В
Рис-
165. Синдром
Халлермана Штранфа (продолжение).
оттопыренные
уши.
Хартнапа
болезнь
Hartnup
disease MIM
234500
Минимальные
диагностические признаки специфическая
гиперамнноацидурия; фотодерматоз;
атаксия: изменения психики.
Клиническая
характеристика
Типичный признак — повышенная
чувствительность кожи к ультрафиолетовым
лучам (гиперемия, шелушение, пузыри);
кожные изменения могут напоминать
пеллагру. Отмечаются атаксия,
хореический гиперкинез, интен- ционный
тремор, повышение периосталь- ных
рефлексов, нистагм, нарушение конвер-
генцин. Изменения психики проявляются
депрессией, фобиями, галлюцинациями;
характерна умственная отсталость,
Имеется склонность к коллаптоидным
состояниям, сильным головным болям.
Возможны боли в животе, диарея. Описаны
также гепато- спленомегалия, остеопороз.
Заболевание начинается в первые
месяцы жизни, с возрастом проявления
его могут уменьшаться. В
основе
болезни лежит дефект транспортной
функции клеток слизистой кишечника и
проксимальных отделов почечных
канальцев, в результате чего нарушается
адсорбция триптофана и ряда других
моноаминокарбоно- вых аминокислот с
нейтральной, ароматической и
гетероциклическими цепями. Развивается
эндогенный дефицит никотиновой кислоты.
При лабораторных исследованиях выявляют
гнперамннацидурию без повышения
концентрации аминокислот в крови,
повышенное выделение с мочой индольных
соединений, отсутствие трнптофана
в крови.
Тип
наследования
— аутосомно-рецессив- ный.
Дифференциальный
диагноз:
пеллагра.
ЛИТЕРАТУРА
Shih
У
, Bnb\
Е.
U.,
Alpers D. Н.
el.
at. Studies of
intestinal transport defect in Hartnup disease Gastroenterology.
1971. v. 61, p.
445—453
К
ilcken
В
Yu
J. S.. Brown D. A
Natural history of Hartnup disease. Arch Dis. Child., 1977,
v. 52, p. 38 40.
6, В микрофтальмия. Косоглазие, птоз, узкий нос. Микростомия.
Хиппеля—Линдау
синдром
Синоним:
церебро-ретинальный анпюматоз.
Опнсан
в 1926 г. A. Lindau.
Минимальные
диагностические признаки: ангиоматоз
сетчатки, гемангиобластомы мозжечка.
Клиническая
характеристика.
Поражение гла»проявляется ангиомами
сетчатки. Заболевание прогрессирует
и осложняется отслойкой сетчатки,
катарактой, глаукомой и слепотой.
Гемангиобластомы ЦНС локализуются
обычно в мозжечке и спинном мозге и
проявляются болями в области затылка,
головокружением, рвотой, атаксией,
нистагмом. дизартрией, спастическими
парапаре- зами. Следствием гемангиомы
задней черепной ямки может быть
гидроцефалия. Возможны гемангиомы
лица, надпочечников, легких, печени;
множественные кисты поджелудочной
железы, почек; рак почек; фео- хромоцитома.
Тип
наследования
— аутосомно-доми- нантный Ген локализован
на Зр26-р25.
Дифференциальный
диагноз:
опухоли мозжечка.
ЛИТЕРАТУРА
Fill
И
L
. Lamiell J U
.
Polk
Л/
О
The radiographic manifesiations of von Hippel Lindau
disease. Radiology. 1979, v. 133.
p. 289 -285
Shokeir
А/.
H
K. Von Hippel
Lindau syndrome: a repori on ihree kindreds. - J.
Med. Gener. 1970. v. 7, p.
155 157.
Холт—Орама
синдром
Holt—Oram
syndrome MIM 142900
Синоним:
синдром руки—сердца.
Описан
в 1960 г. М. Holt и S.
Oram.
Минимальные
диагностические признаки: порок
развития верхней конечности; врожденный
порок сердца.
Клиническая
.характеристика.
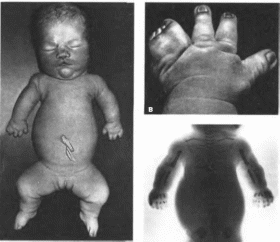
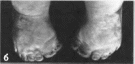
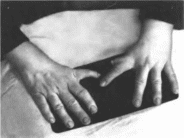
Пороки развития верхних конечностей
варьируют от изменений I пальца кисти
(гипоплазия, трех- фаланговый I палец)
(рис. 166) и
гипоплазии лучевой кости (50"")
до фокомелии Левая рука поражается
чаще У 50" « больных I палец не
противопоставлен остальным. Из скелетных
аномалий наблюдаются также гипоплазия
лопаток и ключиц, сколиоз, воронкообразная
деформация грудины, клинодак- тилия.
