

im
Reed
U..
Dekler
R 4.
Cor
lei С.
C.
Tisli С
Congenital
lipodystrophy diabetes with acanthosis
nigricans. The Seip
Laurence syndrome. Arch.
Dermatol.. I965. v. 91.
p.
326
334.
Лиссэнцефалии
синдром
Lissencephaly
syndrome
MIM: 247200
Синонимы:
синдром Миллера -Днкера;
агирия.
Заболевание
описано в 1963 г. J.
Miller.
\1инг-.1а.и,::ыс
диагностические признаки:
микроцефалия,
характерный внешний вид.
Клинически»
характеристики.
Наиболее
важный диагностический
при так - микро-
Рис.
85.
Лпсоинсфалпя. Выступающим юбпыи
шов,
riincpiciopniM.
«кармин
рог», мпкрорстро-
гнатия. ни |ко
расположенные сформирован-
ные ушные
раковины
Фото! рафии любешо
предоставлены
проф. A.
Schinzel
hSbs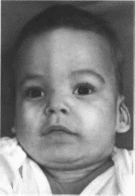

In three siblings. I. Disorders of carbohydrate meta- bolism. — j. Pcdiatr.. 1978. V. 92. P. 221 226.
цефалня
(100%). Типичен внешний вид больных:
высокий лоб. суженный в височных
областях, выступающий затылок,
ротированные назад ушные раковины
со сглаженным рнсунком, антнмонголондный
разрез глаз, гнпертелоризм. мпкрогнатня,
«карпин рот» (рис.
85, а, 6, в),
усиленное оволосение лица. Отмечаются
помутнение роговицы, полидактилия,
камптодактилия, неполная кожная
синдактилия II III стоп,
поперечная ладонная складка,
морщинистая кожа Кроме того, описаны
врожденные пороки сердца, агенезия
почек, атрезня двенадцатиперстной
кишки, крнпторхнзм. паховые грыжи.
Характерны мышечная гнпотония.
затруднение глотания, эпизоды апноэ
с цнанозом. повышение сухожильных
рефлексов, опн- стотонус и децеребрацнонная
ригидность, судорожные приступы с 1-й
недели жизни, выраженное отставание в
психомоторном развитии. Имеются
неспецнфическне изменения на
пневмоэнцефалограммах. Больные погибают
в раннем детстве. На аутопсии обнаруживают
отсутствие борозд и извилин в больших
полушариях головного мозга, не- доразвнтие
серого вещества: возможны так
же
расширение IV желудочка и гипоплазия
средних отделов мозжечка.
Тип
наследования
— аутосомно-рецессив- нын. Ген локализован
на 17р13.3.
Дифференциальный
диагноз:
синдром три- сомии 18-й хромосомы: синдром
трисомии 8-й хромосомы; церебро-гепато-ренальный
синдром.
ЛИТЕРАТУРА
Dohym
И.
В.. Curry
С.
J..
Н
уте И Е. elal.
Clinical and molecular diagnosis Miller Dieker syndrome.
Am. J. Hum. Genet.. 1991.
v.
48.
p.
584—594.
Miller
J. Q.
Lisscncephaly in 2
siblings.
Neurology. 1963.
v.
13.
p.
841
850
Лицевые
расщелины
Facial
clefts
Минимальные
диагностические признаки поперечная
или косая расщелина лица.
Клиническая
характеристика.
Поперечная расщелина лица располагается
сбоку от угла рта. может быть одно- и
двусторонней. Косая расщелина (обычно
односторонняя) встречается редко.
Различают ротоглазную и носоглазную
формы расщелины лица
(рис.
Рис.
86.
Косая расщелина лица.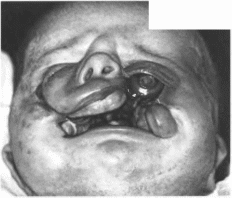
Косая
расщелина нередко сочетается с
расщелинами губы и неба, гидроцефалией,
гипертелоризмом, микрофтальмией,
мозговыми грыжами, артрогрипозом.
аномалиями пальцев.
Популяционная
частота
неизвестна.
Соотношение
полов
— МI :Ж I.
Тип
наследования
неизвестен.
Дифференциальный
диагно :
амниотичес- кие перетяжки.
ЛИТЕРАТУРА
Boo-Chai
К.
The
oblique facial clefl: A report of 2
cases
and review of 41
cases.
—
Br.
J. Plast Surg.. 1970.
v.
23.
p.
352—359.
Kubacek
V., Penkava J.
Oblique clefts of the face.— Acta Chir Plast., I974, v. 16,
p.
I52 I63
Лоуренса—Муна
синдром
Laurence—Moon
syndrome
MIM: 245800
Описан
в 1866 г. J. Laurence и R.
Moon.
