Вопросы для итогового контроля (экзамен в 5 семестре)
1. Сущность гравиметрического анализа, преимущества и недостатки метода.
2. Общая схема определений. Требования к осаждаемой и гравиметрической формам.
3. Условия получения кристаллических осадков. Гомогенное осаждение. Старение осадка.
4. Причины загрязнения осадка (совместное осаждение, соосаждение, последующее осаждение).
5. Методы титриметрического анализа. Классификация. Требования, предъявляемые к реакции в титриметрическом анализе.
6. Виды титриметрических определений: прямое и обратное, косвенное титрование.
7. Способы выражения концентраций растворов в титриметрии. Эквивалент. Молярная масса эквивалента. Первичные и вторичные стандарты, требования к ним. Фиксаналы.
8. Виды кривых титрования. Скачок титрования. Точка эквивалентности и конечная точка титрования.
9.Кислотно-основное титрование. Построение кривых титрования.
10. Влияние величины констант кислотности или основности, концентрации кислот или оснований, температуры на характер кривых титрования.
11.Кислотно-основные индикаторы. Погрешности титрования.
12.Окислительно-восстановительное титрование. Классификация.
13. Построение кривых титрования. Факторы, влияющие на характер кривых титрования.
14. Способы определения конечной точки титрования; индикаторы.
15.Осадительное титрование. Построение кривых титрования.
16. Способы обнаружения конечной точки титрования; индикаторы. Примеры практического применения.
17.Комплексометрическое титрование.
18.Неорганические и органические титранты в комплексометрии.
19. Построение кривых титрования.
20. Металлохромные индикаторы и требования, предъявляемые к ним.
21.Способы комплексонометрического титрования: прямое, обратное, косвенное.
22. Селективность метода и способы ее повышения. Погрешности титрования. Примеры практического применения.
23. Общая характеристика электрохимических методов. Классификация.
24. Электрохимические ячейки. Индикаторный электрод и электрод сравнения.
25. Поляризационные кривые и их использование в различных электрохимических методах.
26. Прямая потенциометрия. Измерение потенциала.
27. Индикаторные электроды. Ионометрия. Классификация ионселективных электродов.
28.Потенциометрическое титрование. Изменение электродного потенциала в процессе титрования.
29. Вольтамперометрия классификация вольтамперометрических методов.
30. Полярография. Уравнение Ильковича. Полярографическая волны Потенциал полуволны.
31. Амперометрическое титрование. Сущность метода. Индикаторные электроды.
32. Амперометрическое титрование с одним и двумя поляризованными электродами. Виды кривых титрования.
33. Спектр электромагнитного излучения. Основные типы взаимодействия вещества с излучением: эмиссия, поглощение, рассеяние. Классификация спектроскопических методов.
34. Спектры атомов. Основные и возбужденные состояния атомов, характеристики состояний. Энергетические переходы. Правила отбора. Законы испускания и поглощения. 35. Спектры молекул; их особенности. Основные законы поглощения электромагнитного излучения (Бугера) и закон излучения (Ломакина-Шейбе). Связь аналитического сигнала с концентрацией определяемого соединения.
36. Аппаратура. Способы монохроматизации лучистой энергии. Классификация спектральных приборов.
37. Методы атомной оптической спектроскопии. Атомно-эмисионный метод. Источники атомизации и возбуждения
38. Метод эмиссионной спектрометрии пламени. Подготовка пробы к анализу. Пламенные фотометры и спектрофотометры.
39. Атомно-абсорбционный метод. Атомизаторы (пламенные и непламенные). Источники излучения , их характеристики.
40.Молекулярная абсорбционная спектроскопия (спектрофотометрия). Связь химической структуры соединения с абсорбционным спектром.
41. Основной закон светопоглощения. Отклонения от закона.
42. Монохроматизация излучения. Понятие об истинном и кажущемся молярном коэффициенте поглощения.
43. Инструментальные погрешности, оптимальный интервал измеряемых значений оптической плотности.
44. Способы определения концентрации веществ. Измерение высоких, низких оптических плотностей (дифференциальный метод). Анализ многокомпонентных систем.
