ца,
микро- и макроцефалию, интракрани-
альные кальцификаты, гипертрофию тканей
орбиты Наблюдаются глаукома, катаракта,
колобома и гетерохромия радужки.
Встречаются висцеромегалия. гемангиомы
желу- дочно-кишечного тракта мочевой
системы, добавочные крупные сосуды,
увеличение наружных половых органов,
липодистрофия. Возможны умственная
отсталость и судорожный синдром.
Нарушение кровотока в конечности может
быть вызвано гипо- или аплазией вены,
сдавлением вены эмбриональными
тяжами или артериями, врожденным
недоразвитием клапанов глубоких вен.
Рентгенологически выявляются внутри-
и подкожные кальцификаты, истончение
коркового слоя костей. Заболевание
проявляется с рождения и неуклонно
прогрессирует.
Дифференциальный
диагноз:
синдром Штурге- Вебера лимфедема:
синдром Протея: нейрофиброматоз,
тип I.
ЛИТЕРАТУРА
Harper
P. S.
Sturge Weber syndrome with Klip- pel Trenaunaj Weber syndrome. The
Clinical De- line. tion of Birth Defects. XII. Sk i. Hair and Nails.
Birth Defects, 1971,
v.
VII<8). p. 314—3I7.
Senelle
M.
Klippel and Trenaunay's syndrome 768
operated
cases.- Am. Surg., 1985.
v.
20l,p. 365
373.
Книста
болезнь
Kniest
dysplasia MIM: 156550
Клиническая
характеристика.
Типична непропорциональная карликовость.
С рождения отмечаются укорочение и
деформация конечностей, тугоподвижность
суставов. Конечности укорочены за счет
проксимальных отделов
(рис. 66).
Длинные трубчатые кости укорочены и
искривлены, суставы увеличены.
Ограничение подвижности суставов
приводит к контрактурам. Пальцы кистей
длинные, движения в суставах пальцев
ограничены. больные не могут сжать
кисть в кулак. В результате
диспропорционального укорочения
туловища развиваются пояснич
ный
гиперлордоз и кифосколиоз. Дети поздно
начинают ходить и испытывают трудности
при ходьбе. Встречается косолапость.
Лицо плоское с выпуклыми, широко
расставленными глазами, уплощенной
переносицей и большим ртом. Отмечается
миопия высокой степени, нередко
сочетающаяся с отслойкой сетчатки. В
50%случаев наблюдается расщелина
неба; часто развивается проводящая
и нейросенсорная глухота. Нередки
пупочные и паховые грыжи. Моторное и
речевое развитие может быть замедлено,
интеллект обычно сохранен.
Рентгенологически выявляется
следующая картина: плати-
Рис.
66.
Болезнь Кннста. Укорочение туловища и
проксимальных отделов конечностей
Viljoen d. L. Klippel Trenaunay— Weber syndrome (angio-osteohypertrophy syndrome). J. Genet. Hum., 1988, V. 25. P. 250 252.
Синоним метатропная дисплазия, тип II. Заболевание описано в 1952 г. W. Kniest. Минимальные диагностические признаки: непропорциональная карликовость, миопия, плоское лицо.
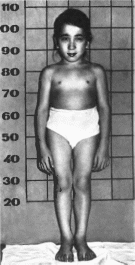
Рис.
67.
Синдром вялой кожи а,
б
новорожденный; в
больная 6 лет; г — больная 13 лет.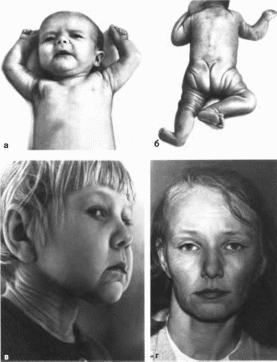
Коккейна
сннд >м
117
споилилня;
длинные трубчатые кости короткие и
тонкие, с уменьшенными, уплощенными
эпифизами, расширенными, неравномерно
разреженными метафизами; кости кисти
разрежены, концы коротких трубчатых
костей кисти расширены; кости таза
укорочены и расширены в поперечнике:
ядра окостенения головок бедренных
костей появляются поздно или
отсутствуют.
