

Наследственные
ПСЙНДГОМЫ
и
медико-генетическое
консультирование
п
р а
к
т
и
к а
С.И.Козлова,
Н.С.Демикова, Е.Семанова, О.Е.Блинникова


С.И.Козлова
И.С.Демикова Е.Семанова О.Е.Блинникова
Наследственные
СИНДРОМЫ
и
медико-генетическое консультирование
2-е
издание, переработанное
и дополненное
£93
Пензенского
государственного
университета
практика
Москва
1996Научная библиотека
ББК
57.3
К 45
УДК
в16-055.5/.7-084(035)
Научный
рсыктор чл.-корр. РАМН профессор Е. К.
Гннтер
Рекомендовано
Управлением учебных и велении Министерства
иравоохранения н мешншнекой
промышленности Р|ШиГ1екой Фе терапии
в качестве учебного пособия для студентов
медицинских ну юн интернов, ординаторов,
аспирантов н врачей ротных специальностей
С
И
Коимна. Н. С\ Демнкова. Е Семанова. О Е
Блинникова Наследственные
синдромы и медино-генетичесное
консультирование
А
тлас-справочннк
Ни
2-сдополн М Практика. IWA
416стр.. 392нл. ISHN S-«H001-tX>K-2
К
45 В книге
описцно около MX) наиболее
распространенных наследственных
синдромов. Прнводягся минимальные
диагностические прижаки. клиническая
характеристика, популяиионная частота,
тип настеюваиия. дифференциальная
диагностика. сведения о юкалнкшии генов
Описана методика медико-генегнческо!
о консультирования. Для генетиков,
акчшеров. псшатров, невропатологов
н врачей других специальностей.1 С II Котлова. Н с Демнкова. Е. Семанова. О. Е. Ь.Шнникова. 1w6. 1 Оформ lemic. Пилю 1ьсгво «Практика». IWft.
Предисловие
авторов к первому изданию
Успехи
в области профилактики наследственных
болезней особенно заметны в последние
годы. Это связано с широким внедрением
в практику здравоохранения
медико-генетического консультирования
и пренатальной диагностики, что
стало возможным благодаря интенсивному
изучению этиологии и патогенеза
наследственных заболеваний. Ежегодно
в литературе появляются несколько
сотен новых описаний генетически
обусловленных аномалий. Поданным
последнего каталога V. McKusick
(1986) известно более 2000 наследственных
синдромов.
Большинство
наследственных синдромов диагностируется
только на основании характерной
клинической картины. В этой связи
синдромологиче- ский анализ приобретает
первостепенное значение в практике
врача-генетика. Однако многие
наследственные заболевания встречаются
довольно редко и поэтому врачи не
имеют достаточного навыка в диагностике
наследственной патологии. Кроме
того, трудности в распознавании
наследственных синдромов связаны
с тем, что нередко решающее значение в
постановке диагноза имеет выявление
у больных микроаномалий, на которые
врачи не всегда обращают внимание.
Например, такие незначительные признаки,
как насечки на мочке уха при синдроме
Беквита Видемана. ямочки на слизистой
нижней губы при синдроме ван дер Вуда,
являются очень ценными диагностическими
критериями. Весьма информативны гакие
признаки, как масса тела ребенка при
рождении и масса плаценты. При синдроме
панцито- пении Фанкони масса тела
новорожденного практически никогда
не бывает более 3000 г. а плаценты — не
превышает 500 г. Иногда пренатальная
гипотрофия является одним из основных
признаков синдрома. Так. при снндроме
Дубовица при доношенной беременности
дети рождаются с низкой массой тела (в
среднем 2260 г). Это касается и синдрома
Корнелии де Ланге (средняя масса тела
при рождении 2300 г), синдрома Смита—Лемли
Опица и др. С другой стороны, при синдроме
Беквита Видемана одним из главных
диагностических признаков является
гигантизм при рождении. Таким образом,
в практике врача-генетика не может быть
«мелочей».
В
диагностике наследственной патологии
важны также знания патогене за пороков
развития. Например, при преаксиальных
аномалиях (гипоплазия или отсутствие
первых пальцев) следует искать аномалии
большого мозга, при декстракардии —
неполный поворот кишечника, при пороках
конечностей — исследовать почки и
мочевыводящие пути. И. наоборот, при
синдромах ADAM и Поланда
не следует ожидать аномалий внутренних
органов.