45. Фотометрические аналитические реагенты, требования к ним. Примеры практического применения метода.
1. Гравиметрия (весовой анализ) — метод количественного 0%90%D0%BD%D0%B0%D0%BB%D0%B8%D0%B7" \o "Анализ" анализа в 0%90%D0%BD%D0%B0%D0%BB%D0%B8%D1%82%D0%B8%D1%87%D0%B5%D1%81%D0%BA%D0%B0%D1%8F_%D1%85%D0%B8%D0%BC%D0%B8%D1%8F" \o "Аналитическая химия" аналитической химии, который основан на измерении массы определяемого компонента, выделенном в виде веществ определённого состава.
Используется уравнение химической реакции типа: aX + bR = XaRb для получения осадка XaRb.
Гравиметрический анализ (весовой) один из важнейших методов количественного анализа. Он сыграл большую роль при установлении законов постоянности состава, кратных отношений, периодического закона. Его применяют при определении химического состава разнообразных естественных и технических объектов, горных пород и руды, минералов, металлов, сплавов, силикатов и других неорганических и органических веществ.
Все многочисленные гравиметрические определения можно разделить на три большие группы:
1. Методы выделения;
2. Методы осаждения;
3. Методы отгонки.
Гравиметрические методы применяют редко. Основное их достоинство — исключается построение калибровочных графиков (построение графика при анализе многокомпонентных смесей затруднительно, из-за невозможности приготовления стандартной смеси, точно моделирующей пробу, не зная заранее состава пробы). Гравиметрические методы применяют в качестве арбитражных при определении магния, натрия, кремнекислоты, сульфат-ионов, суммарного содержания нефтепродуктов, жиров.
Гравиметрический анализ - один из наиболее универсальных методов. Он применяется для определения почти любого элемента. В большей части гравиметрических методик используется прямое определение, когда из анализируемой смеси выделяется интересующий компонент, который взвешивается в виде индивидуального соединения.
Наиболее существенным достоинством гравиметрического метода является высокая точность анализа. Обычная погрешность гравиметрического определения составляет 0,1...0,2%. При анализе пробы сложного состава погрешность возрастает до нескольких процентов за счет несовершенства методов разделения и выделения анализируемого компонента. К числу достоинств гравиметрического метода относится также отсутствие каких-либо стандартизации или градуировок по стандартным образцам, необходимых почти в любом другом аналитическом методе. Для расчета результатов гравиметрического анализа требуется знание лишь молярных масс и стехиометрических соотношений.
Существенным недостатком гравиметрического метода является длительность определений.
Чаще всего гравиметрический метод применяют для определения основных компонентов пробы, когда на выполнение анализа отводится несколько часов или десятков часов, для анализа эталонов, используемых в других методах, в арбитражном анализе.
2.
Используется уравнение химической реакции типа: aX + bR = XaRb для получения осадка XaRb
Требования к осаждаемой форме.
1. Осаждаемая форма должна владеть достаточно маленькой растворимостью, без чего невозможно практически полное осаждение определяемого иона. Как известно, растворимость малорастворимых электролитов характеризуется величиной их ПР. Опыт показывает, что практически полное осаждение имеет место, если ПР осадка не превышает 110-8. Поэтому соединения с ПР10-8 в качестве осаждаемой формы в гравиметрии, как правило не применяются.
2. Осадок должен быть по возможности как можно больше крупнокристаллическим для возможности быстрого фильтрования и промывания от примесей.Крупнокристаллические осадки, как правило, почти не забивают поры фильтра и, имея слабо развитую поверхность, мало адсорбируют посторонние вещества из раствора и легко отмываются от них. Аморфные осадки, в особенности гелеподобные, например: Al(OH)3, имеют сильно развитую поверхность и потому значительно адсорбируют посторонние вещества из раствора и очень трудно отмываются от них. Кроме того, фильтрование проходит тоже очень медленно.Мелкокристаллические осадки, например: BaSO4, CaС2O4, тоже мало удобны, так как забивают поры фильтра, имеют большую площадь поверхности. Кроме того, такие осадки легко проходят через поры фильтра, что не допустимо в весовом анализе.