Соотношение
полов — Ml : Ж1.
Тип
нас ледовиты
— предположи гельно аутосомно-домннант
ный
Диффсрснциильньы
диагноз.
спондшюэнифи- зарная дисцлазня:
мукополисахаридоз. тип IV
ЛИТЕРАТУРА
Chen
N..
Yang
S. Е
. Gonzales
Z. E.
Kniesi dysplasia: neonatal death with necropsy —
Am.
J. Med. Genet,
1980. v.
6.
p.
171
178.
Sianescu
1
. Sianesiu
R Vtarolcau
\
P.
Kniest syndrome Am J. Hum. Genet.
1976. v.
28.
p.
527
528.
Кожа
ёялая
Cutis
laxa
M1M
123700. 2191 DO,
304150
Минимальные
диагностические признаки: генерализованная
вялость кожи, типичные изменения
эластических волокон в биопта- тах
кожи.
Клиническая
характеристика.
Кожа свисает крупными складками,
поверхность ее i рубая и
слегка пигментированная. Подкожная
жировая клетчатка практ нчески
отсутствует. Изменения кожи обычно
проявляются с рождения
(рис. 67, а, 6)
или в раннем детстве и могут
прогрессировать
(рис. 67, в, г). Встречаются
аномалии сердца, эмфизема легких,
дивертикулы кишечника и желудка,
блефарохалазня. эктропион. грыжи,
выпадение прямой кишки и влагалища,
слабость голосовых связок. В биоптатах
кожи уменьшено количество эластических
волокон; волокна фрагментированы и
гранулированы
Тип
наследования
— существуют формы с аут осомно-доминантным,
аутосомно-рецес- сивиым и Х-сцепленным
рецессивным типами наследования.
Дифференциальный
диагноз синдром
Элер- са—Данлоса; эластоз.
ЛИТЕРАТУРА
Beighrom
P. Н
The
dominant and recessive forms of cutis laxa. J. Med. Genet.. 1972.
v.
9.p 216-221.
Кожи
аплазия локальная
Localized
absence of skin MIM 107600
Минилииьные
диагностические признаки: локальное
отсутствие кожи.
Клиническая
характеристика
У новорожденных обнаруживается
локальное отсутствие кожи на
волосистой част и головы. Пораженные
участки изъязвляются и покрываются
серозной пленкой В первые недели жизни
образуются рубцы без последующего
роста волос. Туловище и нижние конечности
поражаются реже; в этих случаях
аплазня кожи может сопровождаться
отсутствием подкожной жировой
клетчатки и мышц.
Попу
ляционная частота
— 1 : 10 000.
Тип
наследования
— аутосомно-доми- нантиый.
Дифференциальный
диагноз:
буллезный эпидермолиз с врожденной
локальной аплазией кожи и деформациями
ногтей; дсрмаль- ная фокальная гипоплазия.
ЛИТЕРАТУРА
Pup
G. S.
Congenital defect of scalp and skull in three generations of one
family: case report Plast. Reconstr. Surg.. 1970.
v
46,
p.
194-
196
Коккейна
синдром
Cockayne
syndrome MIM 216400
Впервые
описан в 1946 г. Е Cockayne
Минимальные
диагностические признаки: пнзкорослость.
старческое лицо, шн чент- ная дегенерация
сетчатки, микроцефалия, снижение слуха,
умственная отсталость, повышенная
чувствительность кожи к солнечному
свету.
Клиническая
характеристика.
При рождении масса тела нормальная,
в дальнейшем наблюдается отставание
в массе и росте, атрофия подкожной
жировой клетчатки, в результате чего
кожа становится сухой, тонкой,
дряблой. Глаза запавшие, лицо старческое.
узкое, нос тонкий: отмечаются прогнатизм
(рис. 68, а),
высокое арковидпое нёбо, .множественный
кариес. У 65% больных обнаруживается
патология зрения пигментная дегенерация
сетчатки (множественные черные и
белые точки на глазном дне), атрофия
зрительных нервов, шпоплазия сетчатки,
помутнение роговицы, катаракта,
косоглазие, нистагм У 2/3
больных отмечается
сни-
118
Коккейна
сипл ром
жение
слуха вплоть до глухоты. Повышенная
фоточувствительность кожи приводит к
развитию эритематозного дерматита.
