120
Комплемента
компонента 5 дефицит
лироваппые
колобомы радужки обычно имеют другое
происхождение Колобомы глаза сочетаются
с микрофтальмией, зрачковой мембраной,
дисплазией макулы, миопией высокой
степени, помутнением хрусталика,
атрофией зрительного нерва, задним
лентиконусом.
Тип
наследования
аутосомно-доми- нантный.
Дифференциальный
диагноз:
аниридия
ЛИТЕРАТУРА
Philips
С.
I.
Hereditary macular coloboma. —
J.
Med Genet., 1970,
v.
7.
p.
224
226.
Sa\i
II J.. Cook J. R.
Optic nerve colobomas of autosomal dominant heredity Arch.
Ophthalmol.. 1976.
v.
94.
p.
395
400.
Комплемента
компонента 5 дефицит
Complement
component 5 deficiency of MIM: 120900
Синоним:
дефицит C5.
Минимальные
диагностические признаки нарушение
фагоцитоза вследствие дефицита С5,
себорейный дерматит, рецидивирующие
инфекции.
Клиническая
характеристика.
Заболевание проявляется вскоре
после рождения повторными инфекциями.
Характерны хронические кишечные
инфекции, резистентные к антибиотнкотерапин
и диете, и приводящие к кахексии,
генерализованный себорейный дерматит.
Лабораторные исследования обнаруживают
нейтрофилез, гинерглобулине- мию,
повышение СОЭ.
Бактериологически
выявляется грамот- рицательная флора,
из фамположительных бактерий — только
золотистыи стафилококк. В основе
нарушения фагоцитоза лежит недостаточность
пятого компонента комплемента.
Тип
наследования
— аутосомно-доми- нантный
Дифференциальный
диагноз: другие
первичные иммунодефицитные состояния.
ЛИТЕРАТУРА
Snrdernum
R.. Durack D. Т..
МсСапу С. A.
ei al. Deficiency
of the fifth component of complement in human subjects: clinical,
genetic and immunologic studies in a large kindred Am. J. Med.,
1979,
v.
67,
p.
638
645.
Контрактура
Дюпюитрена
Dupuytren
contracture MIM: 126900
Синоним:
ладонный фиброматоз.
Минимальные
диагностические признаки флексорные
деформации пальцев.
Клиническая
характеристика
Наблюдаются подкожные узелки на
ладонях и контрактуры проксимальных
межфаланговых и мета- карпофалангеальных
суставов. Контрактуры приводят к
нарушению функции кисти и в 90" о
случаев бывают двусторонними.
Тип
наследования
- аутосомно-доминант- пый с неполной
ненет рантностью.
ЛИТЕРАТУРА
YongJ.
D.. ForiI R. U .
Familial fibromatosis. Clin. Genet.. 1981,
v.
20,
p.
211—216.
Корнелии
де Ланге синдром
Cornelia
de Lange syndrome MIM: 122470
Впервые
описан в 1933 С. de Lange.
Минимальные
диагностические признаки задержка
психомоторного развития, микроцефалия.
синофриз, длинный фильтр, вывернутые
наружу ноздри, гонкая, загнутая внутрь
верхняя i уба, микромелия,
гипертрихоз, отст аваннс в росте.
Клиническая
характеристика.
Наблюдаются микробрахицефалия
(93%). деформированные ушные раковины
(58%). синофриз (99%), тонкие брови, длинные
загнутые ресницы (100%)
(рис. 70),
маленький нос с открытыми вперед
ноздрями (100%), атрезия хоан, длинный
выступающий фильтр (94%), микрогення
(97%), тонкая верхняя губа (94%), рот в виде
полумесяца (94%), высокое арковидное небо
(86%), позднее прорезывание зубов,
большие промежутки между зубами
(86%), иногда — расщелина неба. Описаны
аномалии глаз миопия, микрокорнеа,
астигматизм, атрофия или колобома
зрительного нерва, косоглазие. К
характерным порокам конечностей
относятся акромикрия (93%), офаничение
подвижности локтевых суставов (64%),
фокомелия и олигодактилия (30%),
клинодактилия V пальца
(697ч); реже
встречаются гипоплазия лучевои кости,
короткие I метакарпальные кости.
Наблюдаются гипертрихоз (78%), мраморная
кожа (60%), гипоплазия сосков (55%). У всех
больных отмечаются отставание в
росте, глубо-1'Ифферс тршлылый диагноз: камптодактилня.
Корнелии
де Лаше синдром
Рис.