3. Осаждаемая форма должна достаточно легко превращаться в гравиметрическую (весовую) форму.
Требования к гравиметрической (весовой) форме:
1. Точное соответствие ее состава химической формуле. Если такого соответствия нет, вычисление результатов невозможно.
2. Химическая устойчивость весовой формы.
3. Содержание определяемого в весовой форме должно быть как можно меньшим, тогда погрешности опреде ления меньше скажутся на окончательном результате анализа.
3. Осаждаемая и весовая формы должны быть химически инертными, чтобы не приводить к количественным ошибкам.
Пример.
1)


 2)
СаС12 +
Н2С2О4 -
СаС2О4 +
НСl;
2)
СаС12 +
Н2С2О4 -
СаС2О4 +
НСl;
|
|
|
|

СаО + 2СО2.
СаО — высокореакционное вещество, это означает, что оно может «захватить» пары воды или углекислый газ:
СаО + Н2О →Са(ОН)2 СаО + СО2 - >СаСО3
Требования, предъявляемые к осадкам, в значительной степени определяют выбор осадителя. Осадок увлекает за собой некоторые ионы, присутствующие в растворе. Обычно осадок полностью отмыть от примесей не удается, поэтому очень важно, чтобы примеси были летучими и удалялись при прокаливании осадка. Например, осаждение бария ведут раствором серной кислоты, а не раствором Nа2SО4, хотя и в том, и в другом случае получился бы осадок ВаSО4.
3.
Условия получения кристаллических осадков.
Условия формирования крупных чистых кристаллов вытекают из механизмов образования и загрязнения осадков, рассмотренных нами в предыдущих разделах:
1. Нужно уменьшить относительное пересыщение.
2. Избегать затравок, вызывающих индуцированную нуклеацию.
3. Замедлять осаждение.
4. Оставлять осадок под маточным раствором для старения.
условия осаждения кристаллических осадков можно сформулировать следующим образом: медленное добавление при интенсивном перемешивании к горячему разбавленному (при необходимости подкисленному) раствору осаждаемого вещества разбавленного раствора осадителя.
Гомогенное осаждение
Старение осадка
После образования осадка с ним происходит ряд необратимых физико-химических процессов, приводящих к уменьшению энергии и структурным изменениям и называемых старением осадка. Важнейшими из этих процессов являются перекристаллизация первоначально получившихся частиц, переход метастабильных состояний в стабильные, термическое старение вследствие теплового движения ионов, химическое старение в результате изменения состава осадка. Все эти процессы играют важную роль при проведении гравиметрического анализа и в большинстве случаев благоприятно влияют на гравиметрические свойства осадков.
При перекристаллизации кристаллы растворяются и осаждаются снова.
4.
Осадок, образующийся в процессе гравиметрического определения, всегда содержит то или иное количество посторонних примесей. Примеси могут попадать в осадок по различным причинам. Вид примесей, загрязняющих осадок, и их количество зависят от условий выполнения анализа и характера образующегося осадка.
Загрязнение осадка может быть вызвано соосаждением примесей либо, реже, совместным или последующим их осаждением.
При соосаждении образование осадка приводит к выпадению в осадок соединений, которые в данных условиях либо хорошо растворимы, либо находятся в таких малых концентрациях, что не достигается величина их произведения растворимости.
Совместное осаждение — это одновременное образование двух и более осадков. При послеосаждении сначала образуется чистый осадок, а примесь выделяется при стоянии. Соосаждение заключается в захвате примесей из раствора в процессе осаждения [15—17].
В аналитической химии соосаждение, как правило, играет отрицательную роль, что влечет за собой ошибки при гравиметрическом определении элементов, с одной стороны, и потерю соосаждаемого элемента — с другой.
Последующее осаждение
5.
