108
Камптодактилня
Hull
В..
Spranger
J. W
Campomelic dysplasia, further elucidation of a distinct entity —
Am.
J.
Dis. Child .
1980,
v.
134.
p.
285—289.
Maroieaux
P,
Spranger
J. M. Opii-J. M el at.
Le syndromecampomelique Presse Med.. I97l, t. 22,
p
1157
-II62.
Камптодактилня
с гипоплазией мышц, скелетной дисплазией
и аномальными ладонными складками
Camptodactyly
with muscular hypoplasia, skeletal dysplasia, and abnormal palmar
creases MIM: 219960
Синоним:
синдром камнтодактилии Тель Хашомера.
Рис.
60.
Камптодактилня с гипоплазией мышц,
скелетной дисплазией и аномальными
ладонными складками. Гипоплазия мышц
верхних конечностей.
нообразная
форма пальцев, аномальная дерматоглифика.
Клиническая
характеристика
Наиболее типичные признаки —
камптодактилня (100%), гипоплазия мышц
верхних и нижних конечностей (93%) (рис.
60), низкий рост (71%) Отмечаются жалобы
на боли в ногах, трудности при ходьбе.
Помимо камптодак- тилии наблюдаются
следующие скелетные изменения:
синдактилия (79%), крыловидные лопатки
(71%), косолапость (71%), сколиоз (71%),
клинодактилия (64%), деформации пальцев
стоп. Черепно-лицевые особенности
включают брахицефалию, выступающий
лоб, гипертелоризм (86%), асимметрию лица
(86%), микростомию (86%), длинный фильтр,
аномальную форму зубов, в отдельных
случаях птоз и косоглазие. У 93% больных
изменены дерматоглифическне показатели,
в частности, отмечается гипо- или аплазия
дистальных межфаланговых складок.
В некоторых случаях выявляют симптомы
поражения соединительной ткани:
гиперподвижность и вывихи суставов,
мягкая лишняя кожа, пролапс митрального
клапана, паховые грыжи.
Тип
наследования
— аутосомно-рецессив- ный.
ЛИТЕРАТУРА
Pagnan
N
A..
Gollop Т
R
Ledermun Н
The
Tel Hashomer camptodactyly syndrome: report of a new case and review
of the literature. —
Am.
J.
Med. Genet , 1988.
v.
29,
p
411
417
Toriello
H. V HigginsJ. И
Malvitz
T. Waterman D К
Two
siblings with Tel Hashomer camptodactyly and mitral valve
prolapse. —
Am.
J. Med. Genet,
1990. v.
36.
p
398
-403.
Карликовость
Ларона
Pituitary
dwarfism Laron type MIM 262500
Синонимы:
семейная карликовость с повышенным
уровнем иммунореактивного гормона
роста в плазме; пнтуитарная карли
ковость, тип II.
Минимальные
диагностические признаки пропорциональная
карликовость, повышенный уровень
гормона роста, отсутствие реакции
на введение гормона роста, дефицит
рецепторов гормона роста
Клиническая
характеристика
Дети рождаются с незначительно
сниженной длиной тела и нормальным
весом; пропорциональОписан в 1972 г. R Goodman с соавт Минимальные диагностические признаки: камптодактилня, гипоплазия мышц, верете-
Карликовость
Левн
109
ное
отставание в росте начинается в раннем
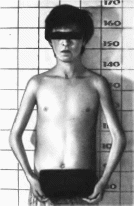
детстве. Отмечаются диспропорция череп-
но-лицевого скелета за счет гипоплазии
верхней и нижней челюсти, седловидный
нос (рис.
61) Кисти и
стопы относительно небольшие. Описаны
хрупкость, дистрофия и преждевременное
разрушение зубов. Волосы редкие,
растут медленно. Характерны тучность,
высокий голос, задержка полового
созревания, медленное развитие моторных
функций и несоответствие костного
возраста паспортному. Умственное
развитие обычно нормальное. Отмечаются
высокий уровень иммунореактивного
гормона роста, гиперчувствительность
к инсулину, спонтанная гипогликемия
в детстве.
Тип
наследования
— аутосомно-рецессив- ный. Ген локализован
на 5pl3-pl2.
Дифференциальный
диагноз:
изолированный дефицит гормона роста;
пангипопитуи-
таризм;
пропорциональная карликовость другой
этиологии.
ЛИТЕРАТУРА
Guerara-Aguirre
J.. Roscnbloom A. L.. Vuccaretto А/.
A.
el al.
Growth hormone receptor deficiency (La- ron syndrome): clinical and
genetic characten ucs Acta Paediatr. Scand..
199I,
v. 377.
p.
96—103.
Saldanha
P. H . Toledo S. P.
Familial dwarfism with high IR-GH: report of two affected sibs with
genetic and epidemiologic considerations. —
Hum.
