336
Эпидермолиз
буллезныи
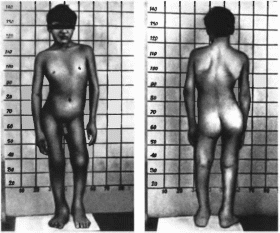
Рис.
200. Поражение
кожи при буллезиом эпидермолизе.
коже
и слизистых
в
месте давления или травмы. при
нагревании или спонтанно. Выделяют
четыре формы буллезного эпидермолиза.
Простой
буллезный эпидермолиз (доминантная
форма). Поражаются стопы, кисти, шея,
реже колени, лодыжки, туловище, локти
(рис. 200),
слизистая ротовой полости, ногти
(20%). Величина пузырей различна. Рубцы
на месте заживления не образуются.
Заболевание проявляется на 1-м году
жизни, когда ребенок начинает ползать.
Теченне простого буллезного эпидермолиза
нетяжелое, к пубертатному периоду
его проявления значительно уменьшаются.
Доминантная
дистрофическая (гипертрофическая)
форма буллезного эпидермолиза (тип
Коккейна—Турена). Пузыри при этой
форме эпидермолиза плоские, розоватые
и оставляют после себя рубцы (иногда
келоидные). Локализация поражений та
же, что при простом буллезном эпидермолизе.
В 80% случаев ногти тонкие и дистрофичные.
Конъюнктива и роговица не поражаются.
Заболевание сочетается с ихтиозом,
ладоно- подошвенным гиперкератозом,
фолликулярным кератозом гипергидрозом,
генерализованным гипертрихозом.
ются
вскоре после рождения. Наиболее часто
поражены стопы, ягодицы, лопатки, локти,
пальцы, затылок, слизистая ротовой
полости. При разрыве пузырей часто
присоединяется вторичная бактериальная
инфекция. После заживления, как правило,
остаются келоидные рубцы, приводящие
к контрактурам, депигментации или
гиперпигментации. В области рубцов
образуются милиаподоб- ные кисты. Ногти
дистрофичные, могут полностью
отсутствовать. Возможен гипертрихоз.
гипергидроз. Поражение глаз проявляется
неспецифическим блефаритом, симбле-
фароном. кератитом с помутненнем
роговицы и формированием везикул.
Образование пузырей на слизистой
гортани и глотки может приводить к
охриплости голоса, афонии, дисфонии. а
в дальнейшем — к стенозу гортани и
полной обструкции пищевода. Отмечаются
гипоплазия эмали, позднее прорезывание
зубов.
4.
Летальный буллезный эпидермолиз
(злокачественный буллезныи эпидермолиз
новорожденных). Это наиболее тяжелая
форма эпидермолиза. Заболевание
проявляется вскоре после рождения
и приводит к смерти в первые 3 мес жизни
Характерны отслоение больших участков
эпидермиса поражение слизистых оболочек,
в том числе бронхиол; рубцевание, милии
и пигментные изменения кожи отсутствуют.
Пузыри локализуются на туловище,
лице, волосистой части головы, конечностях.
Ладони и подошвы никогда не поражаются.
Содержимое пузырей обычно геморрагическое.
Ногтевые пластинки отторгаются.
Потляционная
частота
доминантных форм — 1 : 50 000.
Тип
наследования
— аутосомно-доми- нантнын при простом
буллезном эпидермолизе и дистрофической
доминантной форме (ген картирован на
Iq21-q24);
аутосомно-
рецесснвнын — при дистрофической
рецессивной форме и летальном
буллезном эпидермолизе.
ЛИТЕРАТУРА
Hashimilo
1,
Anlon-Lamprechl
/..
Hofbauer
M Epidermolysis
bullosa dystrophica unversa: Berichi uber zwei Geschwistergalle. —
Hautarzt, 1976, Bd. 27.
S. 532—537.
Pearsson
R. IV, Poll: В.
Slrauss
F.
Epidermolysis bullosa hereditaria lethalis. Clinical and
histological manifestations and course of the disease. Arch. Derm..
1974. v. 109. p. 349
355.
Рецессивная дистрофическая форма буллезного эпидермолиза. Пузыри появля
Эпифизарная
дисплазия множественная 337
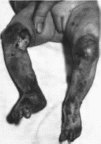
Рис.
201. Буллезный
эпидермолиз с врожденной локальной
аплазией кожи и деформациями ногтей
Эпидермолиз
буллезный с врожденной локальной
аплазией кожи и деформациями ногтей
Epidermolysis
bullosa with congenital localized absense of skin and deformity
of nails MIM: 132000
Синоним:
дистрофический буллезный эпидермолиз.
тип Барта
Минимальные
диагностические признаки участки
отсутствия кожи на нижних конечностях,
аномалии ногтей, буллезные изменения
кожи.
Клиническая
характеристика.
