Миеломенингоцеле
Myelomeningocele
MIM: 182940
Синонимы:
врожденный дефект невраль- ной трубки;
спинномозговая грыжа.
Минимальные
диагностические признаки: кистозная
расщелина позвоночника.
Клиническая
характеристика.
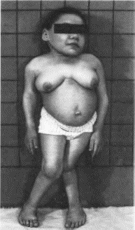
Миеломенингоцеле — грыжевое
выпячивание спинного мозга через
расщелину позвоночника. Грыжевой
мешок представлен кожей и мягкой
мозговой оболочкой, а его содержимое
— спинным мозгом и спинномозговой
жидкостью. Чаще всего дефект локализуется
в пояснично- крестцовой области
(рис. 94) либо
в шейном отделе позвоночника. Клинически
порок проявляется нарушением функции
моторных нейронов спинного мозга,
отсутствием рефлексов и потерей
чувствительности. Миеломенингоцеле
можетсочетатьсяс врожденными пороками
сердца, аномалиями мочеполовой системы,
расщелиной неба. Этиология мультифактори-
альная.
Популяционная
частота
дефектов нев- ральной трубки— I : 1000.
Соотношение
полов — МI :Ж
I. Дифференциальный
диагноз:
менингоцеле; липоменингоцеле;
гидроцефалия.
ЛИТЕРАТУРА
Carter
С.
О.. Evans
К..
Till
К.
Spinal
dysraphism genetic relations to neural tube malformations —
J.
Med. Genet., 1976.
v.
13.
p.
343
350.
Микросомия
гемифациальная
Hemifacial
microsomia MIM: 164210
Синонимы:
односторонняя гипоплазия лица; синдром
первой жаберной дуги.
Минимальные
диагностические признаки: односторонняя
аномалия ушной раковины и гипоплазия
нижней челюсти
Клиническая
характеристика.
Поражение
Рис
95.
Черепно-лицевые дизморфии при геми-
фациальной микросомииVannier j. P.. Lefort j., Cavelier b. Ei al. Spina bifida cystica families X-ray examinations and hla typing. — Pediatr. Res., 1981, V. 15. P. 326—329.
обычно
одностороннее. В 100% случаев отмечаются
аплазия, гипоплазия или другие аномалии
ушной раковины
(рис. 95);
наружный слуховой проход может
отсутствовать. У 95% больных обнаруживаются
преаурику- лярные папилломы Аномалии
глаз включают микрофтальмию, кисты
и колобомы радужки и сосудистой
оболочки, косоглазие. Лицо асимметрично,
глазная щель на пораженной стороне
расположена ниже, чем на здоровой.
Гипоплазия лицевых мышц создает
впечатление макростомии
(рис. 95)
Наблюдаются нарушение прикуса (90%),
гипоплазия нижней и верхней челюстей
(95%). Описаны случаи агенезии легких на
пораженной стороне
Попуяяциотшя
частопш— 1 ;
5600. Соотношение
полов — МЗ :
Ж2 Дифференциальный
диагноз,
окуло-аури- куло-вертебральная дисплазия,
ннжнечелю- стно-лицевой дизостоз;
гемнфациальная атрофия.
ЛИТЕРАТУРА
Conner
J М.
Fernandez
С
Genetic
aspects of hemifacial microsomia -
Hum
Genet, 1984,
v
68.
p.
349.
Gellis
S. S.. Feingold H
Hemifacial microsomia (picture of the month) -
Am.
J. Dis. Child.. 1971,
v.
122.
p.
57
58
Rollinck
В.
Kaye
С
Hemifacial
microsomia and variants, pedigree data -Am. J Med. Genet, 1983.
v.
15,
p.
233—283.
Thomas
P.
Goldenhar syndrome and hemifacial microsomia: observations on three
patients. —
Eur.
J. Pediatr 1980,
v.
133,
p.
387
297
Микротия
с атрезией наружного слухового прохода
и проводящей глухотой
Microtia
with meatal atresia and conductive
deafness
MIM:
251800
Минимальные
диагностические признаки. различные
деформации или отсутствие ушной
раковины с атрезией слухового прохода
или без нее; проводящая глухота.
Клиническая
характеристика
Встречаются различные аномалии
ушной раковины — от небольшого изменения
в размерах до ано- тии. В большинстве
случаев ушные раковины деформированы
(рис. 96)
Имеются остатки хрящевой ткани,
преаурикулярные выросты и фистулы
Атрезия наружного слу-
¥
i
Рис.
96. Деформация ушной раковины при синдроме
микротии с атрезией наружного слухового
прохода и проводящей глухотой.
хового
прохода чаше односторонняя, иногда
на противоположной стороне наблюдается
микротия в сочетании с атрезией или
без нее. Почти у всех больных определяется
снижение слуха по проводящему типу
(даже при неизмененном наружном слуховом
проходе) Глухота обусловлена патолотей
среднего уха (деформация, фиксация
молоточка и наковальни, отсутствие
наковальни, аномальные костные
выступы) Иногда глухота связана с
патологией внутреннего уха
Тип
наследования—
аугосомно-рецеосивный. Дифференциальный
диагноз:
окуло-аури- куло-вертебральная дисплазия;
гемнфациальная микросомия; синдром
хромосомы 18q ; фетальный
синдром краснухи.
ЛИТЕРАТУРА
Orslavik
К
Н. MedhoS..
MairJ.
И
Right-sided
microtia and conductive hearing loss with variable expressivity in
three generations Clin Genet,
1990, v
38,
p
117—120.
PreifferR
Heterogeneity of microtia —In
Abstracts Konf Klin. Genet Munchen, 1982.
p
28
Zankl
M ZangK.D
Inheritance of microtia and aural atresia in a family with five
affected members. —
Clin.
Genet,
I979.
v. 16,
p.
33I—334
Микрофтальмия
Microphthalmia
MIM >51600
Синоним
клиническая анофтальмия Минимальные
диагностические признаки: уменьшение
глазного яблока.
Клиническая
характеристика
Наблюдается изолированное уменьшение
глазного яблока Степень микрофтальмии
может быть различной, вплоть до
клинической картины анофтальмии.
Тип
наследования
— аутосомно-рецессив- ный
Дифференциальный
диагноз:
синдром Ленца, анофтальмия
ЛИТЕРАТУРА
Oliveira
da Silva Е.
Saniana
de Sousa S.
Clinical anophthalmia. —
Hum.