Минимальные
диигностичес кие признаки:
умственная
отсталость, пигментная ретино-
патия,
гипогенитализм, спастическая пара-
плегия.
Клиническая
характеристика.
Типична
умственная отсталость,
сочетающаяся с нев-
рологическими
симптомами (спастическая
параплегия,
судороги, мозжечковые и экст-
рапирамидные
нарушения). Наблюдаются
патология
сетчатки (пигментный ретинит),
гипогонадизм
и гипогенитализм. Описаны
пороки
мозга (атрофия извилин, гидроцефа-
лия,
асимметрия полушарий, агенезия
мозо-
листого тела).
Тип
шследования
- аутосомно-рецессив-
ный.
Дифференциальный
диагноз
синдром Аль- стрема.
ЛИТЕРАТУРА
Farag
Т.
/., Teebi
S.
Bardet- Biedl and Laurence—Moon
syndromes in a mixed Arab population. —
Clin.
Genet.. 1988.
v.
33.
p.
78—82.
Лучевой
кости дефекты
Radial
defects
Минимальные
диагностические признаки. гипоплазия
I пальца кисти или I метакар- пальной
кости.
Клиническая
характеристика.
Проявления этого порока развития
крайне разнообразны. Наиболее легкая
форма — гипоплазия I метакарпальной
кости, часто с гипоплазией I пальца.
В более тяжелых случаях отсутствует I
метакарпальная кость, а гипо- плазированный
I палец прикрепляется к указательному.
Кроме того, возможна гипоплазия или
аплазия лучевой кости в сочетании с
гипоплазией I пальца и I метакарпальной
кости. В наиболее тяж( ых случаях
отсутствуют лучевая и локтевая
кости, плечевая кость нередко
гипоплазирована. Часто встречаются
аномалии позвоночника. В 20% случаев
дефект лучевой кости изолированный,
причем в 2—3%— односторонний.
Тип
наследования
неизвестен. Большинство случаев
спорадическое.
Дифференциальный
диагноз.
синдром Холт—Орама; синдром панцитопении
Фан- кони: тромбоцитопения с отсутствием
лучевой кисти: синдром радиальной
дисплазии и краниосиностоза; ассоциация
VACTERL.
ЛИТЕРАТУРА
Len:
IV.
The
Genetics of Hand Malformations. —
Birth
Defects, I980, v. XV (3),
p.
40.86). Если в процесс вовлечены края орбит, то отмечается недоразвитие века.
Маннозидоз
Mannosidosis
MIM: 248500
Синоним:
дефицит маннозидазы В. Впервые описан
в 1967 г. P. Ockerman. Минимальные
диагностические признаки грубые
черты лица, скелетные аномалии,
отставание в психомоторном развитии,
недостаточность а-маннозидазы В.
Клиническая
характеристика.
Заболевание проявляется в возрасте
нескольких лет. Лицевые аномалии
включают выступающие лоб и нижнюю
челюсть, запавшую переноси
цу,
макроглоссию, большие оттопыренные
уши
(рис. 87, а, б).
Со стороны костно-мы- шечной системы
отмечаются генерализованная умеренная
гипотония, утолщение свода черепа,
остеопороз длинных трубчатых костей,
широкие деформированные локтевая и
лучевая кости с тонким кортикальным
слоем, трапециевидная форма поясничных
позвонков, поясничный лордоз, большие
кисти и стопы
(рис. 87, в).
Наблюдаются большой живот, пупочная
грыжа, гидроцеле. Печень и селезенка
не увеличены. Сухожильные рефлексы
повышены. Частый признак — глубо-
Рис.
87. Маннозидоз. а, б грубые черты лица.
10
173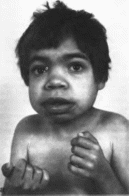
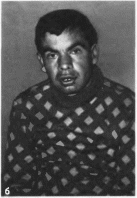
Рис.
87.
Маннозидоз (продолжение), в поясничный
лордоз, изменения кистеи и стоп.
кая
нейросенсорная глухота. В некоторых
случаях обнаружено кольцевидное
помутнение хрусталика. Лабораторные
исследования крови выявляют
вакуолизацию лимфоцитов и грубые
темные гранулы в неитрофи- лах. В клетках
печени и селезенки снижена активность
а-маннозидазы В.
Тип
наследования
— аутосомно-рецессив- ный Ген локализован
на I9pl3.2-ql2.
Дифференциальный
диагноз:
мукополиса- харидозы муколипидозы.
ЛИТЕРАТУРА
Autio
S.. Louhimo Т..
Helenins
М.
The
clinical course of mannosidosis. -
Ann.
Clin. Res., 1982,
v.
14.
p.
93—97.
Mitchell
V/.
/ ..
Erickson
R. P.. bchmid D. et al. Mannosidosis:
lwo brothers with different degrees of disease severity. -
Clin.