Методы титриметрического анализа можно классифицировать по характеру химической реакции, лежащей в основе определения веществ, и по способу титрования. По своему характеру реакции, используемые в титриметрическом анализе, относятся к различным типам — реакциям соединения ионов и реакциям окисления — восстановления. В соответствии с этим титриметрические определения можно подразделять на следующие основные методы: метод кислотно-основного титрования (нейтрализации), методы осаждения и комплексообразования, метод окисления — восстановления. Метод кислотно-основного титрования (нейтрализации). Сюда относятся определения, основанные на взаимодействии кислот и оснований, т.е. на реакции нейтрализации: H++ ОН = H2O Методом кислотно-основного титрования (нейтрализации) определяют количество кислот (алкалиметрия) или оснований (ациди-метрия) в данном растворе, количество солей слабых кислот и слабых оснований, а также веществ, которые реагируют с этими солями. Применение неводных растворителей (спирты, ацетон и т. п.) позволило расширить круг веществ, которые можно определять данным методом. Методы осаждения и комплексообразования. Сюда относятся титриметрические определения, основанные на осаждении того или иного иона в виде малорастворимого соединения или связывания его в малодиссоциированный комплекс.
Методы окисления — восстановления (редоксиметрия). Эти методы основаны на реакциях окисления и восстановления. Их называют обычно по применяемому титрованному раствору реагента, например: перманганатометрия, в которой используются реакции окисления перманганатом калия KMnO4; иодометрия, в которой используются реакции окисления иодом или восстановления I-ионами; бихроматометрия, в которой используются реакции окисления бихроматом калия К2Сr2О7; броматометрия, в которой используются реакции окисления броматом калия КВrO3. К методам окисления — восстановления относятся также цериметрия (окисление Се4+-ионами), ванадатометрия (окисление VO3-ионами), титанометрия (восстановление Т13+-ионами). По способу титрования различают следующие методы.
Метод прямого титрования. В этом случае определяемый ион титруют раствором реагента (или наоборот). Метод замещения. Этот метод применяют тогда, когда по тем или иным причинам трудно определить точку эквивалентности, например при работе с неустойчивыми веществами и т. п.
Метод обратного титрования (титрование по остатку). Этот метод применяют, когда нет подходящего индикатора или когда основная реакция протекает не очень быстро. Например, для определения CaCO3 навеску вещества обрабатывают избытком титрованного раствора соляной кислоты:
Реакции, применяемые в титриметрии, должны удовлетворять следующим основным требованиям:
1) реакция должна протекать количественно, т.е. константа равновесия реакции должна быть достаточно велика;
2) реакция должна протекать с большой скоростью;
3) реакция не должна осложняться протеканием побочных реакций;
4) должен существовать способ определения окончания реакции.
Если реакция не удовлетворяет хотя бы одному из этих требований, она не может быть использована в титриметрическом анализе.
6.
В титриметрии различают прямое, обратное и косвенное титрование.
В методах прямого титрования определяемое вещество непосредственно реагирует с титрантом. Для проведения анализа этим методом достаточно одного рабочего раствора.
В методах обратного титрования (или, как их еще называют, методах титрования по остатку) используются два титрованных рабочих раствора: основной и вспомогательный. Широко известно, например, обратное титрование хлорид-иона в кислых растворах. К анализируемому раствору хлорида сначала добавляют заведомый избыток титрованного раствора нитрата серебра (основного рабочего раствора). При этом происходит реакция образования малорастворимого хлорида серебра.
Не вступившее в реакцию избыточное количество вещества AgNO3 оттитровывают раствором тиоцианата аммония (вспомогательного рабочего раствора).
Содержание хлорида легко рассчитать, так как известно общее количество вещества (моль) серебра, введенное в раствор, и количество вещества AgNO3, не вступившее в реакцию с хлоридом.
Третьим основным видом титриметрических определений является титрование заместителя, или титрование по замещению (косвенное титрование). В этом методе к определяемому веществу добавляют специальный реагент, вступающий с ним в реакцию. Один из продуктов взаимодействия затем оттитровывают рабочим раствором. Например, при иодометрическом определении меди к анализируемому раствору добавляют заведомый избыток KI. Происходит реакция 2Cu2++4I-=2CuI+ I2. Выделившийся иод оттитровывают тиосульфатом натрия.