Genet.. 1981.
v.
57,
p.
115—
116
Микроцефалия
Microcephaly
MIM: 251200
Минимальные
диагностические признаки уменьшение
объема головы, уменьшение массы и
размеров мозга, умственная отсталость
Клиническая
характеристика.
Микроцефалия клинически проявляется
уменьшением объема головы более чем
на 2 стандартных отклонения для
данной возрастной ipyniibi Лоб
скошен, затылок уплощен, отмечается
выраженная диспропорция между лицевым
и мозговым черепом
(рис. 97). У
95% больных отмечается неврологическая
симптоматика: нарушение мышечного
тонуса, спастические парезы, судороги,
нарушение функции черепно-мозговых
нервов, хо- риоретинит Постоянный
признак микроцефалии — умственная
отсталость. Рентгенологически
обнаруживаются открытые черепные
швы, толщина костей черепа может быть
увеличена. На аутопсии находят уменьшение
массы и размеров головного мозга
(микроэнцефалия), аномальное строение
больших полушарий (микро- и полигирия,
Рис.
97.
Диспропорция мозговой и лицевой частей
черепа, скошенный лоб при микроцефалии.
нарушение
расположения извилин, структурные
нарушения цитоархитектоники коры
и др.). Различают истинную микроцефалию
с аутосомно-рецессивным типом
наследования и вторичную микроцефалию,
развивающуюся в результате
органического поражения головного
мозга разной этиологии (токсоплазмоз,
родовая травма, внутриутробная
гипоксия, фетальный цитомегалови-
русный синдром, фетальный радиационный
синдром, фетальный синдром краснухи).
Микроцефалия встречается также при
многих хромосомных и генных синдромах.
Соотношение
по лов — Ml Ж1.
Дифференциальный
диагноз:
краниосте- ноз; вторичная микроцефалия
ЛИТЕРАТУРА
Quart
Q Н.
Reed
Т
Е
A
problem in diagnosis of primary versus secondary microcephaly. Clin.
Genet, 1973.
v
4,
p
46—52.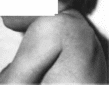

Рис.
98. Синдром микроцефалии и спастической
диплегии.
а
больной 4 лет (брахимнкроцефалия.
плоский затылок, низко расположенные
ушиые раковины): б — больной 9 лет
(тетраплегия, микроцефалия).
Микроцефалии
и спастической диплегии синдром
Microcephaly
with spastic diplegia M1M: 311400
Синонимы:
синдром Семановой 1 синдром Пейна.
Описан
в 1973 г. Е. Seemanova с соавт.
Минимальные
диагностические признаки: микроцефалия,
глубокая умственная отсталость,
судороги спастическая диплегия или
тетраплегия.
Клиническая
характеристика.
При рождении масса тела нормальная
или немного увеличена. имеется
микроцефалия
(рис. 98, а). В дальнейшем
отмечаются отставание в росте и
психомоторном развитии, судороги
(вначале petit mal, затем
grand mal). Развивается
спастическая диплегия или тетраплегия
(рис.
98, б);
сухожильные рефлексы повы
шены,
брюшные и кремастерный рефлексы
отсутствуют С возрастом частота
судорожных припадков нарастает.
Смерть наступает в возрасте 3—8 лет.
При патологоанато- мическом исследовании
обнаруживают ат- рофические изменения
большого мозга Аномалии внутренних
органов не описаны.
Соотношение
полов—
Ml
:
Ж1.
Тип
наследования
— предположительно Х-сцепленный
рецессивный.
Дифференциальный
диагноз, другие
формы микроцефалии; другие типы
Х-сцеплен- ной умственной отсталости.
ЛИТЕРАТУРА
Seemanova
Е
. Lesnv
L Hyanek J. el al
X-chro- mosomal recessive microcephaly wilh epilepsy, spastic
tetraplegia and absent abdominal reflex New variety «Paine
syndrome»9
Human Genetics, 1973,
v
20.
p.
113
117.
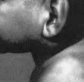

Рис.
99. Синдром микроцефалии, нормального
интеллекта, иммунного дефицита и риска
малигнизации лимфоретикулярнои системы.
Девочки 3, 6 и 10 лет.
Микроцефалии,
нормального интеллекта, иммунного
дефицита и риска малигнизации лимфо-
ретикулярной системы синдром
Microcephaly
with normal intelligence, immunodeficiency and lymphoreticular
malignancies MIM: 251260
Описан
в 1982 г. E. Seemanova с соавт.
Минимальные
диагностические признаки: тяжелая
врожденная микроцефалия, нормальное
психомоторное развитие, частые
респираторные инфекции, иммунная
недостаточность, характерное лицо
с выступающей средней частью,
повышенная частота зло
качественных
заболеваний лимофретику- лярной системы,
отставание в росте.
Клиническая
характеристика.
Дети рождаются с низкой или нормальной
массой тела и выраженной микроцефалией
(окружность головы у новорожденных
— 27— 31 см). Психомоторное развитие
соответствует возрасту. Характерны
отит, мастоидит, частые респираторные
инфекции, приводящие к бронхоэктазам.
Возможно, это связано с недостаточностью
иммунной системы: уровни иммуноглобулинов
снижены, нарушен клеточный иммунитет
(изменена реакция бласттрансформации
лимфоцитов снижен уровень T-лимфоцитов).
Характерны скошенный лоб, гипоплазия
нижней челюсти, монголоидный разрез
глаз, увеличенные
II
173
уши,
короткая шея У детей старшего возраста
отмечаются выступающая средняя часть
лица, большой нос
(рис. 99). Уже
в дегстве встречаются злокачественные
опухоли лим- форетикулярной системы.
Введение у-глобу- лина не снижает рнска
инфекций и малш ни- зации. Аномалии
внутренних органов, за ио ключенисм
мнкроэнцефалии. не характерны.
Тип
наследования
— аутосомно-рецессив- ный.
Дифференциальный
диагноз:
синдром Блу- ма: атаксия-телеангиэктазия;
синдром Сек- келя. микроцефалия другой
этиологии
ЛИТЕРАТУРА
Seemanora
Е..
Passarge
Е..
Benesovu
D. etal.
Familial microcephaly with normal intelligence,
immunodeficiency and risk for lymphoreticular malignancies.
—
Am
J. Med. Genet.
I985.
v 20,
p
40
TeebyS..
Al-Awadi S.. White О
Autosomal
recessive nonsyndromal microcephaly with normal intelligence.