Наблюдается врожденное отсутствие
кожи на нижних конечностях с развитием
буллезных изменений
(рис. 201),
отсутствие или деформация ногтей на
стопах, прежде всего на I пальцах Тип
наследования
— аутосомно-доми- нантный с полной
пенетрантностью и различной
экспрессивностью
Дифференциальный
диагноз:
локальная аплазия кожи; буллезный
эпидермолиз.
ЛИТЕРАТУРА
Barl
В
J.
Congenital localized absense of skin, blistering and nail
abnormalities, a new syndrome. — Birth
Defects. 1971, v
VII (8),
p. 118—120.
Эпифизарная
дисплазия множественная
Epiphyseal
dysplasia multiple MIM: 132400
Синоним:
болезнь Фенрбанка.
Описана
в 1974 г. Т. Fairbank.
Минимальные
диагностические признаки: уменьшенные,
неправильной формы эпифизы трубчатых
костей с фрагментацией ядер окостенения:
болезненность и тугоподвиж- ность
тазобедренных суставов; низкий рост
Клиническая
характеристика
Типично нарушение энхондрального
окостенения (задержка развития ядер
окостенения крупных костей). Отмечаются
болезненность суставов (в основном
тазобедренных), низкий рост, ограничение
подвижности в тазобедренных и
плечевых суставах, брахидакти- лия,
нарушение походки, гиперподвижность
межфаланговых суставов Основной
диагностический критерий —
рентгенологические изменения: маленькие,
деформированные, уплощенные эпифизы
длинных трубчатых костей (чаще всего
бедренных и большебер- цовых, а также
карпальных и тарзальных); фрагментация
ядер окостенения эпифизов; в единичных
случаях — варусная деформация
22
173
338
Эрнтрокератодсрмия
вариабельная
шейки
бедра. Метафизы длинных трубчатых
костей обычно не изменены, фаланги
пальцев укорочены. Заболевание
проявляется в 2—5 лет
Тип
нас лсдования
— аутосомно-доми- нантный с вариабельной
экспрессивностью.
Дифференциальный
диагноз:
хондродис- плазия точечная;
артроофтальмопатия наследственная
прогрессирующая; спондило- эпифизарная
дисплазия врожденная; спон- днлоэпифизарная
дисплазия поздняя.
ЛИТЕРАТУРА
Lie
S О.,
Siggers
D С.
Dorsi
J P.. KopUs S E Unusual
multiple epiphyseal dysplasias. — In:
Skeletal dysplasias/Ed. D Bergsma Amsterdam, 1974,
p. 165—185.
Эритрокератодермия
вариабельная
Erythrokcratodermia
variabilis MIM: 133200
Минимальные
диагностические признаки гиперкератозные
бляшки и эритематозные пятна, обширный
пластинчатый гиперкератоз, акантоз
и папилломатоз.
Клиническая
характеристика.
Размер, расположение и количество
зритематозных
пятен
варьируют Выраженность симптомов
зависит от внешних воздействий,
эмоционального фона. В области
эритемы развиваются стойкие очаги
гиперкератоза желто-коричневого
цвета с четко ограниченными неровными
контурами. Заболевание проявляется,
как правило, с рождения или на 1-м году
жизни, но иногда может начаться и в
более позднем возрасте Поражаются
преимущественно лицо, разгибательные
поверхности конечностей, ягодицы
Ладонно- подошвенная кератодерма
встречается почти в половине случаев;
волосы, ногти и зубы не поражаются.
Кожные изменения иногда сопровождаются
неврологической симптоматикой
(снижение сухожильных рефлексов,
нистагм, дизартрия, в отдельных случаях
— моторная атаксия).
Тип
наследования
— аутосомно-доми- нантный с различной
экспрессивностью. Ген картирован на 1
р36.2-р34.
Дифференциальный
диагноз:
эритродер- мия ихтиозоформная врожденная.
ЛИТЕРАТУРА
Macfarlane
А
И
, Chapman
L J Verlov J. L
Is erythrokeratoderma
one disorder? A clinical and ul- trastructural study of two
siblings. — Brit. J. Derm., 1991,
v 124, p 487—491
Язвенного
поражения кожи, акроостеолиза, кератита
и олигодонтии синдром
Синоним:
киргизский дерматоостеолиз
Описан
в 1983 С И Козловой с соавт.
Минимальные
диагностические признаки кожные
изменения в виде папул с последую
щим
эрозированием и рубцеванием, остео-
пороз, акроостеолиз, сколиоз, деформация
суставов, вторичные рецидивирующие
инфекции, кератит, олигодонтия.
Клиническая
характеристика
На первом году жизни на слизистой
оболочке рта, коже лица, туловища и
конечностей появляются множественные
папулы, которые в после-
Рис.
202. Синдром язвениого поражения кожи,
акроостеолиза. кератита и олигодонтии.
а,
б - деформации коленных и голеностопных
суставов, парциальная акромегалия стоп
и кистей, сколиоз