70.
Синдром Корнелии де Ланге. Длинные
загнутые ресницы, синофриз, маленький
нос с открытыми впред ноздрями, длинный
фильтр: аномалии конечностей.
кая
задержка умственного развития, мышечный
гипертонус, у 74% слабый высокий голос,
у 20% судороги. Описаны пороки внутренних
органов: врожденные пороки сердца
(17%). в отдельных случаях — поли- кистоз
почек, гидронефроз, удвоение или
неполный поворот кишечника,
нилоростеноз, паховые и диафрагмальные
грыжи, крип- торхизм (73%). гипоплазия
гениталий (у мальчиков— 57%). Типичны
рецидивирующие респираторные инфекции.
В рамках фенотн- пической вариабельности
выделяют два варианта синдрома:
первый (классический) характеризуется
выраженной пренатальной гипоплазией,
значительной задержкой физического
и умственного развития и грубыми
пороками развития. Второй (доброкачественный)
сопровождается аналогичными ли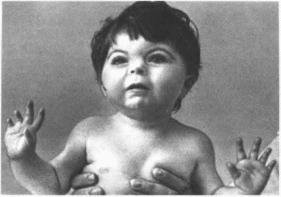
цевыми
и малыми скелетными аномалиями
пограничной задержкой психомоторного
развития: крупные пороки развития
отсутствуют.
Пот
ляционная частота
—I : 12 ООО.
Соотношение
полов
— Ml
:
Ж1.
Тип
наследования
неизвестен. Большинство случаев
спорадические. У некоторых больных
обнаружены мнкроструктурные хромосомные
перестройки, вовлекающие участок
3q26.3.
Дифференциальный
диагноз:
синдром Коф- фина Сириса.
ЛИТЕРАТУРА
Beik
В
, Mikkelson
М
Chromosomes
in the Cornelia de Lange syndrome. —
Hum.
Genet., 1981,
v.
59,
p.
271—276.
I
an Allen \f. J., Filippi G.. Siegel-Barieli J el ai Clinical
variability within Brachmann -de Lange syndrome a proposed
classification system Am J Med. Genet.. 1993,
v.
47,
p.
947
958
Костелло
синдром
Costello
syndrome MIM: 218040
Минимальные
диагностические признаки: низкий
рост, избыточная кожа на шее. кистях
и стопах, курчавые волосы, характерное
лицо, умственная отсталость.
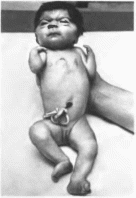
Клиническая
характеристика.
Типичные черенно-лицевые аномалии —
макроцефалия, грубое лицо, низко
расположенные ротированные назад
ушные раковины, эпи- кант. запавшая
переносица, толстые губы (рис.
71, а). Отмечаются
короткая шея. ot- раниченне
движения в локтевых суставах, широкие
днетальные фаланги, избыточная кожа
(рис. 71, б, в, г, д),
глубокие кожные складки на ладонях и
подошвах, повышенная пигментация
кожи, курчавые, редкие волосы.
Наблюдаются постнатальная задержка
роста, умственная отсталость, врожденные
пороки сердца. Описан папилломатоэ в
области рта и носа.
Тип
наследования
предположительно аутосомно-рецессивный.
Дифференциальный
диагноз:
ленречаунизм.
ЛИТЕРАТУРА
Cornelia
J.
V/
A
new syndrome mental subnorma- lity and nasal papillomata. J. Aust.
Pediatr.. I977, v. I3.p.l 14
l
IS.
KalouMian
V Vi
Not a new MCA/MR syndrome but probably Costello syndrome. Am. J Med
Genet.. 1993.
v.
47.
p.
170
I7I

124
Коффипа
-Лоури синдром |
iг |
|
|
|
|
|
|
|
■jYjw |
'Я |
|
|
|
1 Я) |
|
|
Ж |
Ш я |
|
|
|
|
\ |
|
|
|
/1 |
он |
|
|
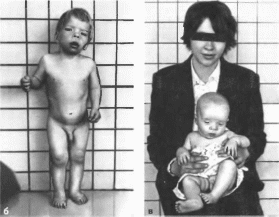
Коффина—Лоури
синдром
Coffin—
Lowry syndrome MIM. 303600
Описан
в 1966 г. G. Coffin с соавт.
Минимальные
диагностические признаки: антимонголоидный
разрез глаз, луковицеобразный нос,
низкий рост, конусовидные пальцы,
умственная отсталость.
