

32
Анкилоблефарон
Тип
наследования
— предположительно аутосомно-рецессивный
Дифференциальный
диагноз:
синдром пан- цитопении Фанкони;
тромбоцитопения с отсутствием
лучевой кости; енндром Холт— Орама.
ЛИТЕРАТУРА
A
(Lie J.. Smith D.
Congenital anemia and Iripha langeal thumbs: a new syndrome. —J.
Pediatr., 1969. v. 74. p.
471- 474.
Alter
B. P. Thumbs
and anemia. Pediatrics, 1978. v. 62.
p. 613 614.
Jones
В.
Thompson
// Triphalangeal thumbs as- sc lated with
hypoplastic anemia. — Pediatrics. 1973,
v. 52. p. 609 612.
Muis
N..
Beemer
F. A., van Dijken P. el at.
The Aase syndrome: case report and review of the literature.
Eur. J. Pediatr.. 1986, v. 145,
p. 153 157
Анкилоблефарон
Ankyloblepharon
MIM: 123570
Синоним:
криптофтальм изолированный. Минимальные
диагностические признаки: частичное
сращение краев век.
Клиническая
характеристика.
Сращения краев век имеют вид плотных
рубцов или тонких тяжей Сращение
внутреннего угла глазной щели сочетается
обычно с эктопией слезных точек и
канальцев. Анкилоблефарон часто
сочетается с расщелиной губы и неба,
анофтальмией, микрофтальмиен, птозом,
микроцефалией и другими пороками.
Соотношение
полов — Ml
: Ж1. Тип
наследования
— предположительно аутосомно-доминантный.
Дифференциальный
диагноз:
криптофтальм с другими пороками
развития.
ЛИТЕРАТУРА
Thomus
I.
Т.. Frias
L.. Felix V.
et
al. Isolated
and syndromic cryptophthalmos — Am. J Med.
Genet., 1986. v. 25. p.
85 89.
Анорхия
семейная
Anorchia
familial MIM: 273250
Синонимы:
агенезия яичек; синдром тести- кулярной
регрессии.
Минимальные
диагностические признаки:
отсутствие
яичек у лиц мужского пола с
дифференцированными наружными
половыми органами. Кариотип 46.XY.
Клиническая
характеристика.
Анорхия может быть одно- и двусторонней.
Последняя форма встречается
значительно реже. Наружные половые
органы дифференцированы правильно,
но могут быть недора шиты. Рост
высокий Других соматических аномалий
нет. Снижено выделение 17-кетосте- роидов
с мочой. Диагноз подтверждается при
хирургическом обследовании мошонки,
пахового канала и пути опускания яичка
в мошонку, а также при гистологическом
исследовании. Возможна агенезия
придатка яичка и семявыносящего протока.
В первом десятилетии жизни развитие,
как правило, проходит нормально, и
диагноз может быть поставлен только в
пубертатном возрасте (при двусторонней
анорхии) или еще позднее (при односторонней
анорхии) на основании недостаточного
развития вторичных половых признаков.
Пациенты с односторонней анорхией
могут быть фертильны.
Тип
наследования
— предположительно аутосомно-рецессивный.
Дифференциальный
диагноз:
гипогонадо- тропный гипогонадизм;
мужской псевдогермафродитизм.
ЛИТЕРАТУРА
Bohroи
М.. Gourh
М.
Bilateral absence of testes. Lancet. 1970,
v. I, p. 336.
Hall
J.. Morgan A.. Blizzard R.
Familial congenital anorchia Birth Defects. 1975. v.
Xl(4). p. 115- 119.
Josso
N..
Brian!
U Embryonic
testicular regression syndrome ari ble phenotypic expression in
siblings. - J. Pediatr., 1980,
v. 97. p. 200 204.
Анофтальмия
Anophthalmia
MIM: 206900
Минимагьные
диагностические признаки: клинические
признаки анофтальмии, подтвержденные
данными микроскопического исследования.
Клиническая
характеристика.
Истинная анофтальмия представляет
собой полное отсутствие тканей глаза
(как правило, с обеих сторон), что связано
с отсутствием глазного зачатка.