синдактилия, гипоплазия или аплазия
других пальцев кисти и костей запястья
(40%). В 85% случаев обнаруживают врожденные
пороки сердца: дефект межпредсерд- ной
перегородки (наиболее часто), дефект
межжелудочковой перегородки (50'У!.),
открытый артериальный проток,
коарктация аорты, стеноз легочной
артерии, пролапс митрального клапана.
Описаны гипертело- ризм. отсутствие
большой грудной мышцы, расщелина неба.
Интеллект, как правило, сохранен.
Тип
наследования
— аутосомно-доми- нантный с различной
экспрессивностью. Ген локализован
предположительно на I4q23-
q24.2.
Рис.
166. Аномалии
больших пальцев рук при синдроме Холт
Орама.
Von Hippcl Lindau syndrome mim 193300
Дифференциальный
диагноз:
дефекты лучевой кости; тромбоцитопения
с отсутствием лучевой кости; синдром
панцитопении Фанкони: VACTERL-ассоциация.
ЛИТЕРАТУРА
Gladstone
/..
Syhert
I P.Holt Oram
syndrome penetrance of ihe gene and lack of maternal effect. —
Clin. Genet.. 1982, v. 21.
p. 99 103.
Hurst
J. A.. Hall С.
M
.
Bur,
ser W. The Holt
Oram syndrome. — J. Med. Genet., 1991,
v 28. p. 406—410.
Хондродисплазия
метафизарная, тип Мак-Кьюсика
Metaphyseal
chondrodysplasia, type McKusick MIM: 250250
Синоним:
гипоплазия волос и хрящей. Впервые
описана в 1964 . V. McKusick Минимальные
диагностические признаки карликовость
с преимущественным поражением
дистальных отделов конечностей; редкие
тонкие волосы.
Клиническая
характеристика.
Отмечается внутриутробное отставание
в росте. Конечности (в основном кисти
и стопы) короткие, пальцы короткие
и толстые. Иногда отмечаются небольшое
искривление ног, разболтанность
суставов, подвывих или вывих головки
лучевой кости. Волосы, брови и ресницы
редкие, тонкие и светлые Возможны
анемия, нарушение всасывания в кишечнике,
аганглиоз кишечника, снижение клеточного
иммунитета. Описаны лимфомы. рак кожи
и двенадцатиперстной кишки, первичный
рак печени. Чрезвычайно тяжело протекает
герпетическая инфекция. Осложнения
включают килсвидную деформацию грудной
клетки, эквиноварусные деформации
стоп, артриты.
К
характерным рентгенологическим
признакам относятся расширение
метафизов в области коленных суставов
и средних фаланг кистей и стоп,
умеренное уплошение и недоразвитие
тел позвонков (у взрослых эти изменения
исчезают).
Тип
наследования
— аутосомно-рецессив- ный.
Дифференциальный
диагноз:
хондродисплазия метафизарная, тип
Шмида; хондродисплазия метафизарная,
тип Янсена; гипо- фосфатемия; гипофосфатазия;
гипохондро- плазия; дисхондростеоз.
ЛИТЕРАТУРА
Seige
М.
Metaphysare Chondrodysplasei vom type McKusick (Knorpel +
Haar Hypoplasie). — Mschr
Kinderheilk.. 1980. Bd. 128.
S. 157 159.
Van
der Burgt J.. Haraldsson A.. Ooslerwijk J. С
et
ul. Cartilage
hair hypoplasia, metaphyseal chondrodysplasia type McKusick:
description of seven patients and review of the literature. —
\m. J Med. Genet. 1991. v. 41.
p. 371- 380.
Хондродисплазия
метафизарная, тип Шмида
Metaphyseal
chondrodysplasia type Schmid MIM: 156500
Синоним:
метафизарный дизостоз. тип Шмида.
Описана
в 1949 г. F. Schmid. в 1958 г. P.
Maroteaux и М. Lamy.
Минимальные
диагностические признаки: низкий
рост, признаки метафизарной хонд-
родисплазии.
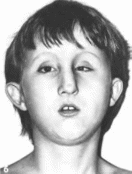
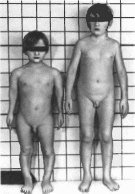
Рис
167. Слева —
больной с метафизарной хондроднеплазней.
тип Шмида (ннзкнй рост, варусная
деформация голеней). Справа — здоровый
мальчик того же возраста.
Клиническая
характеристика.
Типичны отставание в росте, варусная
деформация голеней, варусная установка
стоп
(рис. 167), «утиная»
походка. Средний рост взрослых--130
160 см. Отмечаются выступающие лобные
бугры, выраженный поясничный лордоз.