Клинтес
кия характеристика.
Наблюдаются специфические черты
лица: квадратный лоб. выступающие
надбровные дуги, антимонголоидный
разрез глаз, гипертело- ризм, нериорбитальная
полнота тканей, широкая спинка носа,
открытые вперед поздри. полные выступающие
губы, открытый рот. массивный подбородок,
оттопыренные уши (рис.
72, а,
б, в). Кисти
большие, мягкие.
Рис.
72.
Синдром Коффина Лоури.
а,
б
— антимонголоидный разрез глаз, широкая
спннка
носа, открытые вперед ноздри.
открытый
рот. полные губы:
в
мать и сын с синдромом Коффина Лоури.
Коффина—С
и] иса снндром
125
гиперподвижные,
толстые, пальцы рук конусовидные.
Отмечаются короткая растепленная
грудина, килевидная грудная клетка,
то- раколюмбальпый сколиоз, плоскостопие,
низкий рост. Рентгенологически выявляют
утолщение костей лицевого черепа, кнфо-
сколиоз. узкие межпозвоночные промежутки,
короткие дистальные фаланги, укорочение
длинных трубчатых костей нижних
конечностей. coxa valga Больные
умсгвенно отсталые, коэффициент
интеллекта ниже 50 Тип
наследования
— Х-сцепленный домн- нант ный с выраженными
проявлениями у мужчин и более стертыми
у женщин Ген локализован на Хр22.2-р22
1.
Дифференциальный
диагноз:
синдром Коф- фнна Сириса.
ЛИТЕРАТУРА
Демина
Н А , Блинникова О. Е.. Додали Е. Л. Козлова
В. М.
Диагностика синдрома Коффина Лоури.
Теоретические и прикладные проблемы
мед. генетнки, М.. 1993. стр. 119 124.
Mallei
J. F.
Coffin Lowry syndrome in sibs. —
Am.
J. Med. Genel.. 1981.
v.
8.
p.
315
319.
Gilgenkraiitz
S.
Mujica
P. Gruel P elol
Co (Tin Lowry syndrome: a mullicenter study Clin. Genet.. 1988,
v.
34,
p
230
245.
Young
J D
The Coffin Lowry syndrome -
J.
Med Genet.. 1988.
v.
25.
p.
244
248.
Wilson
W
G.. A///i
Т.
E.
Early recognition of Ihe Coffin Lowry syndrome. —
Am.
J. Med. Genel., 1981,
v.
8,
p.
215-220.
Коффина—Сириса
синдром
Coffin
—Siris syndrome M1M 135900
Синоним:
синдром пятых пальцев Описан в 1970 г. G.
Coffin и Е. Siris. Минимальные
диагностические признаки: грубые
черты лица, гипоплазия или аплазия
пальца
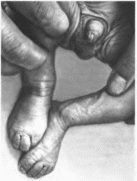
и попей на пальцах стоп. Клиническая
характеристика.
Отмечаются внутриутробное отставание
в росте, умеренная или тяжелая
мышечная i ипотония.
незначительная микроцефалия, грубые
черты лица, густые брови, полные
губы
(рис. 73).
умственная отсталость, обычно глубокая.
Характерны гипоплазия или отсутствие
пальца
и ногтей на пальцах стоп (ногти на
пальцах кистей гипоплазированы в
меньшей степени), разболтанность
суставов с подвывихом лучевой кости
в локтевом суставе. Редкие волосы на
голове сочетаются с генерализованным
гирсутизмом Возможны ги-
потелоризм,
птоз, преаурикулярные выросты,
гемапгиомы, крнпторхнзм. сколиоз,
короткая грудина, пороки сердца,
короткое предплечье, аномалии позвонков,
расщелина неба, гидроцефалия.
Рентгеноло1
ическн выявляются coxa valga,
гнпопла зия ключиц и надколенника,
отставание костного возраста.
Тип
наследования
неизвестен. Все описанные случаи
спорадические.
Дифференциальный
диагноз:
снндром Кор- нелни де Лаиге; частичная
чрнсомия 9р; фе- тальный гидантоиновый
синдром.
ЛИТЕРАТУРА
Coffin
G
5.
Siris
Е.
Menial
retardation with absent fingernail and terminal phalanx. Am. J.
Dis Child.. 1970.
v.
119,
p.
433
439
Lew
P.
Barailser
M
Coffin Siris syndrome. J Med Genet.. 1991.
v.
28.
p
338
341
Qazi
Q H . Heckman F . Enions D. el al.