7.
Все расчеты в титриметрии связаны с законом эквивалентов и понятием эквивалент. Эквивалентом называют реальную или условную частицу вещества, которая в данной реакции равноценна (эквивалентна) одному иону водорода или одному электрону. Например, эквивалент NaOH, НСl, NaCl - реальная частица, соответствующая молекуле этих веществ. Эквивалент Н3PO4 , в зависимости от числа участвующих в реакции протонов, может представлять реальную молекулу Н3PO4 и условную часть молекулы: 1/2Н3PO4 или 1/3Н3PO4. Дробь, показывающую, какая часть молекулы или иона является эквивалентом, называют фактором эквивалентности fэкв. Фактор эквивалентности рассчитывают на основе стехиометрии реакции.
Титр раствора — масса растворённого вещества в 1 мл раствора.
 ,
,
где:
m1 — масса растворённого вещества, г;
V — общий объём раствора, мл;
Молярная концентрация C(B) показывает, сколько моль растворённого вещества содержится в 1 литре раствора.
C(B) = n(B) / V = m(B) / (M(B) · V),
где М(B) - молярная масса растворенного вещества г/моль.
Молярная концентрация измеряется в моль/л и обозначается "M".
Массовая доля растворённого вещества w(B) - это безразмерная величина, равная отношению массы растворённого вещества к общей массе раствора m :
w(B)= m(B) / m

Массовую долю растворённого вещества w(B) обычно выражают в долях единицы или в процентах.
Нормальность раствора обозначает число грамм-эквивалентов данного вещества в одном литре раствора или число миллиграмм-эквивалентов в одном миллилитре раствора. Эоснования = Моснования / число замещаемых в реакции гидроксильных групп Экислоты = Мкислоты / число замещаемых в реакции атомов водорода Эсоли = Мсоли / произведение числа катионов на его заряд
Молярная масса эквивалента вещества - это масса одного моля эквивалента этого вещества, равная произведе-нию фактора эквивалентности на молярную массу вещества. На-пример, для вещества В:
М(fэкв В) = fэкв(В) * М(В) = 1/z*(В)*М(В)
Молярная масса эквивалента вещества может быть различной, в зависимости от протекающей реакции с его участием.
Первичным стандартом называют стандартный раствор, приготовленный по точной навеске.
Вторичным стандартом называют стандартный раствор, характеристики которого установлены по первичному стандарту.
Стандартные растворы можно также приготовить с помощью фиксаналов (стандарт-титров). Фиксаналы предствляет собой запаянные стеклянные ампулы, содержащие количество вещества, необходимое для приготовления 1 л раствора.
8.
Точка эквивалентности (конечная точка титрования) в 0%A2%D0%B8%D1%82%D1%80%D0%B8%D0%BC%D0%B5%D1%82%D1%80%D0%B8%D1%87%D0%B5%D1%81%D0%BA%D0%B8%D0%B9_%D0%B0%D0%BD%D0%B0%D0%BB%D0%B8%D0%B7" \o "Титриметрический анализ" титриметрическом анализе момент 0%A2%D0%B8%D1%82%D1%80%D0%BE%D0%B2%D0%B0%D0%BD%D0%B8%D0%B5" \o "Титрование" титрования, когда число0%AD%D0%BA%D0%B2%D0%B8%D0%B2%D0%B0%D0%BB%D0%B5%D0%BD%D1%82_%D1%8D%D0%BB%D0%B5%D0%BC%D0%B5%D0%BD%D1%82%D0%B0" \o "Эквивалент элемента" эквивалентов добавляемого 0%A2%D0%B8%D1%82%D1%80%D0%B0%D0%BD%D1%82" \o "Титрант" титранта эквивалентно или равно числу эквивалентов определяемого вещества в образце. В некоторых случаях наблюдают несколько точек эквивалентности, следующих одна за другой, например, при титровании многоосновных 0%9A%D0%B8%D1%81%D0%BB%D0%BE%D1%82%D0%B0" \o "Кислота" кислот или же при титровании раствора, в котором присутствует несколько определяемых0%98%D0%BE%D0%BD" \o "Ион" ионов.