-
Am
J. Med Genet., I987. v. 26,
p.
355
359.
Миопатия
дистальная наследственная с поздним
началом
Myopathy,
late distal hereditary MIM 160500
Синонимы:
дистальная миопатия Велан- дер; дистальная
миопатия. шведский тин.
Описана
в 1951 г. Welander L.
Минимальные
диагностические признаки: слабость
в дистальных мышцах конечностей. на
ЭМГ и биопсии — признаки миопа- тии или
мышечной дистрофии.
Клиническая
характеристика.
Заболевание проявляется в возрасте 30
60 лет Процесс начинается с дистальных
мышц конечностей. В 89% случаев сначала
появляется слабость в мелких мышцах
кистей, распространяющаяся затем
проксимально. Исчезают ахиллов,
радиальный и ульнарный рефлексы
Чувствительность не нарушена.
Заболевание npoipeccupy- ет
медленно Причиной смерти можег быть
кардиомиопатия.
Тип
наследования —
аутосомно-доминшггньш.
Дифференциальный
диагноз:
мышечная дистрофия плечевого и
тазового пояса: перонеаль- ная мышечная
дистрофии Шарко— Мари— Туса; другие
формы мышечных дистрофий.
ЛИТЕРАТУРА
Marketberv
If
R . Griggs R С.
Leach
R P . Lap- ham L W.
Late onset hereditary distal myopathy. Neurology. 1974.
v.
24.
p
127
134.
Миотоническая
дистрофия
Myotonic
dystrophy MIM: 160900
Синоним:
болезнь Стейнерта.
Минимальные
диагностические признаки: быстрая
утомляемость и слабость лицевой и
шейной мускулатуры, дистальных мышц
конечностей: признаки мнотонии и
миопатии при ЭМГ.
Клиническая
характеристика.
Основные симптомы — слабость в руках,
затруднения при ходьбе, склонность к
падениям Миотония встречается почта
у 1/3 больных. Процесс начинается с
мыши лица, шеи и конечностей. Почти
всегда отмечаются симметричное
ограничение движений глазных яблок
и птоз Слабость жевательных, височных
и грудино-клю- чично-сосцевидных мышц
придает лицу характерный «измученный»
вид. Слабость мышц лица и языка часто
вы 1ывает дизартрию Слабость в
конечностях обусловлена изменениями
мыши предплечья, передних мышц голени,
икроножных и малоберцовых мышц.
Отмечается слабость мышц передней
брюшной стенки. При црогрессировании
заболевания в процесс вовлекаются
проксимальные мышцы конечносгей.
Генерализованная миотония редка
Снижаются сухожильные рефлексы, на
поздних стадиях развиваются контрактуры
мышц. Bciречаюгея также
катаракта. раннее облысение,
гнногонадизм, патология сердца
(65%). снижение ле! очной вентиляции,
легкие эндокринные нарушения.
задержка умственного развития,
ослабление перистальтики пищевода.
ЭМГ выявляет признаки миотонин.
Иногда несколько повышен уровень
креатинфосфокиназы. В биоптатах мышц
обнаруживают изменения диаметра
мышечных волокон, центральное расположение
ядер в клетках. На поздних сга- диях
мышечные волокна замешаются фиброзной
и жировой тканью.
Тип
наследована
— аутосомно-доми- нантный. Ген локализован
на 19ql3.2-ql3.3-
Дифференциальный
диагноз:
врожденная мнотония; врожденная
парамиотония; периодический t
нпсркалиемический паралич; другие
формы мышечных дистрофий, синдром
Прадера Вилли
ЛИТЕРАТУРА
Bundey
S . Carter С
О
Genetic
heterogeneity for dystrophia myotonica -
J
Med. Genet., 1972,
v.
9,
p
311—315
Gibson
S. L M. Ferguson-Snuih M
The use of
genetii
I nkage in counselling families with dystrophia myotonica. —
Clin.
Genet.. I980, v. 17,
p.
443
448.
Миотония
врожденная
Myotont
i congenita Thomsen MIM: 160800
Синоним:
болезнь Томсена.
Впервые
описана в 1876 г. J. Thomsen.
Минимальные
диагностические признаки: клнннческие
и электромно1 рафическне признаки
миотонии.
Этническая
характеристика.
Основное проявление — миотония
(медленное расслабление мышц с
последующим спазмом). Миотония выявляется
при перкуссии скелетной мускулатуры
в виде коротких (длительностью
несколько секунд) подергиваний или
спазмов мускулатуры. Чаще всего в
процесс вовлекаются только нижние
конечное™, иногда — мышцы глазных
яблок и кистей. Наиболее характерна
генерализованная безболезненная
скованность, усиливающаяся в покое и
уменьшающаяся при работе. Больные
жалуются на затруднение движений после
Отдыха. Движения медленные, при
повторении ускоряются Мш апие не
нарушено, однако сильное зажмуривание
может вызвать спазм, который
продолжается более минуты. Вне lannoe
движение, например чихание. может
спровоцировать длительный спазм мышц
лица, языка, гортани, шеи и 1 рудной
клетки. Обычно наблюдается диффузная
гипертрофия мышц бедра, предплечья,
плеча, которая может распространяться
на жевательную мускулатуру и мышцы
шеи. С возрастом миотония уменьшается,
мышечная сила не снижается Уровень
креатинфосфокинс ы крови в норме или
cjiei ка повышен. Биопсия
мышц патологии не выявляет, иногда
увеличены размеры отдельных мышечных
волокон.
Тип
наследования
— аутосомно-доми- нангный.
Дифференциальный
диагноз:
парамиото- ния врожденная: мнотоническая
дистрофия: паралич t
нперкалнемичеемш периодический;
хондроднстрофнческая миотония.
ЛИТЕРАТУРА
Koch
М
. Hurley
Н..
Surfarazi
М.
et
и!
Myotonia
congenita (Thomsen's disease) excluded from the region of the
myotonic dystrophy locus on chromosome 19.
Hum.
Genet.. 1989.
v.
82,
p.
163
166.
Torbergsen
T.
A family with dominant hereditary myotonia, muscular hypertrophy and
increased muscular irritability, distinct from myotonia
congenita Thomsen. Acta Neurol. Scand.. 1975,
v.
51.
p.
225—232.
Миотония
хондродистрофическая
Chondrodystrophy
myotonia MIM: 255800
Синоним:
синдром Шварца Джампе- ля Аберфельда.