Genet., 1981.
v.
2,
p.
191
- 202.
Okerman
P.
A generalized storage disorders resembling Hurler's syndrome. —
Lancet.
1967,
v.
II. p. 239
241.
Мардена—Уолкера
синдром
Marden
Walker syndrome MIM: 248700
Впервые
описан в 1966 г. P. N. Marden
и W. Walker.
Минимальные
диагностические признаки: блефарофимоз,
контрактуры суставов; мышечная
гипотония.
Клиническая
характеристика.
Синдром проявляется врожденными
множественными контрактурами
суставов (95%), мышечной гипотонией
(100%), кифосколиозом (89%), арахнодактилией
(73%), косолапостью (69%), камптодактилией
(64%), микроцефалией (60%), деформацией
грудины (58%). У всех больных снижена
мышечная масса. Лицевые аномалии
включают блефарофимоз (100%),
гипертелоризм, запавшую переносицу,
поджатые губы, микрогнатию (82%), длинный
фильтр, обвисшие шеки. амимию, косоглазие
(77%)
(рис. 88).
иногда — расщелнну неба. В 87% случаев
ушные раковины деформированы и низко
расположены. Отмечаются пороки
сердца (46%) и почек (20%). С помощью КТ и
МРТ выявляют пороки развития головного
мозга (агене- зия мозолистого тела,
гипоплазия ствола мозга, червя и
полушарий мозжечка). Дети отстают в
физическом (98%) и психомоторном (100%)
развитии.
Тип
нас ледования
— аутосомно рецессивный.
Дифференциальный
диагноз:
хондродист- рофическая миотония.
ЛИТЕРАТУРА
Jaaloul
N.
Haddad
N.
£.. Khoury
L et al.
The Marden- Walker syndrome —
Am.
J. Med. Genet,
1982, v.
11,
p.
259
271.
Giacoia
G. P.. Pineda R.
Expanded spectrum of findings in Marden—Walker
syndrome. —
Am.
J Med. Genet., 1990,
v.
36,
p.
495
499.
Рис.
88.
Особенности лица при синдроме Маряена
Уолкера
Маринеску—Шегрена
синдром
Marinesco—
Sjogren syndrome MIM 248800
Описан
в 1931 г G Marinesco Минимальные
диагностические признаки мозжечковая
атаксия, мышечная гипотония, катаракта,
отставание в росте, умственная отсталость.
Клиническая
характеристика
Ведущие симптомы — задержка роста,
отставание в психическом развитии,
мышечная гипотония, атаксия, катаракта,
нистагм, косоглазие, дизартрия
Поражение головного мозга заключается
в дегенеративных изменениях коры
мозжечка.
Тип
наследования
— аутосомно-рецессив- ный.
Дифференциальный
диагноз:
лейкодистро- фии; аргининянтарная
аминоацидурия, синдром мозжечковой
атаксии, катаракты, глухоты и деменции
или психоза.
ЛИТЕРАТУРА
Alter
М
Talbert
О
R
CruffeaJ G
Cerebellar alaxia. congenital cataracts and retarded somatic and
mental maturation. Report of cases of Marinesco- -
Sjogren
syndrome. —
Neurology,
1962.
v.
12.
p
К
.16
847
Komiyama
A .
Nonuka
J.
Hiraxama
К
Muscle
pathology in Marinesco- Sjogren syndrome. —
J.
Neurol. Sci, 1989.
v.
89.
p
103—113
Марфана
синдром
Marfan
syndrome MIM. 154700
Впервые
описан в 1896 г. В J A Marfan.
Минимальные
диагностические признаки: высокий
рост, арахнодактилия, гиперподвижность
суставов, подвывих хрусталика, аневризма
аорты.
Клиническая
характерш тика.
Больные высокого роста, конечности
(особенно дис- тальные отделы) длинные
и тонкие
(рис. 89, а, в)
Часто встречаются сколиоз (60%). кифоз.
воронкообразная
(рис. 89, б)
или киле- видная грудная клетка,
долихоцефалия, узкое лицо, высокое
дугообразное небо, плоскостопие.
Поражение глаз включает двусторонний
подвывих хрусталика (75%), сочетающийся
обычно с иридодонезом (дрожание радужки),
сферофакию (шаровидная форма хрусталика)
и микрофакию (уменьшение размеров
хрусталика). Отмечаются миопия высокой
степени, отслойка сетчатки, гетеро-
хромия радужки, мегалокорнеа и голубые
склеры Характерны поражения крупных
сосудов и сердца чаще всего обнаруживают
расширение восходящей части аорты
(реже грудного или брюшного ее отдела),
пролапс митрального клапана. Нередко
имеются бедренные, паховые и диафрагмальные
грыжи, гипоплазия мышц и подкожной
клетчатки, мышечная гипотония,
нефроптоз. эмфизема легких,
пневмоторакс.
