

Рис.
106. Мукополисахаридоз. тип VI. а - больная
2 лет: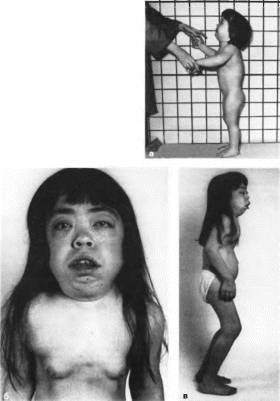
6, А та же больная в возрасте 18 лет. Грубые черты лица, гнпертелоризм, плоская переносица, грубые скелетные деформации, контрактуры суставов.
Мукополисахаридоз,
тип VII
Mucopolysaccharidosis,
type VII Синоним:
болезнь Слая. Описан в 1973 г. W.
Sly с соавт. Минимальные
диагностические признал и низкий
рост, гепатоспленомегалия, прогрессирующие
скелетные деформации, умственная
отсталость, дефицит Р-глюкуронидазы.
Клиническая
характеристика.
Типичны грубые черты лица с гипертелоризмом,
запавшей переносицей, вывернутыми
вперед ноздрями. Отмечаются
гепатоспленомегалия, паховые и
пупочные грыжи, низкий рост, киле- видная
грудная клетка, тораколюмбальный кифоз,
косолапость, повторные легочные
инфекции. Рентгенологически выявляют
клювовидные позвонки, расширенные
длинные трубчатые кости, множественный
дизостоз. Существует клиническая форма
без гепато- спленомегалии. В этом случае
имеются небольшое помутнение
роговицы, фибромы- шечная дисплаэия
аорты с инфильтрацией му- кополисахаридами,
приводящая к аортальной регургитации
и сердечной недостаточности Определяется
умеренная мукополисахариду- рия за
счет всех тр°х фракций гликозаминогли-
канов. Выявляют дефицит р-глюкуронидазы
в лейкоцитах и фибробластах кожи
Тип
наследования -
аутосомно-рецессив- ный. Ген локализован
на 7q? 1. 11.
Дифференциальный
диагноз: другие
типы мукополисахаридоза.
ЛИТЕРАТУРА
Hovme
Н.
Jones
A L.. Higginboiiom М
С. O
'Brien J.
S.
Presentation of mucopolysaccharidosis Vll (beta-glucuronidasc
deficiency) in infancy. —
J.
Med Genet.. 1981.
v.
18,
p.
237
239.
Мышечная
атрофия спинальная
Spinal
muscular atrophy MIM: 253300. 253400
Синонимы:
болезнь Верднига Гоффма- на; болезнь
Кугельберга Веландер
Минимальные
диагностические признаки. мышечная
гипотония, снижение или отсутствие
глубоких сухожильных рефлексов,
характерные изменения ЭМГ и биоптатов
мышц.
Клиническая
характеристика.
Различают несколько форм заболевания.
Инфантильная
форма (болезнь Верднига -Гоффмана)
проявляется внутриутробно снижением
двигательной активности плода или
вскоре после рождения мышечной гипо
тонией,
слабостью, снижением или отсутствием
сухожильных рефлексов Поражаются все
скелетные мышцы; характерна поза
распластанной лягушки — с отведенными
бедрами и согнутыми коленями
(рис. 107, а). Поражение
межреберных мышц и диафрагмы приводит
к возникновению парадоксального
дыхания. Объем активных движений резко
ограничен. Характерны мышечные атрофии.
фасцикулярные подергивания, особенно
в мышцах языка. Течение прогрессирующее.
Смерть наступает через I 2 года вследствие
дыхательной недостаточности н легочных
инфекций. При инфантильной форме с
поздним началом (после 6 мес) дети живут
несколько лет. Интеллект сохранен
Ювенильная
форма (болезнь Кугельберга- Веландер)
проявляется в возрасте от 2 до 18 лет
Характеризуется симметричной атрофией
мышц проксимальных отделов конечностей
(рис. 107, 6),
медленным прогрес- снрованнем. Отмечаются
фибриллярные и фасцикулярные подергивания,
задержка моторного развития,
отсутствие сухожильных рефлексов,
иногда — гипертрофия ягодичных и
икроножных мышц
(рис. 107, в).
Возможны вторичные деформации
позвоночника. У мальчиков заболевание
протекает тяжелее. Прогноз для жизни
благоприятный. На ЭМГ выявляются
снижение частоты разрядов при
произвольных сокращениях, «ритм
частокола», увеличение длительности
и амплитуды моторных разрядов.
Патоморфо- логически обнаруживают
дегенеративные изменения в клетках
передних рогов спинного мозга, в
мышцах — атрофию и дезорганизацию
фибрилл, разрастание соединительной
ткани.
Тип
наследования
— аутосомно-рецессив- ный. По-видимому,
обе формы заболевания являются
аллельными. Ген локализован на
5ql2.2-ql3.3.