При
мнимой анофтальмии, обусловленной
задержкой развития вторичного глазно-Van Weet-Sipman m.. Van Je Kamp j., tie honing j. A female patient with «Aase syndrome» j. Pediatr., 1977, V. 91. P. 753 755.
Анофтальмия
33
Рис.
15. Анофтальмия.
Три
сибса от кровнородственного брака.
го
бокала,
в глубине
орбиты можно обнаружить рудиментарный
глаз.
Придатки
глаз при истинной анофтальмии сохранены,
но их размеры меньше, чем в норме.
Веки небольшие, орбита и конъюнктиваль-
ная полость мелкие
(рис. 15).
Тип
наследования
истинной анофтальмии —
аутосомно-рецессивный
ЛИТЕРАТУРА
Oliveira
da Sriva Е..
Saniana
de Sousa S
Clinical anophthalmia Hum Genet, 1981, v 57,
p. IIS— II6
3
173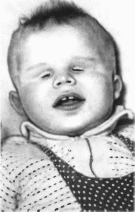

34
Анэнцефалия
Анэнцефалия
Anencephaly
MIM: 206500
Минимальные
диагностические признаки отсутствие
большого мозга, костей свода черепа
и мягких тканей головы
Клиническая
характеристика.
Большой мозг замещен соединительной
тканью с кис- тозными полостями. Часто
повреждается и задний мозг. Отсутствуют
кости свода черепа, глазницы мелкие.
Как правило, имеется выраженная
гипоплазия надпочечников и аплазия
нейрогипофиза. Иногда анэнцефалия
сочетается с расщелиной неба, аномалиями
шейного отдела позвоночника. Этиология
мультифакториальная. Риск для сиб- сов:
при 1 пораженном — 2—5%. при 2 пораженных
— 10%. В части случаев предполагается
аутосомно-рецессивное наследование
Популяционния
частота — 1 :
1000. Соотношение
по лов Ml
: ЖЗ—7. Дифференциальный
диагноз:
гидранэнце- фалия.
ЛИТЕРАТУРА
Farag
Т
J.
Teebi
A S
Nonsyndromal anencephaly possible autosomal recessive variant.
— Am J Med. Genet.. 1986. v
24. p 461—464.
Fuhrmann
1Г,
Seeger
W,
Bolun
R Apparently
monogenic inheritance of anencephaly and spina bifida in a
kindred. Hum. Genet., 1971, v. 13,
p 241 243.
Maslerson
J. G. Empiric
risk, genetic counseling and preventive measures in anencephaly. —
Acta Genet Stat Med . 1962. v 12.
p 219 229.
Арахнодактилия
контрактурная врожденная
Contractural
arachnodactyly congenital MIM: 121050
Синоним:
синдром Билса.
Синдром
описан R Beals и F
Hecht.
Минимальные
диагностические признаки арахнодактилия,
сгибательные контрактуры пальцев
рук, аномальная форма ушных раковин.
Клиническая
характеристика.
Типичные проявления — арахнодактилия.
контрактуры мелких суставов кисти
и других суставов (рис.
16). Нередки
выраженный кифосколи- оз, воронкообразная
или килевилная грудная клетка;
описаны аномалии глаз и сердечно-сосудистой
системы Наблюдаются деформированные,
ротированные назад ушные раковины.
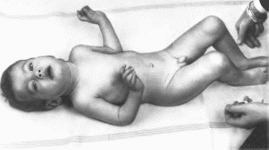
Рис.
16. Арахнодактилия
контрактурная врожденная. Мальчик 8
месс контрактурами суставов кисти,
коленных и локтевых суставов, короткой
шеей и деформацией грудной клетки.
Артрогрипоз
35
Тип
наследования
— аутосомно-доми- нантный с вариабельной
экспрессивностью.
Дифференциальный
диагноз:
артрогрипоз. синдром Марфана.
ЛИТЕРАТУРА
Beats
R. К
Hechi
F Congenital
coniractural ara- ehnodaeiyly: a heritable disorder of connective
iis- sue. — J. Bone Joint Surg.. 1971, v.
53A. p. 987- 993.
Currarino
G.. Friedman J XI
A severe form of congenital coniractural arachnodaclyly in two
newborn infants. Am. J. Med. Genet.. 1986. v.