Попу
ляционная частота
— 4 : 100 000.
Тип
наследования
— аутосомно-доминант- ный с высокой
пенетрантностью и различной
экспрессивностью Обнаружены мутации
в гене фибриллина, локализованном
на 15q21.1.
Дифференциальный
диагноз:
гомоцистину- рия; арахнодактилия
контрактурная врожденная
ЛИТЕРАТУРА
Pveriti
R. Е.
McKusick
V
The
Marfan syndrome '
N Engl
J Med., 1979.
v
300,
p
772
777.
to*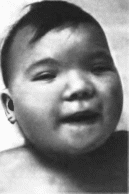
Рис.
89.
Синдром Марфана. Больная 9 лет.
а,
6
- диспропорциональное телосложение,
длинные тонкие конечносги, узкое лицо;
воронкообразная грудная клетка,
плоскостопие; в
арахнодактилня.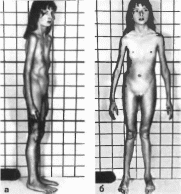
Маршалла
синдром
Marshall
syndrome MIM: 154780
Впервые
описан в 1958 г. D. Marshall.
Минимальные
диагностические признаки. миопия
высокой степени, катаракта, гипер-
телоризм, седловидный нос, снижение
слуха, задержка речевого развития.
Клиническая
характеристика.
Типично лицо больных гипертелоризм,
маленький короткий нос. запавшая
переносица, открытый рот
(рис. 90, а, б),
выступающий лоб, экзофтальм, гипоплазия
средней части лица (рис.
90, в, г). Уже
на первом году жизни может наблюдаться
миопия. В возрасте старше 10 лет
появляется катаракта, спонтанно
рассасывающаяся у взрослых.
В связи с
высокой степенью миопии может
развиться отслойка сетчатки. В раннем
возрасте обнаруживается снижение
слуха по нейросенсорно- му типу,
приводящее к задержке речевого
развития
Интеллектуальное развитие нормальное.
Рост низкий Рентгенологически выявляют
расширение костей черепа, внутричерепные
кальцификаты, гипоплазию нижней челюсти
и фронтальных синусов, аномальную форму
позвонков и костей газа, вальгусную
деформацию бедер, неправильную форму
эпифизов.
Тип
наследования
— аутосомно-доми- нантный.
Дифференциальный
диагноз:
синдром Стиклера; синдром Робинова.
ЛИТЕРАТУРА
Marshall
D.
Ectodermal dysplasia: report of kindred with ocular
abnormalities and hearing defect. —
Am
J Ophthalmol.
1958. v
45.
p.
143
- 156
Zellwegger
И
Smith
J K. Grutzner P
The Marshall syndrome: Report of a new family. —
J.
Pediatr., 1974.
v.4.
p. 868—871.
Stratlon
R. F . Lee В.
Ramirez
F
Marshall syndrome. — Am. J. Med. Genet.. 1991,
v.
41.
p.
35—38.
Рис.
90.
Особенности лица при синдроме Маршалла.
а,
б
гипертелоризм, маленький короткий нос,
запавшая переносица,
открытый
рот.
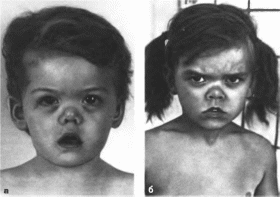
Рис.
90.
Особенности лица при синдроме Маршалла
(продолжение), в, г гипоплазия средней
части лица.
Рис.
91.
Синдром Маршалла Смита. Больной в
возрасте 4 лет. Костный возраст
соответствует 7 годам.
Маршалла—Смита
синдром
Marshall—
Smith syndrome
Описан
R Marshall с соавт в I971
г.
Минимальные
диагностические признаки ускоренные
рост и созреванне скелета, плоские
орбиты, широкие средние фаланги,
умственная отсталость.
Клиническая
характеристика
С рождения ускорены рост и созревание
скелета (рис.
91).
Черепно-лицевые признаки включают
макроцефалию, высокий выступающий
лоб, плоские орбиты, запавшую переносицу,
вздернутый нос, экзофтальм, голубые
склеры. Проксимальные и средние фаланги
пальцев рук широкие, дистальные —
узкие. Отмечаются гипертрихоз, пупочные
грыжи Наблюдаются задержка психомоторного
развития, мышечная гипотония, глубокая
умственная отсталость. Иногда встречаются
атрезия или стеноз хоан. аномалии
гортани, пороки развития головного
мозга (макроги- рия, атрофия мозгового
вешества. агенезия мозолистого тела).
В
связи с затруднением глотания дети
отстают в весе на фоне ускоренного
роста Прогноз определяется поражением
дыхательной системы (стридор.