На графике кривой титрования присутствует одна или несколько 0%A2%D0%BE%D1%87%D0%BA%D0%B0_%D0%BF%D0%B5%D1%80%D0%B5%D0%B3%D0%B8%D0%B1%D0%B0_%D0%BF%D0%BB%D0%BE%D1%81%D0%BA%D0%BE%D0%B9_%D0%BA%D1%80%D0%B8%D0%B2%D0%BE%D0%B9" \o "Точка перегиба плоской кривой" точек перегиба, соответствующих точкам эквивалентности. Точкой окончания титрования (подобна точке эквивалентности, но не то же самое) считают момент, при котором 0%A5%D0%B8%D0%BC%D0%B8%D1%87%D0%B5%D1%81%D0%BA%D0%B8%D0%B5_%D0%B8%D0%BD%D0%B4%D0%B8%D0%BA%D0%B0%D1%82%D0%BE%D1%80%D1%8B" \o "Химические индикаторы" индикатор изменяет свой цвет при колориметрическом титровании.
Если построить график изменения рН раствора по мере возрастания объема добавляемого основания, то в зависимости от того, сильными или слабыми являются кислота и основание, будут получаться кривые четырех типов. Эти четыре типа кривых титрования показаны на рис. 8.2. Следует обратить внимание, что достижение точки эквивалентности характеризуется резким возрастанием рН. Исключением в этом отношении является только титрование слабой кислоты слабым основанием.

Титрование сильной кислоты сильным основанием. Например, H2SO4(водн.) + 2NaOH (водн.) = Na2SO4 (водн.) + 2H2O (ж.). Вертикальная часть кривой этого титрования приходится на область изменений рН от 4 до 10. Следовательно, в точке эквивалентности титрования добавление к кислоте еще одной капли основания вызывает возрастание рН сразу на 6 единиц.
Титрование сильной кислоты слабым основанием.
Например, HCl (водн.) + NH4OH(водн.) -> NH4Cl (водн.) + H2O (ж.)
Вертикальная часть кривой этого титрования приходится на область изменений рН от 4 до 8.
Титрование слабой кислоты сильным основанием. Например, CH3COOH(водн.) + NaOH(водн) -> CH3COONa(водн.) + H2O(ж.)
Вертикальная часть кривой этого титрования приходится на область значений рН от 6,5 до 11.
Титрование слабой кислоты слабым основанием.
Например, CH3COOH (водн.) + NH4OH (водн.) -> CH3COONH4(водн.) + H2O (ж.)
Титрование этого типа характеризуется отсутствием резкого изменения рН в момент достижения точки эквивалентности. Изменения рН происходят плавно во всей области принимаемых значений.
Скачок титрования является самой существенной частью кривой титрования, т.к. на нём всегда лежит точка эквивалентности.
С помощью него осуществляют выбор индикатора, решают некоторые другие вопросы.
Чем больше скачок титрования, тем точнее будут результаты анализа и более широкий выбор индикаторов для его проведения.
На величину скачка титрования влияют, в первую очередь, кислотные и основные свойства исходных веществ, их содержание в растворе, а также температура реакционной среды.
Чем более сильными являются используемые кислота и основание, и чем выше их концентрация в растворе, тем скачок титрования больше.
9.
Кислотно-основное титрование - это экспериментальный метод определения концентрации кислоты либо основания, используемая преимущественно в количественном химическом анализе. основано на реакции нейтрализации:
Н3O+ + ОН− = 2Н2О
Обычно кислоту с известной концентрацией постепенно добавляют из бюретки в щелочной раствор неизвестной концентрации, находящийся в конической колбе. Точка эквивалентности титрования достигается в тот момент, когда к основанию добавлено точно стехиометрическое количество кислоты. В этой точке вся щелочь нейтрализована, и в растворе нет ни избытка кислоты, ни избытка основания. Раствор состоит только из соли и воды.