При
постановке диагноза нужно стремиться
увязать все находки на основе общего
генеза заболевания. Так, тяжелая
гипогликемия при синдроме Беквита
Видемана может нарушить обычно нормальное
умственное развитие. Или в результате
тромбоцитопении при синдроме
тромбоцитопении с отсутствием лучевой
кости могут развиться кровоизлияния,
в том числе и в мозг, приводящие к смерти
или к нарушению умственного развития.
Знание
наследственных синдромов необходимо
врачам для правильного определения
клинического прогноза, прогноза жизни,
а также прогноза в отношении
профессиональной пригодности больного.
Например, при синдроме Патау не имеет
смысла делать сложные операции по
поводу расщелины неба, поскольку
больные дети обычно нежизнеспособны.
Не следует оперировать катаракту у
больных с синдромом Халлермана Штрайфа
или Конради- Хюнермана, так как в
большинстве случаев наступает спонтан
ная
резорбция. Если врач знает, что у больного
с синдромом DIDMOAD после
пубертатного возраста появятся нарушения
зрения, то он уже заранее должен
ориентировать родителей на выбор для
ребенка профессии, в меньшей степени
требующей хорошего зрения То же самое
касается и больных с различными
скелетными аномалиями, которые обычно
усугубляются после 14 —15 лет жизни. Уже
в детстве таких больных следует обучать
работе, не связанной с физическими
нагрузками.
Кроме
того, точный диагноз имеет значение
для определения генетического
прогноза для родственников больного
при медико-генетическом консультировании.
как ретроспективном, так и проспективном.
В этом случае от точной диагностики
зависят судьбы многих людей, а не только
одного больного.
Данная
книга посвящена описанию фенотипическнх
проявлений наследственных болезней.
В нее включены наиболее изученные и
сравнительно часто встречающиеся
синдромы, а также некоторые редкие
синдромы, имеющие значение для
медико-генетического консультирования.
Всего по унифицированной схеме
описано 460 синдромов, из них 148
проиллюстрировано фотографиями. В
книге приведены известные в литературе
популяционные частоты наследственных
болезнен. Для рецессивных синдромов
представлены данные отечественных
авторов, так как диапазон колебаний
этих частот в разных популяциях
вечик.
В
книге содержится раздел по основным
принципам расчета риска при разных
генетических ситуациях. Эти сведения
будут полезны врачам медико- генетических
консультаций на втором этапе
консультирования, после того как диагноз
наследственного синдрома поставлен.
Диагностический
указатель поможет читателям найти
нужный синдром по наличию ряда симптомов
Книга
предназначена для широко круга врачей,
и в первую очередь для медицинских
генетиков, педиатров, акушеров-гинекологов,
невропатологов, ортопедов и др.
Авторы
не претендуют на исчерпывающее описание
наследственной патологии и будут
благодарны за замечания и сообщения о
синдромах, не вошедших в справочник
В
книгу вошли данные многолетних наблюдений
Центра по медико-генетическому
консультированию при Институте
медицинской генетики АМН СССР (Москва)
и медико-генетической консультации
Института по исследованию развития
ребенка педиатрического факультета
Карлова Университета (Прага), а также
отдельные случаи, информацию о которых
нам прислали консультативные кабинеты
по медицинской генетике Минска Еревана,
Кривого Рога, Воронежа, Уфы, Кемерова,
Орла
Предисловие
ко второму изданию
Прошло
уже девять лет после публикации первого
издания этой книги. Справочник получил
множество положи гельных откликов от
тех, для кого он в первую очередь
предназначался от врачей медико-генетических
консультаций. К сожалению, то издание
так и осталось единственной в нашей
стране книгой, посвященной диагностике
редких наследственных синдромов.
Вместе с тем, за рубежом за это время
вышло множество подобных руководств
и атласов.
Автору
предисловия самому приходилось наблюдать
в разных странах, как широко исполыуются
справочники при постановке диагноза.
Наиболее популярной остается книга
Дэвида Смита «Recognizable patterns
of
human malformations», четвертое издание
которой вышло в 1988 г. Впрочем, в ходу и
другие справочники. Все большее
распространение получают компьютер-
ныедиагностическиесистемы, такие как
POSSUM и LONDON DATA
BASE.