The Coffin Siris syndrome. J. Med. Genel.. 1990.
v.
27.
p
333
336
Рис.
73.
Синдром Коффина Сириса.

126
«Кошачьего
глаза» синдром
«Кошачьего
глаза» синдром
Cat
eye syndrome MIM: 115470
Синонимы
колобома глаза и атрезия ануса;
синдром Шмида Фраккаро.
Минимальные
диагностические признаки: колобома
радужки, атрезия ануса, преаури- кулярные
кожные выросты.
Клиническая
характеристика
Колобома радужки обычно двусторонняя
(рис. 74).
колобома сосудистой оболочки иногда
сочетается с микрофтальмией Характерно
лицо: запавшая переносица, гипертелоризм,
эпи- кант
(рис. 74),
антимонголоидный разрез глаз и
микрогнатия. Деформации ушных раковин
часто сочетаются с преаурикулярны- ми
кожными выростами, ямочками или
фистулами. Атрезия ануса и прямой
кишки обычно сочетается с ректовагинальной
или ректоперитонеальной фистулой.
Рис.
74. Г
ипертелоризм, эпикант, двусторонняя
колобома радужки при снндроме «кошачьего
глаза». Фотография любезно предоставлена
проф F.Losan
Отмечаются
аиомалии почек и мочевых путей агенезня.
гидронефроз, подковообразная почка,
пузырно-мочеточниковый реф- люкс.
Описаны аномалии ребер, полупозвон- кн
и вывих бедра. Сердечно-сосудистые
аномалии включают дефекты перегородки
и легочных вен, тетраду Фалло. У 2/3
больных отмечается умственная
отсталость
Тип
наследования
неизвестен Встречается спорадически.
В некоторых случаях обнаружены
микроструктурные перестройки 22
хромосомы, маленькая дополнительная
ак- роцентрическая хромосома
Дифференциальный
диагноз:
синдром Рн- гера.
ЛИТЕРАТУРА
Gerald
Р
S
David С
. Sa\
В
Wdkmg
J
Syndro- mal association of imperforate anus the cat eye syndrome
Birth Defects. 1972, v
VI 11(2).
p. 79 84 Sihachenmunn
G.
Schmid
И
Fracaro
M el at Chromosomes
in coloboma and anal atresia Lancet. 1965. v.
11 p 290.
Wilson
G N..
Baker
D L . SchauJ.
Parker
J
Cat eye syndrome owing to teirasomy 22 pler
ql I J. Med Genet. 1984, v. 21, p.
60—63.
Коэна
синдром
Cohen
syndrome MIM: 216550
Описан
в 1973 г. M. Cohen.
Минимальные
диагностические признаки: мышечная
гипотония, ожирение, выступающие
центральные резцы.
Клиниче<коя
характеристика
Основные признаки синдрома — низкие
масса и длина тела при рождении (61%),
мышечная гипотония (90%). задержка
психомоторного развития с рождения
(96%), умственная отсталость,
незначительная микроцефалия (61%),
ожирение Характерно лицо внтнмонголо-
идный разрез глаз (52%). высокая спинка
носа (94%), короткий фильтр (90%), открытый
рот (84%), выступающие резцы (65%), гипоплазия
верхней челюсти (81%), микрогения (97%).
Аномалии глаз включают миопию, страбизм,
микрофтальмию, колобомы радужки.
Кисти и стопы узкие (90%), с укороченными
метакарпальными и метатарзаль- ными
костями, пальцы длинные, тонкие, те- нар
и гипотенар гипоплазированы Отмечаются
гиперподвижность суставов (58%), поясничный
гиперлордоз, сколиоз или кифоз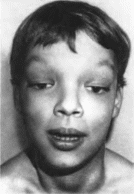
Кранио-карпо-тарзальная
дисплазия
127
(42%).
вальгусная деформация коленных (35%) и
локтевых (58%) суставов, кожная синдактилия
(35%), плосковальгусная позиция стоп,
сандалевидная щель. Характерны высокии
тембр голоса, добродушный характер,
лейкопения.
Тип
наследования
— аутосомно-рецессив- ный.
Дифференциальный
диагноз:
синдром Барде- Бидля.
ЛИТЕРАТУРА
Konsseff
В.
С. Cohen
syndrome: further delineation and inheritance. —
Am.
J. Med. Genet 1981,
v.
9.
p.
25
30.