Дифференциальный
диагноз:
синдром Шарко—Мари Туса: мышечные
дистрофии; структурные миопатии.
ЛИТЕРАТУРА
Ghetti
В
Amati
А..
Типа 14.
I
eia/
Werdnig
-
Hoffmann—Wohlfarl—Kugclberg
Welander disease: nosological unity and clinical variability in
inlra- familial cases. -
Acta
Genet. Med. Gemellol., 1971
v.
20,
p.
43
58
Zcrres
A
Grimm T.
Genetic counseling in families with spinal muscular atrophy type
Kugelberg We lander. —
Hum.
Genet.. 1983.
v.
65.
p.
74—75.
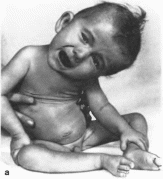
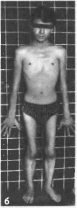
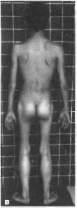
Рис.
107. Спинальная мышечная атрофия
а
больной 18 мес: 6,
а гипотрофия мышц проксимальных
отделов конечностей, гипертрофия
ягодичных и икроножных мышц.
Мышечная
дистрофия аутосомно-рецессивная
Muscular
dystrophy, Duchenne-like MIM: 253700
Минимальные
диагностические признаки: слабость
проксимальных групп мышц, гипертрофия
мышц, особенно икроножных; увеличение
активности креатинфосфокина- зы в
сыворотке; признаки миопатии и дистрофии
в мышечных биоптатах; начало заболевания
в детском возрасте.
Клиническая
харакпи-риспшка.
Заболевание начинается в возрасте от
2 до 14 лет (чаше в 5- Шлет), прогрессирует
сравнительно медленно (больные
утрачивают способность ходить к
20—24 годам).
Мышечная
слабость наблюдается главным образом
в проксимальных отделах конечностей.
Нередки псевдо! ипертрофии икроножных
мышц На поздних стадиях заболевания
умеренно повышается активность
сывороточных ферментов, в том числе
креа- тинфосфокиназы.
Тип
наследования
— аутосомно-рецессив- ный
Дифференциальный
диагноз:
псевдогипертрофическая мышечная
дистрофия; мышечная дистрофия
плечевого и тазового пояса.
ЛИТЕРАТУРА
Ben
Yelloun-Dellagi S.. Cha ffey P. ei al.
Presence of normal dystrophin in Tunisian severe childhood autosomal
recessive muscular dystrophy. —
Neurology,
1990.
v
40.
p.
1903
Hazama
R. Tsujthaia M.
Mori
M Mori
A Muscular dystrophy in six young girls. —
Neurology.
1973,
v.
29,
p.
I486—1491.
Zats
M . Passos-Bueno M R . Rapapori D
Estimate of the proportion of Duchenne muscular dystrophy
with autosomal recessive inheritance. -
Am.
J Med. Genet., 1989,
v.
32,
p
407—410.
Мышечная
дистрофия лице- плече-лопаточная
Facioscapulohumeral
muscular dystrophy MIM: 158900
Синоним:
мышечная дистрофия Ланду- зи - Дежерина.
Описана
в 1885 г. L. Landouzy с соавт.
Минимальные
диагностические признаки: атрофия
мышц лица и плечевого пояса.
Клиническая
характеристика.
Типичны атрофия и слабость лицевой
мускулатуры.
гипомимия,
отсутствие морщин на лбу и сглаженность
носогубных складок. Из-за поражения
мышц плечевого пояса больные не могут
поднять и удержать руки над головой.
Характерны крыловидные лопатки, плоская
грудная клетка и воронкообразная i
рудина При вовлечении мышц тазового
пояса развивается выраженный лордоз.
Описаны поражение мышц верхних и
нижних конечностей, врожденное
отсутствие некоторых мышц, например
грудных. Заболевание проявляется в
возрасте 13— 19 лет
Тип
наследованы)
— аутосомно-доминантный с полной
пенетрантностью. Ген локализован
на 4q34-qter.
ЛИТЕРАТУРА
Becker
Р
Neues
zur Genetik und Klassifikation der Muskeldystrophien. —
Humangenetik,
1972,
Bd
17.
S.
1
22.
Jacobsen
S J Diala E S.
Dorse\
В
t
el al A clinically
homogeneous group of families with facioscapulohumeral
(Landouzy Dejenne) muscular dystrophy: linkage analysis of six
autosomes. —
Am.
J Hum. Genet.. 1990.
v.
47.
p.
376
388.
Morion
N.
Chung
C.
Formal genetics of muscular dystrophy —
Am
J. Hum Genet, 1959
v
II,
p.
360
379.