пневмонии, ателектазы легких).
Тип
наследования
неизвестен. Все описанные случаи
спорадические.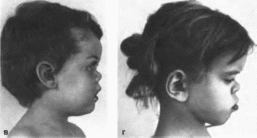
Дифференциальный
диагноз синдром
Со- тоса; синдром Вивера.
ЛИТЕРАТУРА
Ногте
И Е Bull
\1 J.
The Marshall Smith syndrome: Natural history beyond infancy. —
Clin.
Res.. 1987.
v.
35.
p.
225A
Мегалокорнеа
и умственной отсталости синдром
Megalocornea
and mental retardation syndrome MIM: 249310
Описан
в 1975 г. G. Neuhauser с соавт.
Минимальные
диагностические признаки: мегалокорнеа,
низкий рост, умственная отсталость
Клиническая
характеристика.
Рост низкий; степень умственной
отсталости варьирует от умеренной до
тяжелой. Отмечаются признаки центрального
паралича, задержка моторного развития,
мышечная гипотония и атаксия. Иногда
наблюдаются хореоатетоидные движения.
судороги, изменения на ЭЭГ. Постоянный
признак мегалокорнеа с диаметром
роговицы более 12 15 мм в сочетании с
глубокой передней камерой, гипоплазией
радужки и иридодонезом. У большинства
детей с рождения имеется микроцефалия.
Лицевые аномалии включают выступающий
лоб, телекант, эпикант и мнкрогнатию.
Описаны осложнения в виде глаукомы и
катаракты При МРТ выявляется задержка
мислинезации.
Тип
наследования —
аутосомно-рецессив- ный.
ЛИТЕРАТУРА
Frrdman
М.
Berkensiadl
М
Rous-Rothschild.
Goodman R U
Megalocornea. macrocephaly, mental and motor retardation (M
M M M)
Clin.
Genet,
1990. v
38.
p
149
154.
Neuhauser
G.
Kaveggia
E France T. Opuz J Syndrome
of mental retardation, seizures, hypotonic cerebral palsy and
megalocornea, recessi\ely inherited. —
Z.
Kinderhcilk.. 1975,
Bd.
20.
S.
1
18.
Schmidt
R. Rapin I
The syndrome of mental retardation and megalocornea
(abstracts). —
Am.
J Hum. Genet .1981
v.
33,
p
90A
Мезомелическая
дисплазия, тип Лангера
Mesomelic
dysplasia, Langer type MIM: 249700
Синоним:
мезомелическая карликовость с гипоплазией
локтевой, малоберцовой костей и
нижней челюсти.
Минимальные
диагностические признаки диспропорциональная
карликовость с выраженным укорочением
средних частей конечностей; характерная
рентгенологическая картина.
Клиническая
характеристика.
Типична диспропорциональная карликовость
за счет укорочения предплечий и голеней
Средний рост — около 130 см. Отмечается
умеренное ограничение разгибания в
локтевых суставах, выраженная
ульнарная девиация кистей, широкие
кисти, брахидактилия, поясничный
гиперлордоз, иногда — гипоплазия нижней
челюсти. Интеллект в пределах нормы.
Рентгенологически выявляют выраженную
гипоплазию дистальной части локтевой
кости; укорочение, утолщение и искривление
лучевой кости. Большеберцовая кость
короткая и широкая.
Тип
наследования
— предположительно аутосом но-рецесси
вны й.
Дифференциальный
диагноз: другие
формы мезомелической дисплазии,
днсхондро- стеоз; акродизостоз,
хондроэктодермальная дисплазия, синдром
Робинова
ЛИТЕРАТУРА
Fryns
J., ran der Berhe H.
Langer type of mesomelic dwarfism as the possible homozygous
expression of dyschondrosteosis. —
Hum.
Genet., 1979,
v.
46,
p
21
27
Kunze
J KU mm T
Mesomelic dysplasia type Langer —
a
homozygous state for dyschondrosteosis —
Eur.
J. Pediatr., 1980,
v.
134.
p.
269
272.
Мезомелическая
дисплазия, тип Нивергельта
Mesomelic
dysplasia, Nievergelt type MIM: 163400
Впервые
описана в 1944 г К Nievergelt.
Минимальные
диагностические признаки: ннзкнй
рост, укорочение голеней и предплечий,
характерная рентгенологическая картина.
Клиническая
характеристика.
Синдром проявляется непропорциональной
карликовостью с деформацией и
укорочением предплечий и голеней
Типичный признак костные выступы с
кожными ямками на боковых поверхностях
голеней, иногда — на латеральных
поверхностях предплечий. Ограничено
разгибание в локтевых суставах и
суставах пальцев Кисть отклонена в
медиальную сторону. Характерны
вальгусиая деформация локтевых
суставов и отведенное
эквиноварусное
положение стоп. Рост — 135—!47см. Интеллект
сохранен. Рентгенологическая картина
включает специфичную для синдрома
ромбовидную форму больше- берцовой и
малоберцовой костей, дислокацию и
синостоз проксимальной головки лучевой
и локтевой костей, синостоз мета-
тарзальных костей.