Построение кривых титрования кислот и оснований.
Метод кислотно-основного титрования основан на протолитическнх реакциях.
SH2+ + S- === 2SH
в частности, для водных растворов
Н3О+ + ОН- = 2Н2О.
Метод применим для определения концентрации кислот (соляная, уксусная и т.п.), оснований (гидроксид натрия, карбонат натрия, аммиак и т.п.), амфолитов (гидрокарбонат натрия, дигидрофосфат натрия и т.п.).
Изменяющимся параметром при построении кривых титрования служит величина РН раствора.
Вычисление концентрации ионов водорода обычно проводят с точностью до двух значащих цифр. Такая точность вполне достаточна для выбора индикатора и оценки индикаторных погрешностей титрования. Поэтому даже если концентрация ионов водорода может быть вычислена с большей точностью, значения рН можно округлять до второго знака после запятой.

10.
В начале титрования рН среды по сравнению с исходным его значением изменяется очень медленно, вплоть до расходования более 90% вещества в титруемом растворе. Затем наблюдается более быстрое изменение рН и, наконец, вблизи точки эквивалентности добавление буквально одной капли титранта вызывает резкое (взрывное) изменение величины рН сразу на несколько единиц. Этот участок на кривой титрования называется иначе скачком титрования. Дальнейшее добавление титранта опять сопровождается медленным и плавным изменением рН.
Скачок титрования является самой существенной частью кривой титрования, т.к. на нём всегда лежит точка эквивалентности.
С помощью него осуществляют выбор индикатора, решают некоторые другие вопросы.
Чем больше скачок титрования, тем точнее будут результаты анализа и более широкий выбор индикаторов для его проведения.
На величину скачка титрования влияют, в первую очередь, кислотные и основные свойства исходных веществ, их содержание в растворе, а также температура реакционной среды.
Чем более сильными являются используемые кислота и основание, и чем выше их концентрация в растворе, тем скачок титрования больше. Так, при молярной концентрации химического эквивалента НСl и NaOH, равной 0,1 моль/дм3, величина скачка титрования составляет почти 6 единиц рН (рис. 54).
В случае сантимолярных растворов этих же исходных веществ (0,01 моль/дм3) скачок титрования составляет уже 4 единицы рН. Обычно на практике редко используют растворы с концентрацией химического эквивалента вещества больше 0,2 моль/дм3. Это связано с тем, что значительно возрастают ошибки титрования за счёт неточности измерения количественных характеристик (объема, массы растворенной навески) более концентрированных растворов.
Повышение температуры оказывает существенное влияние на скачок титрования в сторону его уменьшения вследствие возрастания константы диссоциации воды. Отсюда следует, что кислотно-основное титрование лучше проводить, не прибегая к нагреванию.
11.Кислотно-основные индикаторы. Погрешности титрования.
ИНДИКАТОРЫ В МЕТОДЕ КИСЛОТНО-ОСНОВНОГО ТИТРОВАНИЯ
Кривая титрования характеризуется резким изменением рН в близи точки эквивалентности, которую называют скачком титрования. Чем больше скачок, тем точнее можно оттитровать определяемое вещество. По величине скачка выбирают индикатор.
Основное правило выбора индикатора. Для каждого данного титрования можно применять только такие индикаторы, показатель титрования которых (рТ индикатора) лежит в пределах скачка рН на кривой титрования
Индикаторами называются вещества, изменяющие свою окраску в зависимости от pH среды. По своей химической природе индикаторы представляют собой с л а б ы е кислоты HJnd илис л а б ы е основания JndOH, у которых молекулярная и ионная формы имеют различную окраску.
Согласно ионной теории индикаторов изменение цвета индикатора вызывается смещением равновесия диссоциации.
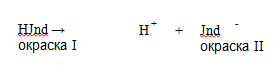
В кислой среде индикатор находится в недиссоциированной форме (HJnd), так как избыток ионов водорода связываетсяс анионами индикатора, — преобладает окраска I.