Минимальные
диагностические признаки. миотония.
отставание в росте, контрактуры суставов,
блефарофимоз.
Клиническая
характеристика.
Ботьные обычно низкого роста: наблюдаю
гея множественные скелетные деформации
и миотония. Миотония появляется в
первые два года жнзни; иногда отмечается
истинная гипертрофия мышц или
гипоплазия и a i рофия
мышечных волокон. Лицо больных
плоское, рот и подбородок маленькие,
выражение лица печальное. Аномалии
глаз включают короткую и узкую
глазную шель. иптермитти- руюший птоз
(рис 100),
неправильный рост ресниц, микрокорнеа.
миопию высокой степени. ювенильиую
катаракту, иногда — мнкрофтальмню.
Скелетные деформации: корот кая шея.
«куриная» t рудь, кнфосколи-
оз, дисплазия та обедренно1 о сустава,
контрактуры суставов. Кроме того,
отмечаются низкий рост волос, низко
распо юженные уши, паховые и пупочные
грыжи, слабость анальною сфинктера,
маленькие яички, транзиторная лактозурня.
Интеллект сохранен.
Тип
нас ледования —
аут осомно-рецессив- нын.
Дифференциальный
диагноз:
арггрогриноз; кранио-карпо-тарзальпая
дисплазия; синдром Секкеля; врожденная
миотония: мнотоническая дистрофия.
ЛИТЕРАТУРА
A
be г
fe
Id D.. Namba Т..
lye
H. К.
Grab
D
Chondrodystrophy myotonia: report of two cases. Myotonic
dwarfism, diffuse bone disease, and unusual ocular and facial
abnormalities. Acrh. Neurol.. 1970.
v.
22.
p.
455-462.
Moodlei
M„ Moosa A.
Chondrodystrophy myotonia (Schwartz- Jampel syndrome) in South
African children. Neuropediatrics. 1990.
v.
21.
p.
206
210.
it*
щ
Митенса—Вебера
синдром
Mietens—Weber
syndrome MIM 249600
Описан
в 1966 г. С. Mietens и Н Weber.
Минимальные
диагностические признаки: ному
гнение роговицы, узкий нос, сгибатель-
ные контрактуры локтевых суставов,
умственная отсталость.
Клиническая
характеристика.
Основные проявления — помутнение
роговицы, горизонтальный или
ротационный нистагм, косоглазие,
узкий нос с гипоплазией крыльев.
Характерны укорочение предплечий,
вывих проксимальной головки лучевой
кости и сгибательные контрактуры
локтевых суставов. Иногда отмечаются
вальгусная деформация стоп, ограничение
разгибания коленных суставов, вывих
бедра, воронкообразная грудная
клетка, аневризмы сосудов. Все больные
умственно отсталые.
Тип
наследования
— предположительно аутосомно-рецессивный.
ЛИТЕРАТУРА
Mietens
С..
Weber
Н.
A
syndrome characterized by corneal opacity, nystagmu ,
flexion
contracture of
Рис.
100. Хондроднстрофичес- кая миотония
Короткая узкая глазная щель. птоз,
маленький рот.
the
elbows, growth failure and mental retardation —
J.
Pediatr.. I966, v. 69,
p.
624
629.
Мозго-глазо-лице-скелетный
синдром
Cerebro-oculo-facio-skeletal
syndrome MIM: 214150
Синонимы:
церебро-окуло-фацио-скелет- ный синдром;
COFS-синдром; синдром
Пена—Шокейра, тип II.
Описан
в 1974 г. S. Репа с соавт.
Минимальные
диагностические признаки: резкая
пренатальная гипоплазия, микроцефалия,
мнкрофтальмия, скошенный лоб, тонкие
губы, кифосколиоз, сгибательные
контрактуры суставов.
Клиническая
характеристики.
Из пороков головного мозга наиболее
часто встречается микроцефалия. Описаны
также внутренняя гидроцефалия; атрофия
зрительных нервов; агенезия мозолистого
тела; гипоплазия верхних теменных
извилин, гиппокампа, зрительных
нервов; микрогирия базиляриых
поверхностей. Отмечаются микрофтальмия,
катаракта, уплощение передней камеры
гла

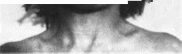
Рис.
101. Мозго-глазо-лице-скелетный синдром.
а
скошенный лоб. микроцефалия, макротия.
ретрогнатия;
6
сгибательные контрактуры суставов
за.
блефарофимоз. Аномалии лица: скошенный
лоб, седловидная переносица, большие
HHiKO расположенные уши,
тонкие губы, ретро- и микрогнатия
(рис. 101, а).
Из аномалий скелета встречаются
кифосколиоз, сгибательные контрактуры
суставов
(рис. 101,6),
камптодактилия. «стопа-качалка», вывих
бедра или дисплазия вертлужной впадины,
coxa valga. узкий таз, остеопороз.
Из внутренних органов чаше поражаются
почки (агене- зия, гипоплазия,
подковообразная почка). В единичных
случаях отмечаются врожденные пороки
сердца, релаксация диафрагмы, подвижная
слепая кишка, гипоплазия селезенки. У
больных короткая шея, широко расставленные
соски, поперечная ладонная складка.
Характерны выраженная пренатальная
гипоплазия, мышечная гипотония,
отставание физического и психомоторного
развития.
Соотношение
полов — Ml
:
ЖI.
Тип
наследования
— предположительно аутосомно-рецесси
вный.
Дифференциальный
диагноз:
артрогрипоз; синдром Халлермана Штрайфа;
окуло-ау- рикуло-вертебральная лисплазия;
церебро- гепато-ренальный синдром,
церебро-косто- мандибулярный синдром,
окуло-церебро- ренальный синдром Лоу,
фетальный синдром краснухи.
ЛИТЕРАТУРА
Gershom-Baruch
R.. Ludalscher R М
ел at
Cereb- ro-oculo-facio-skeletal syndrome: further delineation.
Am J Med. Genei .
I99l,v
41.
p
74—77.
Lourie
I VV.
Chersivoy
E D .
Luzjuk
G. I. el al Further
evidence for ihe autosomal recessive inheri- iance of the
COFS-syndrome Clin. Genet.. 1976,
v.
10.
p.
343
346.
PenaS.
D. J.. Schokeir M N.
K.
Autosomal recessive cerebro-oculo-facio-skeletal (COFS)
syndrome. Clin. Genet., 1974,
v.