Genet., I98l, v. 59,
p.
367—372.
Карликовость
Леви
Dwarfism
Levi type MIM: 127100.223600
Синоним:
карликовость и курносый нос. .1/инимальные
диагностические признаки: низкая
масса тела при рождении, отставание в
росте, курносый нос, гиперстеничное
телосложение.
Рис.
61.
Карликовость Ларона. Внешний вид больно!
о. Седловидный нос, гипопла зия верхней
и нижней челюстей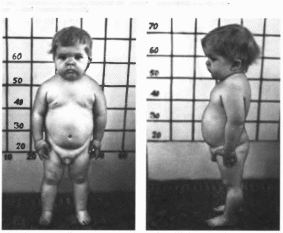
110
Карликовость
пангипопитуитарная
Клиническая
характеристика.
При доношенной беременности дети
рождаются с низким весом. Характерные
проявления - задержка роста, гиперстен
и чное телосложение, курносый нос,
брахицефалия. Встречаются также
паховые грыжи, крипторхи зм. Соотношение
полов -Ml
:
ЖI.
Тип
нас гедования
— предполагается аутосом- но-доминантный
и аугосомно-рецессивный.
Дифференциальный
диагноз: другие
тины карликовости с низким весом при
рождении.
ЛИТЕРАТУРА
Black
J.
Low birth weight dwarfism. Arch. Dis Child.. I96l, v. 36,
p.
633
644.
Карликовость
пангипопитуитарная
Panhypopituitarism
MIM: 262600. 312000
Afunuuaihiibie
диагностические
признаки: низкий
рост; дефицит гормона роста, гона-
дотропинов, адренокортикотропного
(АКТГ) и тнреотропного (ТТГ) гормонов.
Клиническая
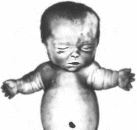
характеристика.
Типичны отставание в росте, ожирение,
высокий голос, мягкая морщинистая
кожа и «детское» лицо
(рис. 62).
Задержка роста появляется в возрасте
6 мес; у взрослых сохраняются пропорции
тела, свойственные ребенку — относительно
длинное туловище и короткие ноги.
Вторичные половые признаки отсутствуют.
У женщин наблюдается первичная аменорея.
у мужчин — гипоплазия яичек и полового
члена. Костный возраст резко отстает
от паспортного. Отмечаются нарушение
толерантности к глюкозе, повышенная
чувствительность к инсулину,
склонность к гипогликемии, снижение
липолиза и уровня соматомедина в плазме.
Дефицит гормона роста может сопровождаться
дефицитом других гормонов гипофиза —
гонадотропи- нов. АКТГ и ТТГ, что влияет
на клиническую картину. Так. дефицит
ТТГ приводит к снижению рефлексов,
основного обмена и к эпифиза рной
дисплазии.
Тип
наследования
— аутосомно-рецессив- ный и X-сцепленный
рецессивный
Дифферетршльный
диагноз:
врожденная аплазия гипофиза:
карликовость Ларона: изолированный
дефицит гормона роста.
ЛИТЕРАТУРА
Donaldson
М. D. С., Tucker S.
A/.. Gram D. В.
Рис.
62.
Больной с паш ипопитунтарной карли
ковостью(ии1кии рост, ожирение, гипогенита-
лизм, характерное лицо). Рядом здоровый
ребенок того же возраста.
Recessively
inherited growth hormone deficiency in a family from Iran. J. Med.
Genet., 1980,
v
17,
p.
288
290.
Карликовость
танатофорная
Thanatophoric
dwarfism MIM: 187600
Синоним:
танатофорная дисплазия.
Минимальные
диагностические признаки: карликовость
за счет укорочения конечностей при
нормальной длине туловища, микроме-
лия, узкая грудная клетка, короткие
ребра; короткие и широкие кости таза
и длинные трубчатые кости; уменьшение
высоты позвонков на рентгенограмме:
гибель в периоде новорожденное™
вследствие дыхательной недостаточности.
Клиническая
характеристика.
Длина тела новорожденных обычно 36—46
см. Размеры

а
Рис.
63.
Танатофорная карликовость,
а
мнкромелия, коническая форма пальцев
рук, макроцефалия, запавшая пере-
носица.
многочисленные кожные складки: б
рентгенограмма новорожденного
вертикальный
диаметр гсл позвонков уменьшен, в
ннжнегрудиом и поясничном
отделах
позвоночника межпозвоночные пространства
увеличены, грудная клетка
узкая,
ребра и длинные трубчатые кости
укорочены.
Карликовость
танатофорная
111
туловища
нормальные, конечности резко укорочены.
Пальцы рук короткие, конической
формы. Череп относительно большой, с
выступающим лбом и запавшей переносицей.
Уменьшение размеров грудной клетки
приводит к дыхательным расстройствам.
