

ЛЕКЦИЯ_03_Кислотно-основное равновесие
Кислотно-основное равновесие
Теории кислот и оснований.
Протолитическая теория: роль растворителя, нивелирующее и дифференцирующее действие.
Равновесие в водных растворах сильных и слабых кислот и оснований.
Буферные растворы.
Гидролиз.
Равновесие в водных растворах гидролизующихся солей.
Вычисление рН в водных растворах солей.
Теория электролитической диссоциации или ионизации
Свойства водных растворов солей, кислот и оснований обнаруживают ряд ненормальностей по сравнению со свойствами других растворов. Приведем следующий пример. Как правило, изменение точки кипения или замерзания данного неводного растворителя будет одним и тем же, если в 1000 г его растворить 1 моль любого вещества (на этом основано приближенное определение молекулярного веса веществ). То же самое справедливо в отношении водных растворов неэлектролитов, т.е. веществ, водный раствор которых проводит электрический ток не лучше, чем сама вода. Если же на основании изменения точки замерзания или кипения водного раствора какого-нибудь электролита (т.е. вещества, водный раствор которого проводит электрический ток значительно лучше, чем сама вода) мы захотели бы определить молекулярный вес последнего, то получили бы слишком низкие величины; раствор ведет себя так, как если бы в нем было большее число молекул растворенного вещества, чем можно ожидать на основании других данных.
К тому же заключению приводят и другие физические свойства растворов электролитов, как, например, упругость паров и осмотическое давление.
М. Фарадей (Faraday) при исследованиях законов электролиза, результаты которых он опубликовал в 1834 г., предполагал, что первое действие электрического тока во время электролиза сводится к расщеплению первоначальных молекул на меньшие частицы, названные им ионами1.
Сванте Аррениус (Svante Arrhenius) в 1887 г. предложил теорию ионизации, согласно которой расщепление молекул на ионы происходит независимо от электрического тока: ионы образуются в водных растворах тотчас по растворении вещества. Эта теория Аррениуса в настоящее время является общепринятой. Исторически теория ионизации была развита на основании изучения чисто физических свойств водных растворов, каковыми являются точка замерзания, точка кипения, упругость паров, осмотическое давление и отношение водных растворов к электрическому току; особенно же важное значение теория ионизации приобрела для объяснения химических свойств растворов электролитов.
Если водный раствор хлористого бария смешать с разбавленным раствором серной кислоты, то образуется белый кристаллический осадок сульфата бария:
BaCl2+H2SO4=2HCl+BaSO4
Такой же осадок идентичного химического состава может быть получен и из любой другой растворимой бариевой соли, причем вместо серной кислоты может быть взят любой растворимый сульфат.
Прибавление небольшого количества нитрата серебра к водному раствору хлористого бария вызывает появление белого творожистого осадка хлористого серебра, темнеющего под влиянием света:
BaCl2+2AgNO3=Ba(NO3)2+2AgCl
Тот же осадок образуется, если вместо хлористого бария пользоваться соляной кислотой или каким-либо другим хлоридом, а вместо нитрата серебра какой-либо другой растворимой солью серебра.
Таким же путем могут быть показаны определенные свойства, которыми отличаются водные растворы всех кислот, как, например, окрашивание синего лакмуса в красный цвет. Этими так называемыми кислыми свойствами они обязаны водороду кислот, который ведет себя совершенно иначе, чем водород других соединений.
Основания также отличаются определенными характерными реакциями, которые приписываются находящемуся в них гидроксилу ОН. Водный раствор какого-нибудь основания окрашивает красный лакмус в синий цвет, реагирует с кислотами, образуя воду и т.д.
Франклин, изучая свойства различных веществ, растворенных в безводном жидком аммиаке, предложил расширить понятие о кислоте, основании и соли, указывая, что NH4+, образующийся из NH3 и H+, может рассматриваться в таком растворе как кислота и что образование NH4+ в среде жидкого аммиака может быть сравнено с образованием Н3О+ при гидратации водородного иона в водном растворе:
H++H2OH3O+
Н.Ф.Голл присоединился к подобным взглядам, применяя их к растворам в безводной уксусной кислоте и в других такого же характера растворителях.
Бренстед3, Лоури4, Бьеррум5 также пытались расширить в подобном направлении понятия о кислотах, основаниях и солях. Сущность реакции между любой кислотой и водой была впервые осознана датским химиком Йоханнесом Бренстедом (1879-1947) и английским химиком Томасом М. Лаури (1874-1936).
Бренстед и Лаури пришли к мысли, что поведение кислот и оснований можно описывать с учетом способности этих веществ к переносу протонов. В 1923 г. Бренстед и Лаури независимо друг от друга предложили рассматривать кислоты как вещества, способные отдавать (донироватъ) протон, а основания как вещества, способные присоединять (акцептировать) протон.,
С такой точки зрения при растворении в воде НСl действует как кислота, отдавая протон растворителю. В то же время растворитель (Н2О) действует как основание, присоединяя протон.
Раньше мы применяли термин основание к веществам, образующим в водном растворе избыток ионов ОН-. Отметим, однако, что ион ОН- представляет собой акцептор протонов; он легко реагирует с гидратированным протоном, образуя воду:
Н+(водн.) + ОН-(водн.) ⇄ Н20(ж.) (15.5)
Подобно этому, нам известно, что водные растворы аммиака обладают основными свойствами, потому что NH3 реагирует с Н2О, образуя NH4+ и ОН-:
Н20(ж.) + NH3(водн.) ⇄ NН4+(водн.) + ОН-(водн.) (15.6)
В этой реакции Н2О отдает протон NH3; следовательно, Н2О играет роль кислоты, а NH3 - основания.
Реакции, которые мы привели выше в качестве примера реакций с переносом протона, являются обратимыми. Так, при смешении ионов NH4+ (например, из NH4C1) и ОН- (например, из NaOH) образуются Н2О и NH3:
NН4+(водн.) + ОН-(водн.) ⇄ NH3(водн.) + Н20(ж.) (15.7)
Это чисто ионное уравнение реакции представляет собой процесс, обратный реакции между NH3 и Н2О, который описывается уравнением (15.6). В обратной реакции, описываемой уравнением (15.7), NH4+ играет роль донора протона, а ОН- - роль акцептора протона.
Таким образом, если реакция протекает в одном направлении, Н2О играет роль кислоты, а NH3 - роль основания. Но в обратной реакции NH4+ играет роль кислоты, а ОН- - роль основания.
Рассмотренный пример показывает, что каждая кислота связана с сопряженным основанием*, которое образуется из этой кислоты в результате отщепления от нее протона. Например, сопряженным основанием для NH4+ является NH3, а сопряженным основанием для Н2О является ОН-. Точно так же каждое основание имеет сопряженную кислоту, которая образуется из этого основания в результате присоединения к нему протона. Например, Н2О является сопряженной кислотой основания ОН-. Кислота и основание, которые, подобно Н2О и ОН-, отличаются только наличием или отсутствием протона, называются сопряженной кислотно-основной парой.
Чем легче какая-либо кислота отдает протон, тем труднее сопряженное ей основание присоединяет к себе протон. Другими словами, чем сильнее кислота, тем слабее сопряженное ей основание, а чем слабее кислота, тем сильнее сопряженное ей основание.
Теория кислот и оснований Льюиса
Для того чтобы вещество могло быть акцептором протонов (т.е. проявляло себя как основание, согласно представлениям Бренстеда-Лаури), оно должно обладать неподеленной парой электронов, которая бы связывала протон. Например, мы уже знаем, что NH3 ведет себя как акцептор протона. Пользуясь валентными (льюисовыми) структурами, реакцию между Н+ и NH3 можно написать следующим образом:

На эту особенность кислотно-основных реакций впервые обратил внимание Г.Н. Льюис. Он предложил определение кислоты и основания, в котором подчеркивается роль неподеленной пары электронов: кислота есть акцептор неподеленной пары электронов, а основание - донор неподеленной пары.
Каждое основание, которое мы обсуждали до сих пор, будь то ОН-, Н2О, какой-нибудь амин или анион, является донором электронной пары. Любое вещество, обладающее свойствами основания в рамках представлений Бренстеда-Лаури (т.е. акцептор протона), с точки зрения Льюиса, также является основанием (донором электронной пары). Однако в теории Льюиса допускается, что основание донирует электронную пару не только ее акцептору Н+. Поэтому определение Льюиса значительно расширяет круг веществ, которые могут рассматриваться как кислоты; Н+ представляет собой отнюдь не единственно возможную, с точки зрения Льюиса, кислоту.
До сих пор мы рассматривали в качестве растворителя только воду, а в качестве носителя кислотных свойств только протон. В таких случаях более удобны определения кислоты и основания, предложенные Бренстедом и Лаури. В самом деле, когда говорят, что вещество обладает кислотными или основными свойствами, то обычно имеют в виду водные растворы, а упомянутые термины применяются в рамках представлений Аррениуса либо Бренстеда-Лаури. Преимущество теории Льюиса заключается в том, что она позволяет рассматривать более разнообразные реакции, включая реакции, не сопровождающиеся переносом протона, такие, как кислотно-основные реакции в водных растворах. Во избежание путаницы вещество, подобное ВF3, редко называют кислотой, но это можно делать в том случае, если из контекста понятно, что данный термин применяется как определение Льюиса. Чаще вещества, обладающие свойствами акцепторов электронной пары, называют «льюисовыми кислотами». К льюисовым кислотам относятся молекулы, подобные ВF3, с незавершенным октетом электронов вокруг какого-либо атома. Помимо этого, свойствами льюисовых кислот обладают многие простые катионы. Например, Fе3+ сильно взаимодействует с цианид-ионами, образуя феррицианид-ион Fe(CN)63-:
Fe3++6:C≡N:- [Fe(:C≡N)6]3-
Соединения с кратными связями также могут вести себя подобно льюисовым кислотам. Например, реакцию диоксида углерода с водой, в результате которой образуется угольная кислота Н2СО3, можно изобразить как взаимодействие донора электронной пары, каким является молекула воды, с акцептором электронной пары, в роли которого выступает СО2:

При этом электронная пара, обобществляемая в одной из -связей С—О, смещается к концевому атому кислорода, в то время как на атоме углерода появляется вакантная орбиталь, придающая ему свойства акцептора электронной пары. Смещения электронов отмечены на рисунке стрелками. Вначале молекулы образуют друг с другом "аддукт", после чего протон переходит от одного атома кислорода к другому, и в результате образуется молекула угольной кислоты:

Подобная льюисова кислотно-основная реакция осуществляется при растворений какого-либо оксида неметаллического элемента в воде с образованием кислого раствора.
Сильные кислоты (НСl, HJ, HNO3 и др.).
В сильных кислотах [Н+] практически считают равной концентрации кислоты. Следовательно, для расчетов применяют следующие формулы:
![]()
![]()
Изменение рН на единицу соответствует изменению концентрации Н+-ионов в 10 раз.
Сильные основания (NaOH, КОН и др.). В сильных основаниях, [ОН-] считаем практически равной концентрации основания КtOH. Следовательно, для расчетов можно применить следующие формулы:

Слабые кислоты.
Из уравнения диссоциации кислоты следует:
НАn ⇄ Н++Аn-.
Н+ и An--ионы образуются в равных количествах, и их концентрации будут равны:
[Н+] = [Аn-].
Если
раствор мало разбавлен, то почти вся
присутствующая кислота находится в
виде недиссоциированных молекул.
Следовательно, можно принять, что
![]() По закону
действующих масс
По закону
действующих масс
![]()
так как [Н+]=[Аn-], то отсюда

Чтобы от [Н+] перейти к рН раствора, прологарифмируем уравнение (12) и переменим знаки у логарифмов на обратные:
![]()
Обозначим
![]() через
через
![]() который называют
силовым
показателем кислоты.
который называют
силовым
показателем кислоты.
Уравнение (13) перепишем в следующем виде:
![]()
Слабые гидроксиды (анилин, NH4OH и др.).
Из уравнения диссоциации слабого гидроксида NH4OH следует:
NH4OH ⇄ NH4+ +OH-
концентрации NH4+ и ОН--ионов равны между собой:
[NH4+] = [OH-].
В разбавленном растворе гидроксид находится в недиссоциированном состоянии.
Следовательно, [NH4OH] = c0. По закону действующих масс
![]()
отсюда
![]()
![]()
отсюда
![]()
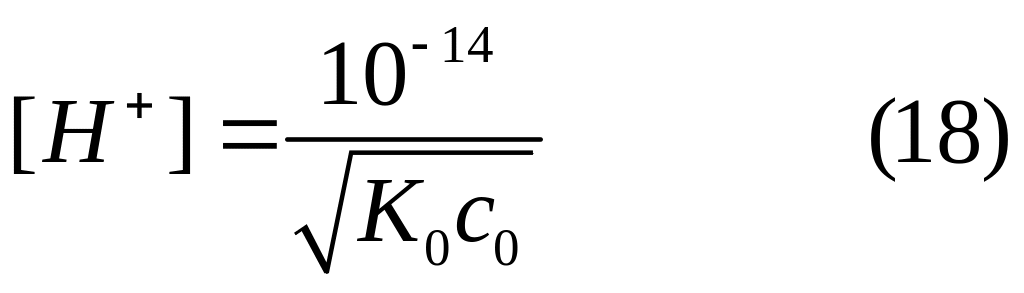
Если прологарифмируем это выражение и изменим знаки у логарифмов на обратные, то получим
![]()