Интеллект сохранен. Рентгенологически
выявляются изменения в метафи- зах всех
трубчатых костей, особенно в проксимальных
отделах метафизов бедренных костей. В
области коленных суставов ростковые
зоны расширены. Длинные трубчатые кости
нижних конечностей укорочены, расширены
и дугообразно деформированы. Эпифизы
трубчатых костей и тела позвонков,
как правило, не изменены.
Тип
наследования
— аутосомно-доми- нантный.
Дифференциальный
диагноз: другие
типы метафизарных хондродисплазий;
гипофос- фатемия; гипофосфатазия;
гипохондропла- зия; дисхондростеоз.
ЛИТЕРАТУРА
SchmidF.
Beiirag zur Dysosiosis enchondralis me- taphysarea. —
Mschr Rinderheilk.. 1949. Bd. 97,
S. 393 397.
Lachman
R.
S.,
Rimoin
D. L.. Spranger J.
Metaphyseal chondrodysplasia Schmid type: clinical and
radiographic delineation wiih a reveiw of the literature. —
Pediat. Radiol.. 1988. v. 18.
p. 93 102.
Хондродисплазия
метафизарная, тип Янсена
Metaphyseal
chondrodysplasia type Jansen M1M: 156400
Заболевание
описано в 1935 г М. Jansen.
Минимальные
диагностические признаки. карликовость;
необычное лицо.
Клиническая
характеристика.
Типично отставание в росте за счет
укорочения и искривления конечностей
(рис. 168).
Средний рост взрослых — около 120 см.
Отмечаются увеличение и контрактуры
суставов, особенно тазобедренных и
коленных, плосковаль- гусная деформация
стоп. В результате дети ходят на согнутых
ногах, туловише наклонено вперед,
руки висят спереди, часто доходя до
колен. Отмечается брахидактилия с
преимущественным укорочением
концевых фаланг. Лицевые аномалии
включают гиперте- лоризм, экзофтальм,
широкую уплошенную переносицу,
микрогению, аномалии прикуса. Уровень
кальция в крови часто повышен
Рентгенологически
обнаруживаются чашеобразное
расширение метафизов, нарушение
энхондрального окостенения. Длинные
трубчатые кости укорочены и дугообразно
деформированы. Встречаются также
склероз основания и истончение
костей свода черепа, недоразвитие
лицевых костей. Возможна глухота.
Тип
наследования
— аутосомно-доми- нантный.
Дифференциальный
диагноз: другие
типы метафизарной хондродисплазии;
энхондро- матоз; гипофосфатазия.
ЛИТЕРАТУРА
Charrow
J.. Poznanski А.
К.
The Jansen type of metaphyseal chondrodysplasia: confirmation
of dominant inheritance and reveiw of radiographic
manifestations in newborn and adult - Am.
J Med Genet.. I984.v. 18. p. 321
327.
Ozonoff
\1. D.
Asphyxiating thoracic dysplasia as a
complication of metaphyseal chondrodysplasia (Jansen type) —
In: Skeletal Dysplasias/Ed. D. Bergenia. Amsterdam,
1974, p. 72 -77.
Рис.
168. Деформация
нижних конечностей при метафизарной
хондродисплазии типа Янсена.

Хондродисплазия
точечная, тип Конради—Хюнермана
Chondrodysplasia
punctata. Conradi Hunermann type MIM: 118650
Заболевание
описано в 1914 г Е. Conradi Минимальные
диагностические признаки: асимметричное
укорочение конечностей, ранняя точечная
кальцификация эпифизов, плоское лицо,
пористая кожа.
Клиническая
характеристика
Основные при жаки синдрома — асимметричное
укорочение конечностей в связи с
точечной кальцификацией эпифизов,
контрактуры крупных суставов (30%), ранний
сколиоз, небольшое отставание в
росте
(рис. 169)
Наблюдаются плоское лицо, запавшая
переносица, гипоплазия скуловых
дуг, антимонго-
Рис.
169. Асимметричное
укорочение конечностей и сколиоз
прн точечной хондродисплалш. тип Конрадн
Хюнермана.
лоидный
разрез глаз, в 20"!■ случаев — катаракта
и ихтиозоформные изменения кожи; кожа
пористая, напоминающая корку апельсина.
Волосы редкие, жесткие: в 25" о случаев
отмечается алопеция. Рентгенологически
выявляется деформация тел позвонков,
укорочение трубчатых костеи (обычно
одностороннее), точечная кальцификация.
в первую очерель появляющаяся в конечных
отделах трубчатых, каргтальных и
тарзальных костей. отростках
позвонков, седалишных и лонных костях.
Отмечается умеренная умственная
отсталость.
Тип
наследования
— аутосомно-доми- нантный.
Дифференциальный
диагноз:
хондродисплазия точечная, ризомелический
тип, множественная эпнфизарная
дисплазия. феталь- ный варфариновый
синдром.