Не
нужно забывать, что больные попадают
к врачу-генетику не сразу; предварительный
диагноз обычно ставят педиатры,
офтальмологи, эндокринологи, ортопеды,
невропатологи, акушеры-гинекологи.
Особую ценность имеют книги, которые
совмешают текстовую и графическую
информацию. Наследственная патология
почти вся редкая, и потому накопить
собственный опыт в диагностике
наследственных синдромов трудно.
Особую роль здесь играет зрительное
восприятие: внешний вид больного
зачастую позволяет распознать
синдром, что называется, с первого
взгляда. Авторы справочника учли
это, и по сравнению с первым изданием
иллюстративного материала стало
значительно больше.
Всего
дано описание около 500 наследственных
синдромов, что по крайней мере в
полтора раза больше, чем в уже упомянутом
руководстве Д.
Смита.
Описание каждого синдрома дано достаточно
кратко: минимальные диагностические
признаки, клиническая характеристика,
тип наследования, перечень состояний,
с которыми надо проводить дифференциальную
диагностику.
Книга
также содержит сведения о том, гены
каких синдромов картированы, что
прозволяет проводить пренатальную
ДНК-диагностику. В разделе, посвященном
медико-генетическому консультированию,
теперь также описан принцип расчета
риска для сцепленных с Х-хромосомой
заболеваний с учетом данных о сцеплении
соответствующего гена с ДНК-полиморфными
маркерами. Дополнен и уточнен
терминологический словарь и
диагностический указатель синдромов
по признакам.
Не
вызывает сомнения, что новое издание
книги будет подспорьем в работе врачей
многих специальностей.
На
учныйредактор, член-hop/
РАМН.
проф. Е. К. Гинтер
А
Аарскога
синдром
Aarskog
syndrome MIM: 100050. 305400
Выделен
в самостоятельную нозологическую
единицу в 1970 г. D. Aarskog.
Минимальные
диагностические признаки: гипертелоризм.
брахидактилия. шалевидная мошонка,
низкий рост.
Клиническая
характеристика.
Наблюдается различная степень
отставания в росте (в 907о описанных
случаев), хотя масса и длина тела при
рождении нормальные. Отставание
в
росте становится очевидным к концу
1-го года. У больных круглое лицо,
клиновидный рост волос на лбу — «мыс
вдовы» — (70%), гипертелоризм (95%), широкая
переносица (85%), короткий нос с вывернутыми
ноздрями (94%), широкий фильтр (97%),
антимонголоидный разрез глаз (55%),
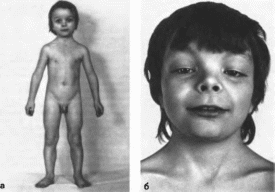
птоз (50%), (рис.
1, а, б). Глазные
аномалии включают офтальмоплегию,
косоглазие, астигматизм, увеличенную
роговицу. Характерны гипоплазия
верхней челюсти (85%), относительная
прогения, легкая складка под нижней
губой (82%), аномалии ушных раковин
Рис.
1. Синдром
Аарскога.
а
внешний вид больного;
6 ■
гипертелоризм. широкая переносица,
широкое округлое лицо, антимонголоидный
разрез глаз, короткий нос.
12
Аарскога
синдром
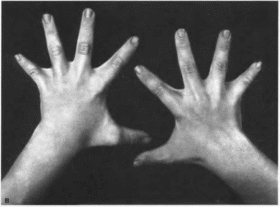
Рис.
1. Синдром
Аарскога (продолжение), а
брахидактилия. неполная кожная
синдактилия; г шалевидная мошонка.
(76%)
Отмечается разболтанность суставов
(70%), брахидактилия, клинодактилия V
пальцев (80%), неполная кожная синдактилия
рук (70°
о)
(рис. 1, в),
переразгибание в проксимальных
межфаланговых суставах с одновременным
сгибанием в дистальных, короткие V
пальцы с единственной сгиба- тельной
складкой (72%), поперечная складка ладони,
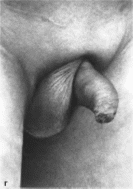
широкие стопы (75%). Наиболее характерное
изменение гениталий — шалевидная
мошонка, складки которой окружают
основание полового члена (81%)
(рис. 1, г). В
75% случаев отмечается крипторхизм.
реже— расщепление мошонки, фимоз
Описаны паховые грыжи (66%). Умеренная
умственная отсталость или затруднения
при обучении наблюдаются у 14% больных
Соотношение
полов
- - М1 . Ж0.