Мышечная
дистрофия плечевого и тазового пояса
Muscular
dystrophy, limb-girdle MIM: 253000
Минимальные
диагностические признаки: симметричная
слабость мышц плечевого или тазового
пояса; симптомы миопатии на ЭМГ и в
биоптатах мышц; повышение активности
креатинфосфокиназы в крови.
Клиническая
характеристика.
Заболевание чаще проявляется в
детстве, иногда — в 30—50 лет. Первоначально
поражаются мышцы тазового пояса, затем
- других областей. Гипертрофия мышц
отмечается редко Тяжесть и быстрота
прогрессировать заболевания варьируют
Тяжелая инвалидность развивается
обычно через 20 лет болезни. Мышечные
контрактуры и скелетные деформации
возникают на поздних стадиях или
отсутствуют. Развивается характерная
покатость плеч, имитирующая сгорбленность.
Из-за слабости мышц тазового пояса
больным трудно вставать с пола и с
низкого стула Деформации скелета
вызваны асимметричным поражением
скелетных мышц. ЭМГ
и
биопсия мышц выявляют признаки миопатии.
Увеличение активности сывороточных
ферментов менее выражено, чем при
псевдогипертрофической мышечной
дистрофии.
Тип
наследования
аутосомно-рецессивный. Ген локализован
на 15ql5-q22.
Дифференциальный
диагноз:
псевдогипертрофическая мышечная
дистрофия; другие формы мышечных
дистрофий.
ЛИТЕРАТУРА
DeCosier
W.
DeReuck
J.
Thiery
E
A laie autosomal dominant form of limb-girdle muscular
dystrophy: a clinical, genetic and morphological study. —
Eur.
Neurol., 1974.
v.
12,
p.
159
-172
Gilchrisi
J Л/
Pericak
Vance M.. Silverman L.. Roses D
Clinical and genetic investigation in autosomal dominant
limb-girdle muscular dystrophy Neurology, 1988,
v.
38,
p.
5—9.
Мышечная
дистрофия псевдогипертрофическая
Muscular
dystrophy, pseudohypertrophic MIM: 310200
Синонимы
■ мышечная дистрофия Дюшен- на; мышечная
дистрофия Беккера.
Минимальные
диагностические признаки: мышечная
слабость, преимущественно в проксимальных
группах мышц; повышение уровня
креатинфосфокиназы в сыворотке крови;
поражение лиц мужского пола.
Клиническая
характеристика.
Выделяют два типа псевдогипертрофичсской
мышечной дистрофии тип Дюшенна с
тяжелым течением и доброкачественный
тип Беккера. Обе формы — результат
мутаций гена, кодирующего белок
дистрофии. Большинство
12
173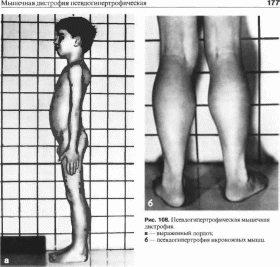
мутаций
представляют собой делеции участков
различной протяженности.
Мышечная
дистрофия Дюшенна начинается обычно
в первые 3 года жизни с неуверенной
походки. Во многих случаях дети поздно
начинают ходить. Иногда до 3— 7 лет
походка не изменена. Ребенку трудно
встать с пола. Для более поздних стадий
характерна утиная походка с широко
расставленными стопами, разведенными
носками отведенными назад плечами и
поднятым подбородком. Нередок
поясничный гиперлордоз (рис.
108, а).
Заболевание неуклонно про- 1 рессирует,
к 10— 11 годам дети уже
прикованы к постели. Прогрессирующая
атрофия начинается с проксимальных
мышц, а затем распространяется на
дистальные; появляется слабость
мышц плечевого пояса (затруднено
поднимание руки до горизонтального
уровня). Развивается псевдогипертрофия
икроножных и ягодичных мышц
(рис. 108, б). В
поздней стадии заболевания часто
возникают сгибательные мышечные
контрактуры бедренных, коленных суставов
и суставов верхних конечностей вследствие
того, что атрофия сгибательных мышц
сильнее, чем разгибательных. Больные
склонны ходить на носках Рано снижаются
глубокие сухожильные рефлексы. У
некоторых детей имеется небольшое
снижение интеллект Деформации
скелета связаны с мышечной атрофией
и включают искривление длинных трубчатых
костей, остеопороз, тяжелое искривление
позвоночника. Средняя продолжительность
жизни — 20 лет. Смерть обычно наступает
от легочных инфекций или сердечной
недостаточности
При
мышечной дистрофии Беккера слабость
развивается преимущественно в
проксимальных мышцах захватывая
тазовый пояс, мускулатуру бедер и в
меньшей степени — мускулатуру верхних
конечностей Забо
1евание
проявляется в возрасте 20—30 лет.