ЛИТЕРАТУРА
Bergslom
К
. Guf/arson
К
-Н.
Jorulf
Н
Chondro- dyslrophia calcificans congenita (Conradfs disease)
in a molher and her child Clin. Genet.. 1972. v.
3, p. 158—161
Silengo
M. С.
Luzzaiii
L..
Silverman
F. /V.
Clinical and genetic aspects of Conradi Hunermann disease: a
report of three familial cases and review of the literature. J
Pediat . 1980. v 97. p.
911 917.
Хондродисплазия
точечная, ризомелический тип
Chondrodysplasia
punctata, rhizomelic type MIM 215100
Минимальные
диагностические признаки: низкий
рост; расщелина тел позвонков; укорочение
плечевой и бедренной костей; точечная
минерализация эпифизов.
Клиническая
характеристика.
Для синдрома типичны отставание в
росте, симметричное укорочение
конечностей за счет проксимальных
отделов (точечная минерализация),
множественные контрактуры суставов
(60" <■). микроцефалия
(рис. 170) Лицо
плоское. с запавшей переносицей,
антимонголо- идным разрезом глаз. В 80%
случаев наблюдается двусторонняя
катаракта, в 28% — их- тиозоформная
дисплазия кожи, алопеция. Рентгенологически
выявляются оссифика- ция вентральной
или дорсальной части позвонков.
симметичное укорочение и деформация
метафизов плечевой и бедренной костей.
Отмечается тяжелая умственная
отсталость. Большинство больных
детей умирают
Рис.
170.
Точечная
хон- дроднсплазия ризомеличес- кого
типа.
на
l-м году жизни. Ризомелический
тип точечной хондродисплазии
относится к группе пероксисомных
болезней.
Тип
наследования
— аутосомно-рецессив- ный.
Дифференциальный
диагноз:
хондродис- плазия точечная, тип Конради
—Хюнерма- на; фетальный варфариновый
синдром; це- ребро-гепато-ренальный
синдром.
ЛИТЕРАТУРА
Gilbert
Е.
F..
Opii: J. М..
Spranger
V.
И . el
at. Chonrodysplasia
punctata— rhisomelic form: pathologic
and radiologic studies of three infants. — Europ.
J. Pediat., 1976. v. 12. p.
89—109.
Happle
R. Cataracts as
a marker of genetic heterogeneity in chondrodysplasia punctata.
— Clin. Genet.. 1981, v.
19. p. 64—66.
Warilinsky
Т.
T.
Pagon R. A.. Powell B. R. et al. Rhisomelic
chondrodysplasia punctata and survival beyond one year: a review of
literature and five cases report. — Clin.
Genet.. 1990. v. 38, p.
84—93.
Хондродистрофия
новорожденных с полидактилией, тип
I
Polydactyly
with neonatal chondrodystrophy, type I
MIM:
263530
Синоним:
синдром коротких ребер- -полидактилии,
тип Салдино—Нунан.
Заболевание
описано в 1972 г. R. Saldino и
С. Noonan.
Минимальные
диагностические признаки: диспропорциональное
укорочение конечностей; постаксиальная
полидактилия кистей и стоп; узкая
грудная клетка; тяжелая дыхательная
недостаточность, приводящая к смерти
вскоре после рождения.
Клиническая
характеристика.
Верхние и нижние конечности резко
укорочены; грудная клетка узкая
из-за коротких ребер, идущих
горизонтально; живот большой Отмечается
полидактилия стоп и кистей, варусная
или вальгусная деформация стоп.
Встречаются пороки сердца и крупных
сосудов (чаще всего транспозиция
крупных сосудов), аномалии почек
(поликистоз, гипоплазия, агенезия),
атрезия или стеноз желудочно-ки- шечного
тракта, неполный поворот кишечника,
гипоспадия, атрезия влагалица. Характерна
умеренная гипотрофия, отеки. Все больные
страдают тяжелой дыхательной
недостаточностью, которая часто бывает
причиной смерти вскоре после рождения.
Нередки мертворождения. Рентгенологически
выявляют короткие горизонтальные
ребра, маленькие лопатки, укорочение
длинных трубчатых костей, изменение
формы мета- физов, горизонтальное
расположение крыши вертлужной
впадины, нарушение осси- фикации костей
черепа, таза и позвонков.
Тип
наследования
— аутосомно-рецессивныи
Дифференциальный
диагноз:
асфиксичес- кая дистрофия грудной
клетки новорожден-
19
173


Рис.
171.
Хондродистрофия новорожденных с
полидактилией, тип
И.
а
- внешний вид больной;
б,
а полиснндактилия
кистей н стоп:
г
■ - рентгенограмма скелета (короткие
ребра, укорочение длинных трубчатых
костей,
особенно большеберцовой).
Фотографин
любезно предоставлены проф F.Majeuski.