Тип
наследования
— аутосомно-доми- нантный с различной
экспрессивностью.
Дифференциальный
диагноз:
мезомелическая дисплазия, тип
Лангера; мезомелическая дисплазия, тип
Реинхардта Пфейффера; дисхондростеоз;
синдром Робинова: акроме- зомелическая
дисплазия; акродизостоз; хон-
дроэктодермальная дисплазия.
ЛИТЕРАТУРА
Hess
О.
Н.. Goebel
N.
Н.. Sirenli
R.
Familiaer mesomeler Kleinwuchs (Nievergelt Syndrom). Schweiz. Med.
Wochenschr, 1978,
Bd.
108,
S.
1202
- 1206.
Мезомелическая
дисплазия, тип Рейнхардта—Пфейффера
Mesomelic
dysplasia, Reinhardt PfeifTer type MIM: I91400
Синоним:
гипоплазия локтевой и малоберцовой
костей.
Минимальные
диагностические признаки: умеренное
снижение роста, мезомелическое укорочение
конечностей, искривление голеней,
кожные ямки на искривленной поверхности.
Клиническая
характеристика.
Рост умеренно снижен, телосложение
диспропорциональное. Предплечья
укорочены, искривлены в латеральную
сторону, ограничены пронация и
супинация в локтевых суставах. Отмечается
умеренная ульнарная девиация кистей.
Голени непропорционально короткие
и искривленные, дистальные их части
утолщены, на искривленных поверхностях
имеются пигментированные кожные ямки.
Стопы плоские и н редко отведены в
стороны. Рост — 150—169 см. Интеллект
сохранен. Рентгенологически выявляется
укорочение дистальнои части локтевой
кости; лучевая кость искривлена, ее
дистальная часть находит на локтевую;
лучезапястный сустав деформирован.
Иногда имеется вывих или подвывих
проксимальной головки лучевой кости
и дисплазия дистальнои суставной
поверхности плечевой кости. Про
ксимальная
часть малоберцовой кости и большеберцовая
кость укорочены и искривлены.
Соотношение
полов — МI
:Ж1.
Тип
наследования
— предположительно аутосомно-доминантный
с различной экспрессивностью.
Дифференциальный
диагноз: другие
формы мезомелической дисплазии;
дисхондростеоз; синдром Робинова;
акромезомеличес- кая дисплазия;
акродизостоз; хондоэктодер- мальная
дисплазия.
ЛИТЕРАТУРА
Reinhardi
К..
Pfeiffer
R.
Ulno-fibulare dysplasie. Eine autosomal-dominant vererbie
Mykromcsomclic aehnlich dem Nievergeli syndrom. Fortschr. Roen-
tgenstr 1967,
Bd.
107.
S.
379
- 391.
Меккеля
синдром
Meckel
syndrome MIM: 249000
Синонимы:
синдром Грубера; синдром Меккеля
Грубера.
Описан
в 1822 г. J. Meckel.
Минимальные
диагностические признаки: черепно-мозговая
грыжа; полидактилия; по- ликистоз почек.
Клиническая
характеристика.
Наиболее важный диагностический признак
— затылочная черепно-мозговая грыжа
(80%). Кроме того, отмечаются микроцефалия
(32%), гидроцефалия (25%), гипо- или аплазия
мозолистого тела (13%), аринэнцефалия
(17%), гипо- или аплазия мозжечка (28%), в
единичных случаях — анэнцефалия,
расширение желудочков мозга.
Встречаются также скошенный лоб, низко
расположенные деформированные ушные
раковины, микрогения, пшертелоризм,
реже — гипотелоризм, капиллярные
гемангиомы на лбу, расщелина губы или
неба (38%), зубы новорожденного, липоматозные
образования на боковых поверхностях
языка. В 25% случаев имеются пороки
развития глаз: микрофтальмия (19,2%), реже
— анофтальмия, колобомы. катаракта.
Поликистоз почек наблюдается у 75%
больных. Обычно обнаруживают гигантские
поликистозные почки, приводящие к
резкому увеличению живота
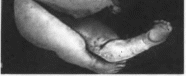
(рис. 92). иногда
— уменьшенные почки с большим количеством
мелких кист. Описаны единичные случаи
других пороков мочевой системы (атрезия
мочеточников, гипо- или аплазия
Рис.
92.
Синдром Меккеля. Живот увеличен
вследствие поликистоза почек; полисин-
дактилия.