NorioR.,
RailaCh.. LindahlE
Further delineation of the Cohen syndrome: report on chorioretinal
dystrophy, leukopenia and consanguinity. —
Clin.
Gen t„ 1984.
v.
25,
p.
I 14.
Young
J. D„ Moore J. R.
lntrafamilial variation on Cohen syndrome. —
J.
Med. Genet., 1987,
v.
24,
p.
488
492.
Кранио-карпо-тарзальная
дисплазия
Craniocarpotarsal
dysplasia M1M: 193700
Синонимы:
синдром Фримена Шелдона; синдром
«свистящего лица».
Заболевание
описано в 1938 г. Е. Freeman и
J. Sheldon.
Минимальные
диагностические признаки: микростомия,
гипоплазия крыльев носа, ульнарная
девиация пальцев кисти, множественные
контрактуры суставов.
Клиническая
характеристика.
Основные признаки синдрома —дефекты
строения лица и скелетно-мышечной
системы. Аномалии лица: гипертелоризм,
энофтальм, эпи- кант, сходящееся
косоглазие и блефарофи- моз маленький
нос с узкими носовыми ходами и
гипоплазированными крыльями, длинный
фильтр, маленькая верхняя губа, две
Рис.
75.
Кранно-карпо-тарзальная дисплазия. а
больной I года:
б
больной 20 лет (выраженный кифосколиоз.
деформация стоп);.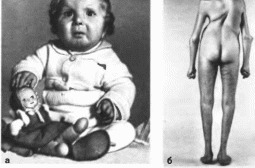
128
Краниоснностоз
Рис.
75.
Кранио-карпо-тарзальная дисплазия
(продолжение), в
ульнарная девиация кистей больного,
изображенного на рис. 75. б.
вертикальные
бороздки на подбородке, микрогения.
Перечисленные изменения придают
больному характерный вид «свистящего
человека»
(рис. 75, а).
Небо высокое, язык маленький. Аномалии
скелета: сколиоз, вывихи тазобедренных
суставов, множественные сгибательные
контрактуры крупных суставов,
камптодактилия, высокий свод стопы,
косолапость
(рис. 75, б).
Характерные рентгенологические
признаки — вертикальная ориентация
таранной кости и смещение
II плюсневои
кости кзади. Постоянный симптом —
ульнарная девиация пальцев
(рис. 75, в) —
связан не с костными нарушениями, а
с поражением локтевого нерва или
соответствующих мотонейронов. Пороки
развития внутренних органов не
характерны. Умственное развитие
обычно нормальное, рост немного ниже
нормы. Продолжительность жизни не
изменена.
Тип
наследования
— аутосомно-доминант-
ный.
Предполагается существование ауто-
сомно-рецессивных форм.
ЛИТЕРАТУРА
Ahes
A. F. P., Azeredo Е.
S.
Recessive form of Freeman- Sheldon syndrome. J. Med. Genet.. I977,v.
14.
p.
139
141
Wettstein
A.. Buchiuger G.. Braun A., Bazau U. B. A
family with whistling face-syndrome Hum Genet.. 1980,
v.
55,
p.
171
189.
Краниосиностоз
Craniosynostosis
M1M:
123100,218500
Минимальные
диагностические признаки: аномальная
форма черепа.
Клиническая
характеристика.
Краниосиностоз — это преждевременное
зарастание черепных швов, приводящее
к асимметрии (плагиоцефалия) и
деформациям черепа (рис.
76). Чаше всего
встречается скафоце-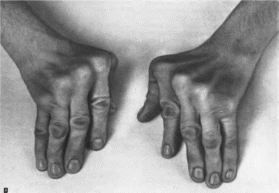
Рис.
76.
Краниосиностоз.
Аномальная
форма черепа с выступающими
метатропным
и коронарным швами.
фалия
(длинный узкий череп с выступающим лбом
и затылком) вследствие преждевременного
зарастания стреловидного шва. Турри-
цефалия (башенный череп) возникает
из-за преждевременного зарастания
венечного и стреловидного швов,
брахицефалия в результате зарастания
венечного шва.
Тип
наследования
— аутосомно-доми- нантный. Возможно
существование ауто- сомно-рецессивных
форм. Ген аутосомно- доминантной формы
локализован предположительно на
7р21.3-р21.2.
Дифференциальный
диагноз:
черепно-лице- вой дизостоз;
акроцефалосиндактилии.
ЛИТЕРАТУРА
Ншиег
А.
С И ..
Rudd
N
L.
Craniosynostosis. I.