Наблюдается прогрессирующий поясничный
лордоз, появляются утиная походка,
затруднение при подъеме с пола («приемы
миопата»), беге, а в поздних стадиях
- при ходьбе. Течение заболевания
доброкачественное. При биопсии мышц
выявляют характерные изменения
(перерождение мышц, некроз отдельных
волокон). В крови повышена активность
ферментов, в первую очередь
креатинфосфокиназы, а также альдолазы,
трансаминазы, лактатдегид-
рогеназы;
на ЭМГ обнаруживают признаки миопатни.
на ЭКГ — признаки поражения миокарда
и нарушения проводимости.
Популяционная
частота
мышечной дистрофии Дюшенна — 1 : 3500
мальчиков, мышечной дистрофии Беккера
— I : 30 000 мальчиков.
Тип
наследования
Х-сцепленный рецессивный. Ген
локализован на Хр21.2.
Дифференциальный
диагноз другие
формы мышечных дистрофий.
ЛИТЕРАТУРА
Beggs
И..
КипкеI
L.
Improved diagnosis of Du- chenne- Becker muscular dystrophy. -
J
Clin. Invest.. 1990.
v.
85,
p.
613
619.
Covone
F Lerone M.. Romeo G.
Genotype-phe- notype correlation and germ line mosaicism in Du-
chenne muscular dystrophy /
Becker
muscular dystrophy patients with deletions of the dystrophin
gene Hum. Genet.. 1991.
v.
87.
p.
353
360
Ron
H D.
Detection the Duchenne carrier by ultrasound and computerised
tomography. Lancet, 1983,
v.
l.p. 1199
Nerman
A Thomas N
kings/on
H.. Harper P Becker
muscular dystrophy: correlation of deletion type with clinical
severity. J. Med Genet.
1990 v.
27,
p.
236—239.
Мышц
глазного яблока и глотки дистрофия
Muscular
dystrophy oculopharyngeal MIM 164300
Описан
в 1962 г. Victor G. с
соавт
Минимальные
диагностические признаки нтоз
век. слабость мышц глотки, признаки
миопатии или дистрофии в мышечных био-
птатах.
Клиническая
характеристика.
Заболевание начинается в возрасте
30—50 лет с двустороннего птоза век
и прогрессирующей дисфагии. Птоз со
временем обычно усиливается и требует
коррекции. Из-за дисфагии больной не
может проглотить сначала твердую,
а затем и жидкую нишу. Возможны осложнения
в виде аспирационных пневмоний. Через
10—20 лет болезни развивается небольшая
слабость в проксимальных мышцах
плечевого и тазового пояса. Достоверных
диагностических критериев, кроме
биопсии мышц. нет. Активность сывороточных
ферментов, таких как креатинфосфокиназа,
в пределах нормы или незначительно
увеличены На ЭМГ- симптомы миопатии.
Тип
наследования
— аутосомно-доминантный.
Дифференциальный
диагноз:
миотоничес- кая дистрофия; другие формы
мышечных дистрофий.
ЛИТЕРАТУРА
Pauzner
R..
BlauJ'.,
Monalleni
M.elal.
Mitochondrial abnormalities in oculopharyngeal muscular
dystrophy.
- Muscle
Nerve. 1991.
v.
14.
p.
947-
952.
Scrimgeour
E. M.. Masiaglia F.
Oculopharyngeal and distal myopathy. Am.
J.
Med. Genet., 1984,
v.
17.
p
763
-771.
Victor
M.. Haves R.. Adams R. D.
Oculopharyngeal muscular dystrophy. A familial disease of late
life characterized by dysphagia and progressive ptosis of the
eyelids. N.
Engl.
J.
Med., 1962,
v.
267.
p.
1267-
1272.
Мюллеровых
протоков персистенция у мужчин
Mullerian
derivatives in males persistent MIM: 261550
Синоним:
мужской внутренний псевдогермафродитизм.
Минимальные
диагностические признаки: развитие
наружных половых органов по мужскому
типу; наличие яичек, матки и фаллопиевых
труб при кариошне 46.XY.
Клиническая
характерисптка.
Кроме нормально развитых мужских
наружных и внутренних половых органов
имеются матка и фаллопиевы трубы.
Характерны кринтор- хизм, вирилизация
в пубертатном периоде. Эндокринных и
соматических аномалий не отмечается.
Заболевание выявляется обычно при
обнаружении матки и фаллопиевых труб
в содержимом паховой грыжи.
Тип
наследования
— аутосомно-рецессив- ный.
Дифференциальный
диагноз:
мозаицизм 45. Х/46, XY.
ЛИТЕРАТУРА
Beheslui
М..
Churchill
В..
Hardy
В.
ei
at.
Familial persistent mullerian duct syndrome J.
Urol..
1984.
v.
131,
p.
968—969.
Solan
W. R.. Walsh P. C.
Familial persistent Mullerian duct syndrome. J. Urol., 1976.
v.
115.
p.
459.