5,
p.
285—295.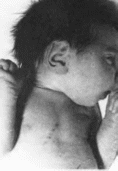

Муковисцидоз
Cystic
fibrosis MIM 219700
Синоним:
кистофиброз поджелудочной железы
Минимальные
диагностические признаки: рецидивирующие
легочные инфекции, повышение
концентрации ионов натрия и хлора
в поговой жидкости, нарушение функции
поджелудочной железы и кишечника.
Клиническая
характеристика.
Муковисцидоз представляет собой
множественное поражение экзокринных
желез, проявляющееся выделением
секретов повышенной вязкости. Вторично
в патологический процесс вовлекаются
легкие, поджелудочная железа и кишечник
Различают следующие формы муковисцидоза:
1) смешанная (ле- гочно-кишечная); 2)
преимущественно легочная, 3)
преимущественно кишечная. 4) мекониальный
нлеус; 5) стертые формы. Легочные
проявления возникают на 1— 3 году жизни
и характеризуются рецидивирующими
пневмониями, бронхитами, эмфиземой
легких, бронхоспазмом, бронхоэк- газамн.
Кишечные проявления связаны с нарушением
активности ферментов поджелудочной
железы. Гнилостные процессы в кишечнике
приводят к вздутию живота, появлению
обильного жирного стула с резко
выраженным гнилостным запахом. У
10—20% больных отмечается выпадение
прямой кишки, у 9—22% — билиарный цирроз
печени. Характерна выраженная гипотрофия,
несмотря на хороший аппетит Мекониальный
нлеус отмечается у 10—20% новорожденных
с муковисцидозом. При этом развивается
картина кишечной непроходимости:
рвота с примесью желчи, неотхожде- ние
мекония, увеличение живота. Возможен
мекониальный перитонит. Все больные
мужского пола бесплодны. При диагностике
муковисцидоза измеряют уровень натрия
и хлора в поте, концентрацию натрия в
ногтях и слюне, определяют активность
дуоденального содержимого и
протеолитическую активность кала,
проводят копрологичес- кое исследование.
Заболевание обусловлено мутациями
гена, кодирующего белок-регулятор
трансмембранной проводимости ионов
(CFTR).
Попуыционная
частота — 1 :
2500 новорожденных
Тин
наследования
- аутосомно-рецессив-
ный.
Ген локализован на 7q. Известно
более 400 мутаций
в гене
CFTR. самая частая из них-
AF508.
Дифференциальный
диагноз:
целиакия. хронические бронхолегочные
заболевания.
ЛИТЕРАТУРА
Кегет
В Sh..
Rommens J. М
. Buchanan
J el al Identification
of the cystic fibrosis gene, genetic analysis. —
Science,
1989,
v.
245.
p.
1073
1080.
Pussarge
E.
Cyslische Fibrose-Genetik und Erbbe- ratung. Mschr Kinderheilk 1978,
Bd
126.
s.
172
-173.
Sferru
Th J
The molecular biology of cystic fibrosis -
Ann.
Rev Med. 1993.
v.
44.
p
133-144.
Муколипидоз,
тип I
Mucolipidosis,
type I MIM: 256550
Синоним:
сналидоз, типы lull.
Впервые
описан в 1968 г. J. Spranger
Минимальные
диагностические признаки: умеренная
умственная отсталость, сипмтом «вишневой
косточки» на глазном дне, грубые
черты лица, снижение активности P-N-
ацетилнейрамннидазы в лейкоцитах
и в фиб- робластах.
Клиническая
характеристика
Заболевание проявляется в конце
первого года жизни отставанием в
психомоторном развитии. Отмечаются
также отставание в росте, диспропорциональное
телосложение за счет укорочения
туловища. Внешне больные му- колнпидозом
напоминают больных с муко- полнеахаридозом
1типа. На глазном дне обнаруживается
симптом «вишневой косточки»: в
некоторых случаях имеется помутнение
роговицы. Неврологические нарушения
появляются после 4 года жизни и
характеризуются медленно прогрессирующей
мышечной гипотрофией и гипотонией,
атаксией. судорогами, тремором,
нистагмом, признаками периферической
нейропагии Скелетные изменения
включают множественные дизостозы.
ограничение подвижности суставов. К
непостоянным признакам синдрома
относятся грыжи, гепатоегшеномега-
лия, снижение слуха. Лимфоциты
периферической крови вакуолизированы,
в костном мозге обнаруживаются
клетки с грубыми гранулами. Экскреция
мукополисахари- дов с мочой нормальная,
а экскреция олиго- сахарндов, содержащих
сналовую кислоту.
повышена.
Отсутствует активность p-N-аце-
тилнейраминидазы в лейкоцитах и фибро-
бластах.
Тип
нас кдования
— аутосомно-рецессив- ный.
Дифференциальный
диагноз:
мукополиса- харидозы. муколипидоз. типы
II. III
ЛИТЕРАТУРА
Kelly
Т..
Banosheski•
L..
Harris D. J. et al.
Mucolipidosis I (acid neuraminidase deficiency): three cases
and delineation of the variability of the phenotype. Am. J. Dis.
Child.. 1981.
v.
135.
p
703—708.
Spranger
J.
Mucolipidosis I: phenotype and nosology. Perspecl. Invest.
Mctabol. Dis., 1981.
v.
4,
p.
303-315.
Spranger
J.. Wiedemann H.
The genetic of mucolipidosis. Diagnosis and differential
diagnosi —
Hu-
mangenelik. 1970.
Bd.
9.
S.
113—139.
Муколипидоз,
тип II
Mucolipidosis,
type II MIM:252500
Синоним:
болезнь I-клеток.
Описан
в 1967 г. J. Leroy и R.
Demars.
Минимальные
диагностические признаки тяжелая
задержка психомоторного развития,
низкий рост, грубые черты лица,
гиперплазия десен.
Клиническая
характеристика.
Заболевание проявляется вскоре
после рождения. Больные резко отстают
в росте и психомоторном развитии.