25. p. 763 773.
Артрогрипоз
Arthrogryposis
MIM: 108110
Минимальные
диагностические признаки: деформации
и ограничение подвижности суставов.
Клииическия
характеристика.
Характерны множественные деформации
суставов. Ограничения движений в
суставах варьируют от умеренных до
анкилоза с фиксацией разных отделов
конечностей в положении сгибания, реже
— разгибания
(рис. 17):
ребенок может находиться в «положении
пло
да».
Предполагаемый патогенетический
механизм — ограничение движений плода
вследствие различных причин: нейрогенных,
миогенных, изменений соединительной
ткани, инфекций, действия лекарственных
препаратов, уменьшения объема матки.
Существует несколько форм артрогрипоза
Кроме того, он входит в состав других
синдромов.
Соотношение
полов ■ - МI : Ж1.
Тип
наследования
не установлен. Большинство случаев
спорадические. Описаны случаи с
аутосомно-рецессивным и аутосом-
но-доминантным наследованием.
Дифференциальный
диагноз:
синдром три- сомии 18-й хромосомы:
арахнодактилия контрактурная врожденная;
синдром множественных птеригиумов;
диастрофическая дисплазия.
ЛИТЕРАТУРА
Hall
J. F. Reed S.. Greene G
The distal arthrogryposis: delineation of new entities review
and nosologic discussion. Am. J. Med. Genet., 1982,
v. 11, p. 85 239.
McCarmack
M. K. Coppola-MiCormack P.. Lee А/
L.
Autosomal-dominant inheritance of distal arthrogryposis. Am. J
Med. Genet., 1980, v. 6, p.
163 169.
Рис
17.
Аргрогрнпоз. Контрактуры суставов
верхних и нижних конечностей.
36
Артроофтальмопатня
наследственная прогрессирующая
Rosenmunn
A.. Arad L.
Arthrogryposis multiplex congenital: neurogenic type with autosomal
recessive inheritance.- J. Med. Genet., 1974, v.
11, p. 91—94.
Артроофтальмопатня
наследственная прогрессирующая
Arthroophthalmopathy,
hereditary
progressive
MIM:
108300
Синоним:
синдром Стиклера Описан в 1965 г. G.
В. Stickler с соавт.
Минимальные
диагностические признаки: миопия,
патология суставов, марфаноидный
фенотип.
Клиническая
характеристика.
К нарушениям зрения относятся
прогрессирующая миопия, аномалии
пигментации сетчатки, атрофия сосудистой
оболочки, ретиноши- зис, катаракта,
спонтанная отслойка сетчатки,
астигматизм, глаукома. У новорожденных
отмечаются увеличение и гиперподвижность
суставов, в раннем детстве — боли и
тугоподвижность в суставах, начиная с
подросткового возраста — прогрессирующий
остеоартрит с периодическими обострения
ми,
у взрослых — дегенеративная артропа-
тия. Чаще поражаются лучезапястные.
коленные, голеностопные суставы.
Рото-лице- вые аномалии включают
микрогению, расщелину неба (в тяжелых
случаях — анома- лад Пьера Робена).
гипоплазию средней части лица,
экзофтальм, эпикант, короткий нос.
Характерен марфаноидный фенотип —
астеническое телосложение,
воронкообразная грудина, кифоз, сколиоз,
(рис. 18)
Выявляется пролапс митрального
клапана (45%). Возможны гиподонтия,
аномалии позвонков, глухота, вальгусная
деформация голени. Рентгенологически
определяется легкая спондилоэпифизарная
дисплазия. Интеллект обычно сохранен.
Тип
наследования
— аутосомно-доми- нантный с варьирующей
экспрессивностью. Обнаруживаются
мутации гена COL2AI,
локализованного на I2ql3.l
l-ql3.2.
Дифференциальный
диагноз:
синдром Марфана, синдром Элерса Данлоса.
спондилоэпифизарная дисплазия.
ЛИТЕРАТУРА
Temple
/.
К.
Stickler's syndrome. J. Med. Ge- net , 1989,
v. 26. p. 119 126.
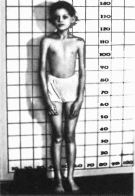
Рис.