У новорожденных отмечаются многочисленные
кожные складки
(рис. 63, а),
мышечная гипотония и арефлексия. Дети
погибают в первые дни жизни, чаще всего
от дыхательной недостаточности.
Наблюдается следующая рентгенологическая
картина: у меньшение высоты тел
позвонков, расширение межпозвоночных
пространств, уменьшение вертикального
и увеличение поперечного размера
подвздошной кости, горизонтальное
положение нижнего края подвздошной
кос
ти,
короткие и широкие седалищная и лонные
кости, узкая грудная клетка с короткими
ребрами, сильно укороченные и относительно
широкие длинные трубчатые кости со
шпорообразным расширением метафизов:
бедренные кости по форме напоминают
«телефонную трубку»: короткие и
широкие кости кистей и стоп
(рис. 63, 6). На
аутопсии находят сдавление спинного
мозга и сужение большого затылочного
отверстия.
Тип
наследования
— аутосомно-доми- нантный. Предполагают,
что все случаи обусловлены свежими
летальными мутациями.
Дифференциальный
диагноз:
ахондропла- зия; ахондрогенез;
асфиксическая дистрофия грудной клетки
новорожденных.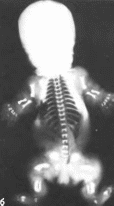
112
Картагенера
синдром
ЛИТЕРАТУРА
Gtedion
A
Thanatophoric dwarfism ■
Helv.
Рае-
dial. Acta.. 1968.
v.
23.
p.
175-
183.
Harris
R .
Potion
J
Achondroplasia and thanatophoric dwarfism in the newborn -
Clin
Genet., 1971,
v.
2,
p.
61
72
Perm
S.. Goodman II
The genetics of thanatophoric dwarfism -
Pediatrics,
1973.
v.
51,
p.
104
109.
Карликовости,
церебральной атрофии, генерализованного
фолликулярного кератоза синдром
Dwarfism,
cerebral atrophy, generalized keratosis follicularis MIM: 308830
Описан
в 1974. J. Cantu с соавт
Минимальные
диагностические признаки карликовость,
фолликулярный гиперкератоз.
Клиническая
характеристика.
Отмечаются микроцефалия, алопеция,
отсутствие бровей и ресниц,
фолликулярный гиперкератоз на волосистой
части головы, эпикант. гинер- телоризм,
длинный фильтр, микрогнатия, аномалия
прикуса. Больные резко отстают в росте
и психомоторном развитии Наблюдается
судорожный синдром, атрофия мозга
различной степени. Рентгенологически
выявляются остеопороз и отставание
костного возраста.
Тип
нас ледовиты
— предположительно Х-сцепленный
рецессивный.
Дифференциальный
диагноз:
синдром Ду- бовица; другие формы
карликовости.
ЛИТЕРАТУРА
Caniu
J Hernandes А
. Larracilla
J el al
A new X-linked recessive disorder with dwarfism, cerebral atrophy
and generalized keratosis follicularis. —
J.
Pediatr., 1974.
v
84,
p.
564
570
Картагенера
синдром
Kartagener
syndrome MIM: 244400
Синоним:
синдром декстрокардии, брон- хоэктазов
и синуситов.
Описан
в 1936 г. М. Kartagener.
Минимальные
диагностические признаки: обратное
расположение органов, синуситы,
бронхоэктаэы.
Клиническая
характеристика.
Характерны неполное или полное
обратное расположение органов,
пансинусит, хронический
отит,
бронхиты, бронхоэктазы Вторично
развиваются аносмия, умеренное снижение
слуха по проводящему типу, полипы
носовой полости. При неполном обратном
расположении органов может наблюдаться
нарушение сердечной деятельности.
Описаны женское и мужское бесплодие.
Тип
наследования
— аутосомно-рецессив- ный.
Дифференциальный
диагноз,
синдром обратного расположения
органов; изолированная декстрокардия.
ЛИТЕРАТУРА
Moreno
A Murphi F A.
Inheritance of Kartagener syndrome —
Am
J Med Genet.. 1981,
v.
8,
p.
305—313.
Roll
H D
Kartagener's syndrome and the syndrome of immobile cilia Hum.
Genet.
1979. v.
46,
p.
49
261
Катаракты
врожденные
Cataracts
MIM
116200,212500.302200.
Минимальные
диагностические признаки: помутнение
хрусталика.
Клиническая
характеристика.
Клиническая картина зависит от
интенсивности и локализации помутнения
в хрусталике. Точечные двусторонние
участки помутнения на зрение не влияют
и не прогрессируют. Полная ядерная
катаракта составляет около 25% всех
врожденных катаракт и приводит к
снижению остроты зрения, иногда
значительному. Слоистая катаракта
представляет собой зону помутнения
между эмбриональным ядром и капсулой
хрусталика и поражает обычно оба глаза.