Тип
нааедования
— предположительно
Агашлиоз
кишечника врожденный
13
аутосомно-доыипантный
или Х-сцетеннын рецессивный В последнем
случае ген лока- лнюван на Xql3.
Дифференциальный
диагноз синдром
Hv- нан. гипертелоризм с
аномалией пищевода и гипоспадией.
ЛИТЕРАТУРА
<jrur
R Autosomal
dominant inheritance of the Aarskog syndrome Am J Med Genet. 1983.
v 15. p 19 46
Hoo
J The Aarskog
(fdcio-digito genital) syndrome Clin. Genet.
1979. v. 16. p. 269
276.
Tichi
1
S
RucquuniJ. A
Mcyn M S
Aarskog syndrome report of a family with review and discussion
of nosology. — Am. J Med. Genet.. 1993.
v 46. p. 501 509.
Агаммаглобулинемия
Х-сцеплен- ная инфантильная
Agammaglobulinemia
X-linked infantile M1M 300300
Синонимы:
агаммаглобу:ишемия Бруто- на: врожденная
агаммаглобулинемия
Впервые
описана в 1952 г О Bruton
Минимальные
диагностические признаки: рецидивирующие
тяжезые бактериальные инфекции,
поражающие дыхательные пути,
желудочно-кишечный гракт и кожу
Отсутствие В-шмфоцитов. отсутствие
или резкое снижение уровнен иммуноглобулинов
основных
К IdCCOB
в крови
Клиническая
характеристика.
Забозева- нис характеризуется тяжело
протекающими воспалительными процессами.
Чаще всего развиваются отит, конъюнктивит,
синусит, i айморит. энтерит,
рециднвиру юшие инфекции верхних
дыхательных путей, бронхит, ппевмоиия.
пиодермия, вы «ванные главным обра юм
стафилококками, пневмококками,
стрептококками. Очень тяжело протекает
гепатит, который может привести к
смерти. Во зможны полиартрит и дерматомио
зит: нередко состояние осложняется
сепсисом. Больные бледны, малоподвижны:
на коже тица. конечностей, туловища
очаги пиодермии В периферической
крови обнаруживаются анемия,
лейкопения, нейтропения. трашиториая
то зинофи лия и моноцитоз. В крови и
лимфоиднон ткани отсутствуют В-
лимфоциты. Резко снижен уровень
иммуноглобулинов. IgM и
IgA отсутствуют, уровень
IgG при рождении нормальный,
к 6 мес сни
жается
до 100 мг%. Иммунимцня не приводит к
положительным результатам: изогем-
агглютинины отсутствуют. Плазматические
клег ки в костном мозге отсутствуют
или их количество резко снижено. При
этой форме иммунодефицита отмечается
высокая летальность в раннем возрасте.
Тип
нас ледоваиии
Х-сцепленный рецессивный Ген
локализован на Xq2l 3-q22
Дифференциальный
диагноз
агранулоцн- тоз Костмана: вторичные
иммунодефицит- ные состояния.
ЛИТЕРАТУРА
Letlernuui
Н
\1
ttuikeLuein J A
X-linked agammaglobulinemia an analysis of 96
patients. Medicine. 1985. v 64.
p 145 156
Meiuink
E J Thompson A.
Si
hoi J D L t'l at Genetic
heterogeneity in X-lmked agammaglobulinemia complicates carrier
detection and prenatal diagnosis. Clin. Genet.. 1987.
v 31. p. 91 96.
Аганглиоз
кишечника врожденный
Megacolon
aganglionic MIM 249200
Синонимы:
болезнь Гиршпрунга; истинный
врожденный мегаколон.
Минима
льные диагностические признаки: стойкие
запоры, динамическая кишечная
непроходимость. агенезия ганглиев
межмышечного и подели зистого нервных
сплетений в различных участках
кишечника.
Клинический
характеристика
Заболевание обусловлено врожденным
отсутствием интрамуратьных
парасимпатических ганглиев и
симпатических сплетений в различных
участках толстой кишки. В зависимости
от локализации аганглиозных участков
выделяют ректальную форму (21.9%).