18
Артроофтальмопатня наследственная
прогрессирующая. Гипоплазия средней
части лица, марфаноидный фенотип.

Ассоциация
VACTERL
37
Viniiner
G.
Si
Temple
I. к
Shddteion-Price
H R
ei al. Genetic
and clinical heterogeneity of Stickler syndrome. —
Am. J. Med. Genet., 1991, v. 41,
p. 44 48.
ZloiogoraJ..
Sagi M.. Sehuper A. ei al.
Van ibility of Stickler syndrome — Am. J.
Med. Genet., 1992, v. 42 p.
337 339.
Асплении
синдром
Asplenia
syndrome MIM: 208530
Синонимы:
синдром асплении Ивемарка; агенезия
селезенки; синдром полисплении.
В
1955 г. В. Ivemark выделил ранее
описанное сочетание асплении и
определенных пороков сердца в отдельную
нозологическую единицу.
Минимальные
диагностические признаки правый
изомеризм (органы левой половины тела
сформированы так же, как правой), ас-
пления, врожденный порок сердца синего
типа, ретгенологически выявляемая
декст- ракардия с признаками гипертензии
малого круга кровообращения.
Клинической
характеристика.
Отмечается аспления. полиспления или
гипоплазия селезенки. симметричное
развитие правой и левой сторон тела
Левое легкое трехдолевое, имеются
двусторонние добавочные бронхи (90%);
левая доля печени значительно увеличена,
в 40% случаев она такого же размера, как
и правая. Часты нарушения поворота
кишечника, в половине случаев желудок
смещен вправо. Сердечно-сосудистые
аномалии включают мезо- и декстрокардию,
атриовен- трикулярный канал, единый
атриовентрику- лярный клапан, единственный
желудочек (или большой дефект
межжелудочковой перегородки),
транспозицию крупных сосудов, атрезию
или стеноз легочного ствола (70%),
аномальное расположение легочных вен,
наличие двух верхних полых вен. Дети
погибают чере несколько дней или недель
после рождения от пороков сердца или
позднее от инфекций
Соотношение
полов — М2 :
Ж1. Тип
наследования
— предположительно аутосомно-рецессивный.
Дифференциальный
диагноз:
врожденная изолированная агенезия
селезенки; синдром Картагенера.
ЛИТЕРАТУРА
ArnoldG.,
BixlerD.. GlrodD.
Probable autosomal
recessive
inheritance of polysplenia, situs inversus and cardiac defects in an
Amish family. — Am. J. Hum. Genet.. 1983.
v. 16. p. 35—42.
Niikawa
N.
el
al. Familial
clustering of situs inversus totalis, and asplenia and
polysplenia syndro mes —Am. J. Hum. Genet., 1983,
v. 16. p 4 -47.
Zlologora
J., Elian E.
Asplenia and polysplenia yndromes wiih abnormaliti :s of
lateralization in a sibship. — J. Med.
Genet., 1981, v. 18. p.
301 302.
Ассоциация
VACTERL
V
ACTER L-association MIM: 1192350
Комплекс
пороков выделен в самостоятельную
нозологическую единицу в 1973 г. L.
Quan и D. Smith.
Минимальные
диагностические признаки: дефекты
позвоночника, трахеопищеводные свищи,
дисплазия лучевой кости, неперфо-
рированный анус, пороки почек.
Клиническая
характеристика.
Название синдрома составлено из первых
букв симп-
Рис.
19. Лучевая
косорукость при VACTERL-ac-
социации.

38
Асфикснческая
дистрофия
груднои
клетки новорожденных
томов,
входящих в его состав: V (vertebral).
A (anal atresia). С (cardiac), ТЕ
(tracheoesophageal). R (radial and renal). L
(limb). Основные проявления: аномалии
позвоночника — полупозвонки, кифосколноз,
менингоцеле (82%); дефекты перегородок
и другие пороки сердца (50" о); атрезия
ануса (70%); трахеопи- щеводный свищ с
атрезией пищевода (78%); дисплазня лучевой
кости
(рис. 19) -
гипоплазия I пальца или лучевой
кости, преакси- альная полидактилия и
синдактилия (65%); аномалии почек —
агенезия (33"/.), дисплазня (25%).