Характерны аномалии лица: мелкие
орбиты, отекшие веки, небольшой
экзофтальм, выраженная подкожная
венозная сеть вокруг глаз и в височных
областях, сглаженные надбровные
дуги, полные щеки с множественными
телеангиэктазиями, выраженная гиперплазия
десен. Скелетно- мышечные аномалии
включают короткую шею и короткую грудную
клетку, деформацию грудины, паховые
и пупочные грыжи, врожденный вывих
бедра, ограничение подвижности
суставов, в частности плечевых и
запястных, широкие и короткие кисти,
тора- колюмбальный кифоз, шишковидные
ребер- но-хрящевые сочленения, признаки
множественного дизостоза. Диафизы
длинных трубчатых костей и свод черепа
поражены меньше, чем при муколипидозе
I типа Иногда встречаются помутнение
роговицы, небольшое увеличение печени.
Больные склонны к респираторным
инфекциям. Лабораторные исследования
выявляют нор
мальную
экскрецию мукополисахаридов с мочой,
необычные цитоплазматические включения
в лимфоцитах и фибробластах, повышение
уровня лизосомальных ферментов (в
частности p-N-ацетилгексозаминида-
зы и арилсульфатазы А) в сыворотке, моче
и спинномозговой жидкости.
Тип
наследования
— аутосомно-рецессив- ный. Ген локализован
на 4q21-q23.
Дифференциальный
диагноз:
мукополиса- харидоз; муколипидозы. типы
I, III и IV: G м
| -га н гл иозидоз.
ЛИТЕРАТУРА
Gilbert
Е.
F..
Dawson G.. Zurhein G. М.
et
al.
I-cell disease, mucolipidosis II. Pathological, hisiochemical.
ultrastructural and biochemical observations in four cases. —
Z.
Kinderheilk.. 173.
Bd.
114.
S.
259—292.
Okada
S.. Owada M.. Sakiyama T. et al.
1-cell disease: clinical studies of 21
Japanese
cases.—
Clin.
Genet., 1985.
v.
28,
p.
207—215.
Муколипидоз,
тип III
Mucolipidosis,
type III MIM: 252600
Синоним:
псевдогурлеровская полидистрофия.
Впервые
описан в 1965 г V. McKusick с
соавт.
Минимальные
диагностические признаки: болезненность
и тугоподвижность суставов, низкий
рост, грубые черты лица отставание
умственного развития, признаки
множественного дизостоза
Клиническая
характеристика.
Заболевание проявляется на втором
году жизни увеличением и ограничением
подвижности суставов. Тугоподвижность
суставов с возрастом медленно
нарастает. Отмечаются низкий рост,
укорочение туловища и верхних конечностей,
короткие и утолщенные ключицы.
Рентгенологически определяются
деформированные позвонки, сколиоз,
прогрессирующая деструкция эпифизов
головок бедренных костей, маленькие,
деформированные. поздно окостеневающие
карпаль- ные кости. Черты лица грубые.
Выявляется помутнение роговицы. Описаны
врожденные пороки сердца (недостаточность
аортального клапана), грыжи Возможна
умеренная умственная отсталость.
Экскреция мукополисахаридов с мочой
в пределах нормы. В костном мозге
обнаруживают вакуо- низированные
клетки. В культуре фибробла-
стов
выявляется дефицит N-ацегилглюкозамни-
I -фосфотрансферазы.
Тип
наследования
— аутосомно-рецессив- ный Ген локализован
на 4q2l-q23
Дифференциальный
диагноз:
мукополнсахарндоз, тип I
ЛИТЕРАТУРА
Honey
N.
К, Mueller
О
Т.. Little
L. Е.
et
al.
Мисс
lipidosis
111 is genetically heterogeneous. Proc. Natl Acad. Sci. A 1982.
v.
79.
p.
7420—7424.
Mt
Kusi к
V
.. Kaplan D,
Wise
D. ei al.
The genetic mucopolysaccharidoses Medicine. 1965,
v
44,
p.
445
—483.
Thomas
C.. Tiller С.
E..
Reynolds L.
И".
el
al.
Sia- lidase deficiency in mucolipidosis II (l-cell disease) and
mucolipidosi 111 (pseudo Hurler polydyslro- phy). —
In
Fifth Int. Cong Hum. Genet., Mexico City. 1976,
p.
40.
Varki.
P., Reitman M. L.. Kornfeld S.
Identification of a variant of mucolipidosis 111 (pseudo Hurler
polydystrophy): a catalitically active N-acetylglu-
cosaminylphosphotransferase that fails to phosphore- lale lysosomal
enzymes. —
Proc.
Natl Acad. Sci. USA. 1981,
v.
78,
p.
7773—7777.
Мукополисахаридоз,
тип I
Mucopolysaccharidosis,
type I MIM: 252800
Синонимы:
синдром Гурлер; синдром LLIefie;
фенотип Гурлер—Ш не
Впервые
описан в 1919 г. G. Hurler.
Минимальные
диагностические признаки задержка
роста, выраженная умственная отсталость,
черепно-лицевые дизморфии, множественный
дизостоз, помутнение роговицы,
повышенная экскреция мукополисаха-
ридов с мочой.
Клиническая
характеристика.
При рождении внешних изменений нет,
масса тела иногда повышена. В первые
месяцы жизни черты лица становятся
грубыми. Характерны запавшая
переносица, помутнение роговицы,
гепатоспленомегалия, тугоподвиж- ность
суставов и тораколюмбальный кифоз. Рот
обычно открыт. К постоянным симптомам,
развивающимся на втором году жизни,
относятся короткая шея, воронкообразная
или килевидная грудная клетка, паховые
и пупочные грыжи, гипертрихоз, особенно
на разгибательных поверхностях
конечностей и спине, макро- и скафоцефалия
(рис. 102, а),
увеличение языка и губ, мелкие редкие
зубы, ринит со слизистым отделяемым,
шумное дыхание, ограничение подвижности
в
межфаланговых,
локтевых, плечевых и тазобедренных
суставах. Кожа сухая, грубая, бледная.
Позже появляются признаки поражения
сердца: шум, кардиомегалия. Развиваются
глухота, слепота. После 1-го года жизни
рост замедляется Психомоторное развитие
до 2 лет нормальное. Для поздних стадий
характерна глубокая деменция. Больные
погибают в возрасте до 10 лет от бронхо-
легочных инфекций и сердечной
недостаточности. Рентгенологически
обнаруживают уплощение и расширение
турецкого седла, расширение диафизов
трубчатых костей, особенно верхних
конечностей и ребер, клювовидную
форму тел позвонков. Лабораторные
исследования выявляют повышенную
экскрецию дерматансульфата и гепаран-
сульфата, метахроматические гранулы
в 10- -60% лейкоцитов, метахроматическую
окраску фибробластов. На аутопсии
обнаруживают отложение мукополисахаридов
в клапанах сердца, хрящевых клетках,
периферических нервных ганглиях,
сетчатке, склере, роговице, почках,
селезенке. Основной дефект — дефицит
a-L-идуронидазы (фермент,
ответственный за катаболизм кислых
мукополисахаридов). Аллельная форма
синдрома Гурлер — синдром Шейе
(мукополисахаридоз типа IS,
ранее выделяемый как мукополисахаридоз
типа V) — проявляется в школьном возрасте;
течение его более доб- рокачественнно.