В щелочной среде гидроксильные ионы связывают протоны в малодиссоциированное вещество — воду. Равновесие смещаетсяв сторону диссоциации кислоты, что приводит к возрастанию концентрации ионной формы (Jnd ), — преобладает окраска II.
К уравнению (I) применим закон действующих масс:

При какой-то определенной реакции среды количество диссоциированных молекул индикатора будет равно количеству недиссоциированных молекул, т.е. [HJnd] = [Jnd ], тогда:
![]()
Это, так называемая, точка перехода окраски индикатора, когда индикатор имеет переходную (смешанную) окраску.
Сдвиг реакции среды в ту или иную сторону от точки перехода окраски сопровождается нарушением соотношения окрашенных форм индикатора и изменением цвета, указывающего на преобладание в растворе одной из форм индикатора. Окраску одной из форм индикатора можно различить, если концентрация ее в 10 раз превосходит концентрацию другой формы, что
объясняется чувствительностью глаза к определенным цветам. Тогда получим:

Таким образом, индикатор меняет свою окраску в интервале приблизительно двух единиц водородного показателя pH .
Согласно хромофорной теории индикаторов, изменение их окраски связано с обратимой перегруппировкой атомов в молекуле органического соединения. Такую обратимую перегруппировку в органической химии называют таутомерией. Если в результате таутомерного изменения строения в молекуле органического соединения появляются особые группировки (обычно с чередующимися двойными и одинарными связями), называемые хромофорами, органическое вещество приобретает окраску. Когда таутомерное превращение ведет к изменению строения хромофора — окраска
изменяется; если же после перегруппировки молекула не содержит более хромофора — окраска исчезает.
Таким образом, один и тот же индикатор может существовать в двух формах с разным строением молекул, причем эти формы могут переходить одна в другую, и в растворе между ними устанавливается равновесие.
О б л а с т ь з н а ч е н и й pH, в пределах которой наблюдается заметное для глаза изменение окраски индикатора, называется и н т е р в а л о м п е р е х о д а окраски индикатора (ИПО).
Титрование с различными индикаторами заканчивают в момент резкого изменения окраски индикатора. Точка перехода окраски каждого индикатора лежит примерно в середине интервала перехода окраски. Индикатор характеризуют показателем титрования рТ.
Показатель титрования рТ – это такое значение рН, при котором резко меняется окраска индикатора. Таким образом рТ определяет рН конечной точки титрования (КТТ).
Индикаторная
ошибка титрования
обусловлена тем, что рТ индикатора
чаще
всего не совпадает с рН в точке
эквивалентности (ТЭ).
Поэтому
титрование заканчивается не в точке
эквивалентности, а несколько раньше
или
позже.
Вследствие этого раствор может быть
либо несколько перетитрован
либо,
наоборот, недотитрован и в конце
титрования содержит избыток рабочего
или анализируемого раствора.
Индикаторную
ошибку титрования находят экспериментальнос
помощью контрольного опыта или вычисляют.
При
вычислении абсолютной ошибки титрования
(∆n)
находят
избыточное или недостающее количество
одного из реагирующих веществ. Чаще
всего
вычисляют не абсолютную (∆n),
а
относительную (δ)
ошибку
титрования и выражают ее в процентах:
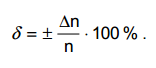
Индикаторные
ошибки титрования
1) в момент изменения окраски индикатора (КТТ) в растворе находится
+
некоторый избыток свободной сильной кислоты (водородная ошибка δН )
-
или сильного основания (гидроксидная ошибка δОН );
2) в момент изменения окраски индикатора (КТТ) в растворе находится
некоторое количество недотитрованной слабой кислоты (кислотная ошиб-
ка), слабого основания (основная ошибка) или недотитрованной соли (соле-
вая ошибка).
Для каждого типа индикаторной ошибки применимы свои формулы рас-
чета. Поэтому предварительно надо установить тип ошибки. Для этого нужно
вычислить pH в точке эквивалентности и на основании сравнения значений
pH в точке эквивалентности и pТ индикатора определить тип ошибки данного
случая титрования.