гидронефроз (15%); единственная пупочная
артерия (22%). Встречаются также
незавершенный поворот кишечника,
дивертикул Меккеля, стеноз
двенадцатиперстной кишки, гипоплазия
половых органов.
Соотношение
по we
-Ml Ж
l.
Тип
наследования
неизвестен. Все случаи спорадические.
Дифференциальный
диагноз,
синдром три- сомии 18-й хромосомы; синдром
хромосомы I3q-; синдром
панцитопении Фанкони.
ЛИТЕРАТУРА
БочконаД.
Н. ПюпшикиваЛ Р.. КушковаГ. В VATER-ассоциация
у больной карднохнрургиче
скогоотделения.—Клин мед. 1981,№8.с.78—79
Auchierlonie
I. A While М
Р
Recurrence of the VATER-association within a sibship. —Clin.
Genet, 1982. v. 21. p.
122 124.
Khourv
M J . CorderoJ. F,
Greenberg
F. el al A population
study of VACTERL association: evidence for its etiologic
heterogeneity. — Pediatrics, 1983.
v.71.p. 815—820.
Seemanova
E . Sevcikova M TosovskyV
VATER syndrom u dvaapulleteho devcatka — Csl.
pedial. 1979. r 34,
1.291-292.
Асфиксическая
дистрофия грудной клетки новорожденных
Asphyxiating
thoracic dystrophy of the
newborns
MIM:
208500
Синоним:
синдром Жена.
Заболевание
описано в 1955 г. М. Jeune
Минимальные
диагностические признаки: деформация
грудной клетки, отставание в росте,
укорочение конечностей, неглубокая
умственная отсталость, почечная
недостаточность, врожденный порок
сердца.
Клиническая
характеристика.
Типичны скелетные дисплазии макроцефалия,
укоро
чение
конечностей, узкая грудная клетка,
короткие ребра. Рост низкий. У части
больных отмечается постаксиальная
полидактилия кистей и стоп. У новорожденных
наблюдается респираторный
дистресс-синдром, который часто
приводит к смерти. Другая распространенная
причина гибели — врожденные пороки
сердца. Если новорожденный выживает,
то в клинической картине начинает
преобладать патология почек (фиброз,
приводящий к почечной недостаточности).
Отмечается горизонтальный нистагм.
Умственное развитие отстает.
Рентгенологически выявляются шишковидные
эпифизы костей верхних конечностей,
гипоплазия тазовых костей, короткие
горизонтально расположенные ребра.
Тип
наследовании
аутосомно-рецессив- ный.
Дифференциальный
диагноз:
хондродист- рофия новорожденных с
полидактилией, типы 1 и II:
хондроэктодермальная дисплазия;
метатропная дисплазия; танатофорная
карликовость.
ЛИТЕРАТУРА
Buloi-Lopeг
Р Ablo\
R С
Ogden
J. A ei. al. Case
of short
rib Polydactyly
— Pediatrics, 1978, v.
36, p. 32 —39.
Cortina
H .
Bel
Iran J .
Olague
R. et al The
wide spectrum of the asphyxiating thoracic dysplasia. —
Pediat. Radiol.. 1979, v. 8.
p. 93 99.
Oherklaid
F. Danks D. M Mayne V.. Campbell P.
Asphyxiating thoracic dysplasia: clinical, radiological and
pathological information on 10 patients —
Arch. Dis. Child.. 1977.
v
52. p. 758 765.
Атаксия-телеангиэктазия
Ataxia-teleangiectasia
MIM: 208900
Синоним:
синдром Луи-Бар.
Заболевание
описано в 1941 г. D. Louis- Bar.
Минимальные
диагностические признаки: атаксия,
телеангиэктазии, рецидивирующие
инфекции верхних дыхательных путей,
снижение уровня или отсутствие IgA.
Клиническая
характеристика.
Заболевание начинается в раннем
детстве и проявляется в первую
очередь мозжечковой атаксией (100%).
Отмечаются дрожание головы и туловища,
нарушение походки, интенционный тремор
и хореоатетоз (90—100%). Характерны
нарушения движений глазного яблока
Атаксня-телеангиэктазия
39