Отставание в росте незначительное,
интеллект обычно не страдает.
Отмечается укорочение шеи и туловища
(рис.
102, б), короткие
и широкие кисти, тугоподвижность
межфаланговых суставов, умеренное
ограничение подвижности локтевых,
плечевых и коленных суставов. В
большинстве случаев развивается
поражение аортального клапана, помутнение
роговицы.
Тип
наследования
— аутосомно-рецессив- ный Ген локализован
на 4pl6.3
Дифференциальный
диагноз: другие
типы мукополисахаридозов; муколипидоз,
тип III: GMi-ганглиозидоз.
тип I.
ЛИТЕРАТУРА
ForiuinJ..
Kleijer
И'.
Hybridization
siudies of fibroblasts from Hurler. Scheieand Hurler Scheiecom-
pound patients: support for the hypothesis of allelic mutants. Hum.
Genet., 1980,
v.
53,
p.
I55—159.
Rouhicek
M Cehler J., Spranger J.
The clinical spectrum of alpha-L-iduronidase deficiency. —
Am.
J. Med. Genet., 1985,
v.
20,
p.
471—481.
Рис.
102. Мукополнсахарндоз, тип I. а— макро-
и скафоцефалня: б внешний вид.
Scheie
Н
Hamhrick
G.
И'.
Barncss
L
A newly recognized form frusle of Hurler's disease (gargoy- lism). —
Am
J. Ophthalmol.. 1962,
v
53,
p
753
- 769.
SprungerJ
И
The
systemic mucopolysaccharidosis —
Ergeb
Inn Med Kinderheilkd,
1972. Bd.
32,
S.
165
265.
Мукополисахаридоз,
тип II
Mucopolysaccharidosis,
type II MIM 309900
Синоним
синдром Хан гера. Описан в 1917 г. С. Hunter.
Минимальные
диагностические признаки: характерный
внешний вид. мукополнсаха- ридурия,
дефицит идуронатсульфатазы в фибробластах,
сыворотке и лимфоцитах.
Клиническая
характеристика.
Течение доброкачественное. До I—2
лет наблюдаются
макро-
и скафоцефалия, паховые и пупочные
грыжи, повторные риниты, шумное дыхание
вследствие обструкции верхних
дыхательных путей В возрасте старше
2 лет появляются утолщение ноздрей,
губ и языка, туго- иодвнжность суставов,
задержка роста, гипертрихоз и
гепатоспленомегалия
(рис. 103).
Частые признаки — утолщенная кожа,
короткая шея, редкие зубы. В некоторых
случаях наблюдаются узелковые изменения
кожи конечностей и спины, пигментация
сетчатки, умеренное воронкообразное
вдав- ленне грудной клетки, полая стопа,
диарея, судороги Интеллект сохранен,
но возможны изменения психики.
Имеются слабо выраженные изменения
клапанного аппарата сердца, снижение
слуха и нарушение подвижности
суставов. Выделяют форму с более тяжелым
течением (IIA), при которой
нарастает умственная отсталость.
Продолжитель
но
130
120 ПО 100 ЭО
во
70
GO
ЭО
40 ЗО 20
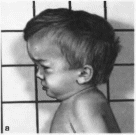
Рис.
103.
Мукополисахаридоз. тип
II.
Грубые черты ища. короткая шея.
гепатосгшеномегалия.
ность
жизни может быть достаточно большой
(описан больной, доживший до 60 лет), но
обычно смерть наступает в возрасте до
20 лет от сердечно-сосудистой
недостаточности. Отмечаются увеличение
турецкого седла, множественный
днзостоз, небольшие изменения
позвонков, остеоартрит тазобедренных
суставов. Повышена экскреция
дерматансульфага и гепарансульфата с
мочой. Накопление мукополисахаридов
связано с дефицитом идуронатсульфатазы
Тип
наследования
— Х-сцепленный рецессивный. Ген
локализован на Xq28.
Дифференциальный
диагноз:
другие типы
мукополисахаридозов;
муколипидоз, тип III.
Gm
-ганглиозидоз. тип I
ЛИТЕРАТУРА
Machill
G'..
Burhujam
G.
Dunielh
G.
Herrmann
F
H Segregation and sporadic cases in families with Hunter's syndrome.
J. Med. Genet., 199I. v. 28,
p.
398
401.
(
atziv
S.
Erickson
R. P.
Epstein
C. J.
Mild and severe Hunter syndrome (MPS II) within lhe same sibships. —
Clin.
Genet,
1977. v.
11,
p. Я19
326.
Мукополисахаридоз,
тип III
Mucopolysaccharidosis,
type III MIM 252900
Синоним
синдром Санфилиппо.
Описан
в 1963 г S
SanfilippoccoaBT
Минимальные
диагностические признаки: умственная
отсталость, относительно легкие
соматические проявления мукополисахари-
доза, экскреция гепарансульфата с
мочой, дефицит N-ацетил-а-глюкозаминидазы
и ге- паран-Ь)-сульфатазы
Клиническая
характеристика.
Раннее развитие детей нормальное.
Заболевание проявляется в возрасте
2 3 лет или позднее — в школьном возрасте
— нарушением психики, изменением
поведения (возбудимость, неспособность
к сосредоточению, агрессивность,
нарушение сна). Вдальнейшем снижается
интеллект, развиваются спастическая
диплегия Больные умирают в возрасте
до 20 лет (в редких случаях доживают до
30 лет) Отмечаются изменения скелета,
гепатосгшеномегалия
(рис. 104)
Черепно-ли- цевые аномалии незначительны.
Слух снижен, иногда значительно.
Рентгенологически выявляют утолщение
костей черепа. Повышена экскреция
гепарансульфата с мочой. Выделяют типы
А, В, С. D. сходные клинически.