Потоотделение снижено. волосы редкие,
конечности холодные, цианотичные.
Обнаруживается ряд аномалий
опорно-двигателыюго аппарата:
конечности непропорционально длинные,
с большими кистями и стопами; сгибательные
деформации суставов, кифоз, килевндная
грудная клетка
(рис. 68, б). Рентгенологически
выявляются диффузный остеосклероз,
утолщение костей черепа и уменьшение
его размеров (микрокрания). увеличение
передне-заднего размера позвонков,
удлинение диафизов длинных трубчатых
косгей, укорочение метакарпальных
костей и фаланг, i ипоплазия
подвздошных костей. Клинически важный
признак — наличие неврологической
симптоматики (прогрессирующие
мозжечковые расстройства, тремор, i
инеркинезы, анорексия). Больные
Рис.
68.
Синдром Коккейна а - старческое лицо,
запавшие глаза, прогнатизм: б больные
снбсы.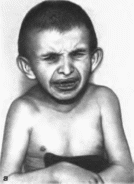
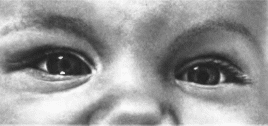
Колобомы
радужки,
сосудистой оболочки и сетчатки
119
Рис.
69.
Левосторонняя колобома радужки.
отстают
в психическом развитии. У мальчиков
отмечаются крипторхизм и i
ипонлазия яичек.
Тип
наследования
— аутосомно-рецессив- ный Ген локализован
на I0q21.1.
Дифференциальный
диагноз:
прогерия; синдром Секкеля; синдром
Блума; синдром Дубовица; лепречаунизм.
ЛИТЕРАТУРА
Fujimolo
W. Y.. Greene М.
L.,
Seegmiller J. Е.
Cockayne's
syndrome: report of a case with hyperlipoproteinemia.
hyperinsulin :mia, renal disease and normal growth hormone. -
J.
PediJtr., 1969.
v.
75,
p.
881
884.
Nance
M. A .
Berry
S. 4.
Cockayne
syndrome: review of 140
cases.
-
Am.
J. Med. Genet.
1992. v.
42.
p.
68
-84.
Колобома
желтого пятна и брахидактилия
Macular
coloboma and brachydactyly MIM: 120400
Синоним:
синдром Сорсби. Впервые описан в 1925 г.
A. Sorsby. Минимальные
диагностические признаки: врожденная
ко юбома желтого пятна и аномалии
развития конечностей.
Клиническая
характеристика.
Типичные признаки—двусторонняя
пигментная колобома желтого пятна
и брахидактилия типа В. Отмечается
гипоплазия ногтей на II пальцах кистей
и I пальцах стоп; I пальцы кистей и
стоп
широкие или удвоенные. Petrrie-
нологически выявляется уменьшение
II фаланг пальцев рук, расщепление
терминальных фаланг I пальцев рук и
ног и значительная гипоплазия всех
терминальных фаланг.
Тип
наследования
— аутосомно-доми- нантный с полной
пенегрантностью.
ЛИТЕРАТУРА
Smith
R. D.. Finenum R. М..
Sillence
D. О.
Congenital
macular coloboma and short-limb skeletal dysplasia. -
Am.
J. Med. Genet., 1980.
v.
5,
p.
365
371.
Колобомы
радужки, сосудистой оболочки и
сетчатки
Coloboma
of iris, choroid and retina MIM: 120200
Минимальные
диа. ностическис признаки: колобомы
разных отделов глаза.
Кшническш
шрактсриспшка.
Колобомы глаза бывают частичные и
полные. На глазном дне выявляется
сегмен гарное отсутствие сосудистой
оболочки и сетчатки с выпадением
нолей зрения (в виде скотом), в некоторых
случаях возможны колобомы зрительного
нерва. Характерны снижение остроты
«рения, косоглазие, нистагм. При
поражении зрительного нерва формируется
задняя киста. При исследовании с
помощью щелевой лампы хорошо видны
колобомы цилиарно- го тела, радужки
(рис. 69),
хрусталика. Изо-