мочевого
пузыря). Характерны критттор- хизм,
микропения, двурогая матка, атрезия
влагашшха, перегородка влагалища,
двойственное строение наружных
половых органов. Отмечаются пороки
сердца (20,8%), кис- тозные и кистофиброзные
изменения печени (30,3%), атрезия ануса
или сигмовидной кишки (в единичных
случаях). Типичный признак — полидактилия
(66%), обычно постаксиальная (чаще
поражаются кисти). В 15% случав полидактилия
сочетается с син- дактилиеи (обычно II-
III стоп). Могут наблюдаться
камптодактилия, клинодакти- лия V,
косолапость и поперечная ладонная
складка. Больные рождаются с умеренной
пренатальной
гипоплазией, погибают в перинатальном
периоде или на первом году жизни.
Пот'ляционпая
частота — от
I : 70 ООО до I : 90 ООО.
Тип
наследования—аутосомно-рецесснвный.
Дифференциальный
диагноз:
синдром три- сомии хромосомы 13, синдром
Смита —Лем- ли Опица.
ЛИТЕРАТУРА
Fraser
F. С..
Lylwyn
A.
Spectrum of anomalies in the Meckel syndrome or: «May be there is a
malformation syndrome with at leasi one constant anomaly».
-
Am.
J. Med. Genet., 1981,
v.
9.
p.
67—73.
Mecke
S.. Passargt E.
Encephalocele, polycystic
kidneys
and Polydactyly
as
an autosomal recessive trait simulating certain
other
disorders.
The Meckel syndrom.
—
Ann.
Genet., 1971.
v.
14,
p.
97—103.
Farag
T. J.. Usha R . Uma R ei al.
Phenotypic variability in Meckel- Gruber syndrome. —
Clin.
Genet.. 1990,
v.
38.
p.
176-
179.
Мелкерсона—Розенталя
синдром
Melkersson
Rosenthal syndrome MIM: 155900
Впервые
описан в 1859 г. Е. Mart.
Минимальные
диагностические признаки: паралич
лицевого нерва, отек лица, складчатый
язык.
Клиническая
характеристика.
Патогно- моничный симптом — отек губ
и щек, который может распространяться
на веки, нос и подбородок. Поражение
бывает одно- и двусторонним. Другой
характерный признак — паралич лицевого
нерва, частичный или полный, иногда
двусторонний, с тенденцией к восстановлению.
Описаны также отеки кистей, грудной
клетки, ягодиц, акропаре- стезии, головные
боли, гипергидроз, блефа- роспазм,
отечность и складчатость слизистой
оболочки рта, снижение слюноотделения,
складчатый язык с атрофией сосочков.
Тип
наследования
— аутосомно-доми- нантный с различной
экспрессивностью.
ЛИТЕРАТУРА
LvgidakisC..
Tsankanikas С..
Ilias.
Vassilopoulos D. Melkersson
—
Rosenthal's
syndrome in four generations -
Clin.
Genet., 1979,
v.
15,
p.
189
192.
Менингоцеле
Meningocele
MIM: 176450
Минимальные
диагностические признаки: черепно-
и спинномозговые грыжи.
Этническая
.характеристика.
При менингоцеле грыжевой мешок
представлен твердой мозговой
оболочкой и кожей, а содержимое —
спинномозговой жидкостью. Грыжи
локализуются чаще всего в поясничном,
шейном отделах позвоночника (spina
bifida) и в местах соединений черепных
костей. На коже, покрывающей грыжевые
выпячивания, могут наблюдаться гемангио-
мы и рост волос. Паралич и нарушения
чувствительности не характерны.
Иногда ме
нингоцеле
сочетается с гидроцефалией. Этиология
порока мультифакториальная.
Попу
гяционная частота
дефектов нев- ральнон трубки — 1 : 1000.
Соотношение
полов
— М1 :Ж1.
Дифференциальный
диагноз:
мнеломенин- гоцеле: энцефалоцеле.
ЛИТЕРАТУРА
Kotzmannova
J.
The empiric risks of recurrence of neural tube defects. Plzen. Lek.
Sb.. 1973,
Suppl.
31,
p.
357
360.
Менкеса
синдром
Menkes
syndrome MIM: 309400
Синоним:
синдром скрученных волос.
Описан
в 1962 г. J. Menkes.
Минимальные
диагностические признаки: выраженная
задержка психомоторного развития.
судороги, скрученные и ломкие волосы.
Клиническая
характеристика.
Характерны задержка психомоторного
развития, дегенеративные изменения
ЦНС, дефект транспорта меди и изменения
структуры волос. Волосы редкие,
ломкие, скрученные, часто светлые
(рис. 93, а).
Неврологические изменения включают
судороги, внутричерепные кровоизлияния,
мышечный гипертонус. При исследовании
головного мозга выявляют дегенеративные
процессы в коре с глиозом и атрофией.