Sagittal
synostosis, its genetics and associated clinical findings in 214
patients
who lacked involvement of the coronal suture(s). —
Teratology,
1976,
v.
14,
p.
185
- 193.
Shillito
J.. Malson D.
Craniosynostosis: a review of 519
surgical
patients. —
Pediatrics,
1968,
v.
41,
p.
829
853.
Криглера—Найяра
синдром
Crigler-
Najjar syndrome MIM: 218800
Синонимы:
дефицит печеночной глюкуро- нилтрансферазы,
тип I; негемолитическая гипербилирубинемия.
Минимальные
диагностические признаки персистенция
желтухи новорожденных в отсутствие
гемолиза; концентрация билирубина
в крови более 340 мкмоль/л; отсутствие
билирубиндиглюкуронида в желчи.
Клиническая
характеристика.
Желтуха появляется с рождения и
интенсивно нарастает. В крови повышен
уровень непрямого билирубина. Печень
и селезенка не увеличены. Развиваются
симптомы ядерной желтухи: судороги,
гиперрефлексия, мышечный гипертонус,
симптом «заходящего солнца». (Ядерная
желтуха может также провоцироваться
недоношенностью и незрелостью,
инфекциями, гипоксией.) В 75% случаев
ядерная желтуха приводят к смерти
в течение первых 5 лет жизни. Физическое
и психомоторное развитие значительно
отстает. Отсутствует активность
глюкуронилтрансфе- разы печени. Выделение
уробилиногена с калом низкое (40—50% от
нормы). Анемия не характерна. Количество
уробилина в моче в пределах нормы,
в желчи отсутствует прямой билирубин;
функциональные пробы печени патологии
не выявляют Морфологических изменений
в печени не отмечается.
Тип
наследования
— аутосомно-рецессив- ный. Ген локализован
на Iq21-q23.
Дифференциальный
диагноз: другие
формы гипербилирубинемии.
ЛИТЕРАТУРА
Blumenschein
A.. KaUen R. J,
Storey
В.
et.
al. Familial
nonhemolytic jaundice with late onset of neurological change. -
■ Pediatrics,
1968,
v.
42,
p.
786—792.
9
173
Криптофтальм
с другими пороками развития
Crypiophthalmos
with other malformations MIM 219000
Синонимы:
синдром Фразера; криптоф- тальм-синдактнлия.
Впервые
описан в 1962 г. С. Fraser.
Минимальные
диагностические признаки: криптофтальм,
аномалии ушных раковин и гениталий.
Клиническая
характеристика.
Криптофтальм - это недоразвитие или
отсутствие глазного яблока, часто
сочетающееся с отсутствием век и
глазных щелей. Криптофтальм может
сочетаться со следующими пороками
развития: низкий рост волос на латеральных
поверхностях лба. гипоплазия крыльев
носа, аномалии ушных раковин, кожные
синдактилии, аномалии гениталнй
(гипоспадия, крипторхизм, двурогая
матка, атрезня влагалища и гипертрофия
клитора) В некоторых случаях имеется
гнпертело- ризм, дефекты носослезного
канала, расщелина неба, срединная
расшелина лица, атрезня наружного
слухового прохода и дефекты
среднего
уха, стеноз или атрезня гортани, аномалии
почек, атрезня ануса.
Тип
нас ледотния—аугосомно-рецесснвный.
Диффсренциальиый
диагноз:
анкнлоблефа- рон.
ЛИТЕРАТУРА
Fraser
С.
R
Our genetical «load». A review of some aspects of genetical
variation. —
Ann
Hum Genet 1962,
v.
25.
p
387
415.
GaitusoJ.
Patton
M A .
Baraitser
M
The clinical spectrum of the Fraser syndrome: report of three new
cases and review —
J.
Med. Genet., 1987,
v.
24.
p.
549
-555.
Ramsing
M .
Render
H Holzgrere W etal
Fraser syndrome (crypiophthalmos with syndactyly) in the fetus and
newborn. Clin Genet., 1990,
v.
37,
p.
84—96.
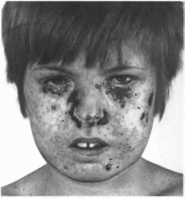
Ксеродерма
пигментная
Xeroderma
pigmentosum MIM 133510.278700
MuHUMatbHbtc'
диагностические
признаки: повышенная
чувствительность кожи к ультрафиолетовым
лучам, светобоязнь, хтока- чественные
опухоли кожи.
Рис.
77.
Пигментная ксеродерма.