Эта форма врожденных катаракт составляет
40% всех случаев, медленно прогрессирует
и приводит к инвалиди- зации по зрению,
Другие формы част ичного помутнения,
особенно в области оси хрусталика,
поражают несколько слоев, бывают обычно
двусторонними, не прогрессируют, на
остроту зрения не влияют Врожденные
катаракты со снижением остроты зрения
в 30% случаев сопровождаются нистагмом
и косоглазием.
В
25% случаев односторонней врожденной
катаракты и в 11% случаев двусторонней
встречается микрофтальмия Иногда
катаракта сочетается с помутнением
роговицы, передним лентиконусом,
колобомой радужки, аниридией, эктопией
хрусталика, перси-
Каудальной
регрессии синдром
113
стирующим
гиперпластическнм первичным стекловидным
телом, васкулярнзованным хрусталиком.
Тип
наследования
— аутосомно-доминант- ный,
аутосомно-рецессивный. Х-сцеплен- ный
рецессивный.
ЛИТЕРАТУРА
Маитепес
/ Hereditary
cataracts. -
Trans.
Am. Ophthalmol. Soc.. 1979,
v.
86.
p.
1554
-1558.
Каудальной
регрессии синдром
Caudal
regression syndrome MIM: 182940
Синонимы:
синдром каудальной диспла- зии;
сиреномелия.
Минимальные
диагностические признаки: гипо-
или аплазия каудального отдела
позвоночника. аномалии развития
соответствующе
го
сегмента спинного мозга, гипо- или дис-
плазия таза и нижних конечностей.
Клиническая
характеристика.
Тяжесть проявлений зависит от характера
и степени поражения поясничных,
крестцовых позвонков и копчика.
Характерны гипоплазия, атрофия и
аномалии строения нижних конечностей
(рис. 64). в
крайне тяжелых случаях — резкое их
недоразвитие и слияние (сиреномелия),
сужение и гипоплазия таза, врожденный
вывих бедра, флексорные контрактуры
тазобедренных и коленных суставов,
косолапость. Нижняя часть туловища
сужена, аномально сформирована. Рост
снижен за счет укорочения туловища и
конечностей. Неврологическая симптоматика
включает нижний парапарез, мышечную
гипотонию, угнетение сухожильных
рефлексов. Отмечается паралич мышц
тазового дна и сфинкте
8
173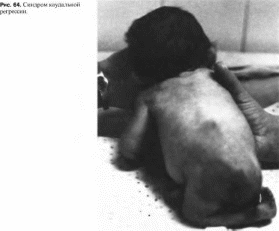
114
Клиппеля—Треноне
Вебера
синдром
ров,
приводящии к недержанию мочи и кала
Описаны пороки развития мочевыводящей
системы, желудочно-кишечного тракта,
гениталий.
Тип
наследования
не установлен. Большинство случаев
спорадические
Дифференциальный
диагноз:
детский церебральный паралич,
врожденные миопатии.
ЛИТЕРАТУРА
Finer
N..
Bower
Р.
Dunbar
L. G.
Caudal regression anomalad (sacral agenesis) in siblings. —
Clin. Gene!.. 1978. v
13, p. 353—359.
Welch
J. P., Aiertnan K.
The syndrome of caudal dysplasia: a review, including etiologic
considerations and evidence of heterogeneity. — Pediat.
Pathol., 1984. v. 2, p.
313—327.
Клиппеля—Треноне—Вебера
синдром
Klippel
Trenaunay Weber syndrome MIM: 149000
Синонимы:
гипертрофическая гемангиэк- тазия;
ангиоостеогипертрофия.
Описан
в 1900 г. М. Klippel ссоавт.
Минимальные
диагностические признаки: асимметричная
гипертрофия конечностей, гемангиомы.
Клиническая
характеристика.
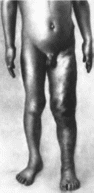
Типичный признак — гипертрофия одной
или нескольких конечностей
(рис 65),
связанная с врожденным пороком развития
артериове- нозной системы и лимфатических
сосудов. Отмечаются гиперплазия мягких
тканей и костей пораженной конечности,
трофические язвы и отеки. Гемангиомы
бывают единичные или множественные;
капиллярные, кавернозные, кистозные;
различающиеся по размерам и локализации
(на любой части тела, но чаще всего на
голенях, ягодицах, животе, нижней части
туловища). Преобладает одностороннее
поражение Описаны гиперпигментация
кожи, телеангизктазии, мраморная
кожа, макродактилия, диспропорциональная
длина пальцев, синдактилия, поли- и
олигодактилия. Черепно-лицевые аномалии
включают асимметричную гипертрофию
ли
Рис.
65.
Синдром Клиппеля Треноне— Вебера
Гипертрофия нижней конечности.