При микроскопическом исследовании
волос обнаруживают pili torti,
trichorexis nodosa, moniletrix
(рис.
93, б). В
сыворотке снижен уровень меди.
Рентгенологически определяются
вормиевы кости. расширение метафизов
с формированием шпорообразных боковых
выростов. Большинство больных
погибает в возрасте от 6 мес до 3 лет.
При большей продолжительности жизни
развиваются дистрофия и тет- раплегия.
Потляционная
частонш — 1 :
298 000 новорожденных в странах Западной
Европы.
Соотношение
полов
— МI :Ж0.
Тип
наследования
— Х-сцепленый рецессивный. Ген
локализован на Xq 13.
Дифференциальный
диагноз:
синдром скрученных волос и глухоты.
ЛИТЕРАТУРА
Dankv
D. М . Stevens В. J..
Campbell P. F. et al. Menkes
kinky-hair syndrome. -
Lancet.
1972.
v.
I, p.
1100—1102.
б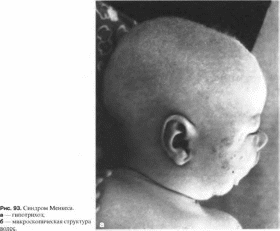
Метатропная
дисплазия
Metatropic
dysplasia MIM: 250600
Синоним:
метатропная карликовость. Описана в
1966 г. P. Maroteaux с соавт.
Минимальные
диагностические признаки: уплощение
тел позвонков, короткие ребра,
прогрессирующий кифосколиоз, гиперплазия
вертельной области бедренной кости и
метафизов трубчатых костей, деформация
зпифи юв и метафизов, дисплазия основания
черепа.
Клиническая
характеристика
У новорожденных длинная узкая грудная
клетка и относительно короткие
конечности. Быстро прогрессирует
кифосколиоз, приводящий со временем к
укорочению туловища. Суставы увеличены,
движения в них ограничены. Рентгенологически
выявляется спонднлоэшадетафн'зарная
дисплазия скелета: платиспондилия
(в грудном отделе позвонки имеют вид
узких «языков»), укорочение ребер
укорочение тела подвздошной кости с
<перетяжкои» на границе между телом
и крылом, укорочение и расширение шейки
бедра, гиперплазия вертельной области,
укорочение шинных трубчатых костей
с симметричным увеличением метафизов.
уплощение и сглаженность контуров
эпифизов в области коленных суставов.
Дисталыгьге эпифизы костей предплечья,
пястных костей и фаланг уменьшены;
запарывает появление ядер окостенения
костей запястья.
Тип
iuu
чедовсишя
аутосомно-рецесснвнын. Дифференциальный
диагноз:
ахондропла- wm.
mj
стслжслхарилот. тип IV-, асфмтси-
ческая дистрофия i рудной
клет ки новорожденных; болезнь
Книста.
ЛИТЕРАТУРА
Beck
М
. Rouhicck
М
. Rogers
J G. eta!
Heterogeneity of metairopic tij plasia. Eur. J. Pediatr.. 1983,
v.
140,
p.
231
237.
Jenkins
P.
Smith
M. В..
Л/<
KinnelJ.
S
Metatropic dwarfism. Br. J Radiol.. I970. v. 43.
p.
561
565 Maroteaux
P.. Sprunger J. И
.. Wiedemann
H.-R. Der
meiairopische Zwergwuchs. ■
Arch
Kinderhe- ilk.. 1966.
Bd.
173.S. 2ll 226.
Метахондроматоз
Metachondromatosis
MIM: 156250
Минимальные
диагностические признаки экзостозы
и энхондроматоз.
Клиническая
характеристика.
Рост низ-
Рис.
94.
Миеломепингоцеле пояспичио- крестцового
отдела по
1Воночника.
кии.
На костях кисти и длинных трубчатых
■копач
шмйчй'ктт'с?! т/клюстг) уы. осл йохондро-
матозный
процесс поражает зоны роста. Со временем
возможно обратное развитие патолог
нческих изменений в костях. Может
наблюдаться ограничение функции
пальцев, связанное с экзостозами. Рент1
енологически выявляют признаки экзостоза
и энхондро- матоза. В патологической
процесс
moi
ут вовлекаться тела по тонкое.
Соотношение
полов — М1
:Ж1. Тип
наследования -
предположительно аутосомно-доминантный.
Дифференциальный
диагноз,
множест венные хрящевые зкзостозы;
энхондроматоз.
ЛИТЕРАТУРА
Bassett
G. S.. СонеП
Н R.
Melachondromalosis: report of four cases. —
J.
Bone Jomi Surg.. 1985.
v.67A.
p. 811—814.
Kennedi
L.
Metachondromatosis. Radiology. 1983.
v.
148.
p.
117—118.