

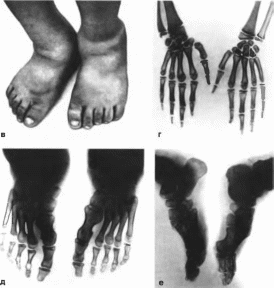
Рис.
202. Синдром
язвенного поражения кожи, акроостеолнза,
кератита и олигодонтии (продолжение).
В
— парциальная акромегалия: г рентгенограмма
кистей
(аномалии,
обусловленные акроостеолизом концевых
фаланг): д,
е рентгенограммы
стоп340 Язвенного поражения кожи, акроостеолнза. Кератита и олигодонтин синдром
Язвенного
поражения кожи, акрооетеолиза. кератита
и олигодонтин синдром
341
дуюшем
изъязвляются и рубцуются. Примерно
в то же время обнаруживаются изменения
костно-суставного аппарата и уже к
5-летнему возрасту развиваются грубые
деформации коленных и голеностопных
суставов. концевых фаланг пальцев
рук с истончением кожи и изменением
формы ногтей по типу когтистой лапы;
парциальная акромегалия стоп и
кистей; грубый сколиоз грудного и
поясничного отделов позвоночннка;
вдавление в области грудины
(рис. 202, а, 6, в).
На коже лица, конечностей и туловища,
в области крупных суставов помимо
многочисленных рубцов полиморфного
характера формируются рубцы со
свищевыми ходами. Отмечается
аномальный рост зубов, частичная
адонтня. Помутнение роговицы вследствие
повторных рецидивов кератита приводит
к резкому снижению зрения. Рентгенологическая
картина характеризуется
значительной
перестройкой костей и наличием
ячеистых полостей в пяточных и таранных
костях; остеонекрозом терминальных
фаланг кистей
(рис. 202, г, д, е):
остео- порозом и искривлением поясничного
отдела позвоночннка.
Тип
наследования—аутосомно-рецессивный.
Дифференциальный
диагноз:
синдром Пу- ретнча; буллезныи эпндермолиз
с акроосте- олизом; синдром Хайду -Чннея.
ЛИТЕРАТУРА
Козлова
СИ..
Кравченко В. А . А ыпш lep
Б.
А Новый
наследственный вариант поражения кожи
и костей. Клиническая медицина. 1983. т.
4. стр. 97 —101.
Kozto\a
S. /..
Kravchenko
l". 4
. Allshuler
В.
А. Self-limited
autosomal recessive syndrome of skin ulceration.
arthroosteolysis with pseudoacromegaly. keratitis and oligodontia in
Kirghizian family. Am. J. Med. Genet.. 1983. v.
15. p. 205—210.