но отличающиеся биохимическими
дефектами. При типе А отсутствует
гепаран- N-сульфатаза,
при типе В — N-ацетил-а-
глюкозаминидаза, при типе С — глюкоза-
минацетилтрансфераза, при типе D
— N-
ацетил-глюкозамин-6-сульфат-сульфатаза.
Гистохнмнческн определяется отложение
метахроматического материала в фибробла-
стах и лимфоцитах.
Тип
наследования
— аутосомно-рецессив- ный
Дифференциальный
диагноз:
другие типы мукополисахаридозов;
муколипидоз, типы I,
II,
III; GM-ганглиозидоз,
тип I.
Рис.
104.
Мукополисахарндоз, тип 111. Запавшая
переносица, короткий нос, гепатосплеиоме-
галия.
ЛИТЕРАТУРА
Siciliani)
L.. Fiumara A.. Pavone L. el al.
Sanfillip- po syndrome lype D in Iwo adolescent sisters -
J
Med Genet 1991.
v.
28.
p.
402
-405.
Van
de KampJ. J. P., Niermever H. F.. von Figura K.. Giesberis M.
Genetic heterogeneity and clinical variability in the Sanfilippo
syndrome (types A. B. C). Clin. Genet.. 1981.
v.
20.
p.
152—160.
Мукополисахарндоз,
тип IV
Mucopolysaccharidosis
type IV MIM: 253000
Синоним:
синдром Моркио. Описан в 1929 г. L.
Morquio и I. Brailsford. Минимальные
диагностические признаки: выраженное
отставание в росте, прогрессирующие
деформации позвоночника и грудины,
короткая шея, дефицит галактозо-6-
сульфатазы.
Клиническая
характеристика.
На 2-м году жизни начинается отставание
в росте и появляются скелетные
деформации (вальгусная деформация
коленных суставов, выбухание
нижних
ребер, кифосколиоз). Туловище укорочено
в большей степени, чем конечности (рис.
105). Интеллект
относительно сохранен. Отмечаются
значительная задержка физического
развития, диффузное помутнение роговицы,
выступающая нижняя часть лица, гипоплазия
эмали зубов, короткая шея (голова
выглядит сидящей прямо на плечах),
скафоцефалия, кипевидная грудная
клетка, выраженный поясничный лордоз,
большой живот, гиперподвижность и
подвывихи суставов (например
запястных), плоскостопие. К 20 годам
обычно развивается недостаточность
аортальных клапанов с регургита- цией.
У большинства больных снижен слух.
Возможны симптомы сдавления спинного
мозга деформированными позвонками,
тет- раплегия, обусловленная дислокацией
I шейного позвонка (в связи с аплазией
зубовидного отростка или слабостью
связок). Рентгенологические данные:
платиспондилия, увеличение расстояния
между позвонками, неправильная форма
эпифизов, отставание костного возраста,
широкие ребра, генера-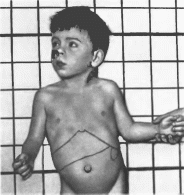
лнзованный
остеопороз. Смерть обычно наступает
до 20 лет от сердечно-легочной
недостаточности. Лабораторные
исследования выявляют повышенную
экскрецию с мочой кератансульфата или
всех кислых му- кополисахарндов. Тип А
характеризуется дефи цигом гала ктоза
м и н -6-сул ьфат-сул ь- фатазы в культуре
фибробластов, а тип В — дефицитом
р-галактозидазы.
Tim
наследования
- аутосомно-рецессивный.
Дифференциальный
диагноз,
другие типы мукополисахаридозов;
муколипидоз, типы I,
II
и III GMi-ганглиозидоз.
тип I; споидн- лоэпифизарные дисплазии
ЛИТЕРАТУРА
Holzgreve
W., Grobe Н . von Figura К.
el ul
Mor
quio
syndrome: clinical findings in 11 patients with
mucopolysaccharidosis IV A and 2
patients
with mucopolysaccharidosis IV B. —
Hum.
Genet.. I98I. v. 57.
p.
360
365
Nelson
J.
Broadhead
D.
Moss/nan
J
Clinical findings in 12
patients
with MPS IV A (Morquio s disease): further evidence for
heterogeneity Part 1:
Clinical
and biochemical findings. -
Clin.
Genet.
1988. v.
33.
p.
Ill
120.
Мукополнсахаридоз,
тип VI
Mucopolysaccharidosis,
type VI MIM 253200
Синоним:
синдром Марото—Ламп
Впервые
описан в 1963 г. P. Maroteaux и
М. Lamy.
Минимальные
диагностические признаки: выраженные
изменения фенотипа (сходные с изменениями
при мукополисахаридозе типа I).
нормальный интеллект, дерматансуль-
фатурия, дефицит арилсульфатазы В
Клиническая
характеристика.
Внешний вид новорожденных обычный.
Отставание в росте начинается к 2 -3
годам (рис. 106, а). Постепенно
прогрессируют помутнение роговицы.
снижение слуха, тугоподвнжность
суставов, огрубление черг лица.
Полностью симптомокомплекс развивается
к школьному возрасту (рис. 106, б, в).
Характерны низкий рост,
гепатоспленомегалня. поясничный кифоз,
вальгусное искривление голеней, грыжи.
аортальный стеноз, сердечно-сосудистая
и дыхательная недостаточность. Умственное
развитие не нарушено Больные погибают
в возрасте до 20 лет Выраженность
клинических проявлений варьирует
Определяется дефицит арилсульфатазы
В (N-аиетнлгалактоза-
мнн-4-сульфат-сульфатазы).
Тип
наследования
— аутосомно-рецессив- ный.
Дифференциальный
диагноз,
мукополнсахаридоз. типы II, IV V;
муколипидозы,
Gm,-
ганглиозидоз,
тип I
ЛИТЕРАТУРА
Barton
Я
И'..
NeufeldE.
F
A distinct biochemical deficit in the Maroteaux Lamy syndrome
(mucopolysaccharidosis). J. Pediatr., 1972,
v.
80.
p.
114
116.
Paterson
D E Rail P Harper J
?t
aI
Maroteaux Lamy syndrome, mild form MPS
vi в
Br
J. Radiol.. 1982.
v
55.
p.
805—812.
Tan
С
Т. SchaffH
У.
Miller
F el al.
Valvular heart disease in four patients with Maroteaux—Lamy
syndrome. Circulation, 1992,
v.
28,
p.
188
- 195.
Рис.
105. Мукополнсахаридоз, тип IV
Множественные
деформации скелета.