирующий
спастический тет-
рапарез, атаксия,
деменция, мышечная гипо-
тония,
сменяющаяся ригидностью и судоро-
гами.
Терминальная стадия характеризуется
децеребрацней.
Продолжительность жиз-
ни — от 3 до
10 лет. Иногда рентгенологиче-
ски
выявляется дизостоз, значительно
менее
выраженный по сравнению с I
типом
Gmi-
ганглиозидоза.
В костном мозге обнаружи-
вают
«пенистые» клетки; в печени, селезенке
и
почечных клубочках — вакуолизирован-
ные
клетки.
В
тканях мозга накапливается GMrraHr~
лиозид,
в печени — кератансульфат-подоб-
ный
полисахарид. В мозге, печени, лейкоци-
тах
и культуре фнбробластов отмечается
де-
фицит р-галактозидазы. Иногда
незначи-
тельно повышен уровень
мукополисахари-
дов в моче.
Тип
на чедования
— аутосомно-рецессив-
ный.
Дифференциачьный
диагноз:
См,-ганглио-
зидоз, тип I; цероидлипофусциноз
невро-
нальный: другие сфинголипидозы.
ЛИТЕРАТУРА
О'Brim
J. S Но
М W,
Veath М
L.
el al.
Juve-
nile GM-gangliosidosis; clinical, pathological,
chemi-
cal and enzymatic attitudies. Clin. Genet., 1972.
v.
3, p. 411-434.
См2-ганглиозидоз,
тип I
Gm
gangliosidosis, type 1 MIM: 272800
Синонимы:
См;-ганглиозидоз с дефицитом
гексозаминидазы А; амавротическая
идиотия Тея -Сакса
Минимачьныс
диагностические признаки: задержка
психомоторного развития; симптом
«вишневой косточкн» на глазном дне;
дефицит гексозаминидазы А в сыворотке
и тканях.
Кчиническая
характеристика.
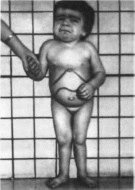
С 4 -5 мес дети начинают отставать в
психомоторном развитии, становятся
апатичными, перестают интересоваться
окружающим, фиксировать взгляд
(рис. 34).
Отмечаются гипераку-
Рис.
34. Gm;
гаиглиозидоз, тип I.