ректосигмо- идальную (69,2""),
субтотальную (3,2"..). тотальную
(0.6%) и сегмешарную (5.1 ) В последнем
случае агаигэиснный участок находится
между двумя здоровыми или наоборо т. В
1% случаев аганглиоз захватывает тонкую
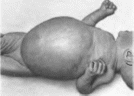
кишку. Клиническая картина характеризуется
стойкими запорами (100 ) и динамической
кишечной непроходимостью. В 45 о случаев
отмечается рвота и в 85%случаев - вздутие
живота
(рис. 2). Отдел
кишечника выше аганглиозного участка
расширяется, стенки ninepiрофируются,
развивается мегаколон. который может
осложниться энтероколитом и
перфорацией кишки. Каловая ин-
14
Аганглиоз
кншечника врожденный
Рис.
2. Вздутие
живота при врожденном аган- глиозе
кишечника
токсикация
может вызвать жировую дистрофию
печени Рентгенологически определяются
сужение участка кишки и расширение
предшествующего ему сегмента, задержка
бария. Этиология мультифакториальная.
При поражении короткого сегмента
кишечника риск для братьев 5%. сестер
Г ».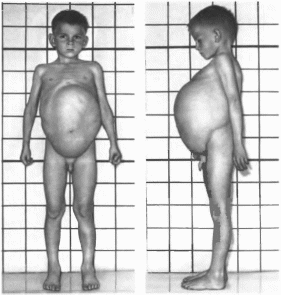
Агранулоцнто'
Костмана
15
При
поражении длинного сегмента кишечника
риск для сибсов обоего пола — 10%.
Соотношение
полов — М2—3
: Ж1. Дифференциальный
диагноз:
мегаколон функциональный: мегакотон
психогенный.
ЛИТЕРАТУРА
BaJncr
J. A.. Sieber И'.
A..
Garrer A. L.. Chakra- varii A.
A genetic study of Hirschsprung disease. — Am.
J. Hum. Genet.. 1990. v. 46.
p. 568—580.
MiKinnon
A.. Cohen S.
Total intestinal aganglio- nosis. An autosomal recessive condition?
Arch. Dis. Child.. 1977. v. 52.
p. 898 900.
Passarge
E. Genetic
heterogeneity and recurrence risk of congenital intestinal
aganglionosis. — Birth Defects, 1972.
v. VIII (2). p. 63 67.
Аглоссии-адактилии
синдром
Hanhart
syndrome M1M: 103300
Синонимы:
синдром гипоглоссии-гипо- дактилии;
синдром Ханхарта; перомелия и микрогнатия.
Впервые
описан в 1950 г. Е. Hanhart.
Минимальные
диагностические признаки микрогения,
микроглоссия или аглоссия: редукционные
пороки конечностей в виде пе- ромелнн.
Клиническая
характеристика.
Наиболее постоянные признаки —
гипоплазия (или аплазия) языка и
гипоплазия нижней челюсти
(рис. 3, а).
Встречаются расшелина неба, синехии
ротовой полости, сигнатия (сращение
челюстей), микростомия, анкилоглоссия
(сращение кончика языка со слизистой
неба), олигодонтия. Возможен одно-
или двусторонний паралич черепно-мозговых
нервов. Пороки развития конечностей
варьируют по тяжести и включают
синдактилию, олигодактилию
(рис. 3,
6). адактилию,
частичную или полную ахейрию (аподию).
частичную гемимелию предплечья или
голени. Интеллект иногда снижен.
Соотношение
полов— Ml
:Ж1.
Тип
наследования
неизвестен, все описанные случаи
спорадические.
Дифференциальный
диагноз:
зктродакти- лия; ахейроподия:
рото-лице-пальцевои синдром;
амниотические перетяжки.
ЛИТЕРАТУРА
Tuncbileke
Е
, Yalcin
С..
Atasu
М.
Aglossia-adac- tylia syndrome (special emphasis on the
inheritance pattern). Clin. Genet.. 1977. v.
11, p. 421 423.
Рис.
3. Синдром аглоссии-адактилии а гипоплазия
нижней челюсти: б олнгодактплпя
Агранулоцитоз
Костмана
Kostmann
agranulocytosis MIM: 202700
Синоним:
нейтропения постоянная. Впервые описан
в 1956 г. R. Kostmann. Минимальные
диагностические признаки: рецидивирующие
гнойные поражения — абсцессы кожи,
легких, отит и т. д.
Клиническая
характеристика.
Проявляет-

