

1. Introduction to the who/tdr (Training Development Recourse)
Handbook on GLP
GENERAL INTRODUCTION
The need to implement quality standards in drug research, development and testing; the situation in developing countries and the role of WHO/TDR
Tropical diseases are a major public health problem in developing countries (Disease Endemic Countries – DECs). For many of these diseases no new, effective and affordable medicines have been developed, while older therapeutic agents are increasingly compromised
by the emergence of resistance. Because multinational pharmaceutical companies have not traditionally focused on tropical disease research and development (R&D), WHO has initiated R&D programmes in a number of priority areas such as malaria.
WHO’s Special Programme for Research and Training in Tropical diseases (TDR) commissions studies to be conducted in the geographical regions most affected by such diseases. If such R&D is to result in marketing approval of effective and safe new drug products, the component studies must comply with current research practice standards ensuring the quality, reliability and integrity of study data. Market authorisation regulations require that quality standards, i.e. Good Manufacturing Practice (GMP), Good Laboratory Practice (GLP) and Good Clinical Practice (GCP), are followed in the respective stages of the development and life-cycle of a drug product.
WHO published standards for Good Manufacturing Practice (GMP)1 in 1999 (covering the manufacture of a drug product) and Good Clinical Practice (GCP)2 in 1995 (covering clinical trials in man). However, until the publication of the first version of this handbook in 2001, WHO had not addressed quality standards for non-clinical testing for the safety of potential products : Good Laboratory Practice (GLP). This handbook, and its associated training volumes, specifically address this gap in WHO recommendations.
The introduction of GLP quality standards in test facilities of developing countries was seen as an urgent issue and, accordingly, WHO convened a working party (Scientific Working Group on GLP issues – SWG) in 1999 and 2000 to address the WHO position on GLP.
During the SWG discussions it became evident that, for test facilities in developing countries, the introduction of GLP could be impeded by resource constraints (e.g. few trained personnel, inadequate facilities and equipment) or by the instability of the infrastructure (e.g. water or electricity supply), either within the testing laboratory itself or in the community as a whole. However, GLP could result in tangible returns through the number of studies placed with research organisations in DECs, resulting in an overall increase in funding. It is clear that as funding is scarce sponsors will not invest in studies if the reliability of results cannot be assured. Specifically, WHO/TDR will be reluctant to allocate their limited funding to non-clinical safety studies unless the results can be reliable on and thus support decisions concerning the progress of products to clinical stages and eventually to product registration.
The deliberations of the Scientific Working Group on GLP issues underlined the following points :
• In DECs, demonstrating compliance with GLP will become a prerequisite for nonclinical safety testing and for drug registration particularly where drug products are projected for markets other than the country of origin;
1 Quality assurance of pharmaceuticals: A compendium of guidelines and related materials. Volume 2
Good manufacturing practices and inspection, WHO Geneva 1999
2 Guidelines for good clinical practices (GCP) for trials on pharmaceutical products. WHO Geneva 1995
• It is essential to avoid the co-existence of two or more international GLP regulatory standards for non-clinical safety testing;
• Guidance is needed for the implementation of GLP.
With such considerations in mind the SWG recommended that WHO/TDR adopt the Revised OECD Principles of Good Laboratory Practice as its official guidance for nonclinical safety testing. The handbook sets forth the OECD Principles in their original text, supplemented by sections on training and the implementation of GLP.
The drug discovery and development process: non-regulated vs. regulated research
The drug discovery and development process can be divided into a number of distinct stages which may overlap in time (e.g. clinical Phase I studies may be started before the completion of toxicology studies of longer duration; oncogenicity studies may not even have been started at this point).
Typically, the process starts with basic discovery activities, the results of which may then be used to define efficacy targets for the potential drug. The discovery phase often involves thousands or even tens of thousands of new molecular entities (NMEs) being screened for activity against a target disease. The ten or twenty successful NMEs are then checked for their potential toxic effects, again in screening-type tests, further reducing the number of potential drug substances taken forward to full development. In countries without an established pharmaceutical industry, the discovery process may be different; the initial identification of potential compounds is likely to come from a medical or scientific research institution, possibly attached to a university or centre of learning. For example, a population may traditionally use a plant remedy for certain indications. After observational studies to ascertain whether the practice is sound, one could set up chemical studies to find the active principles, and perhaps prepare a set of chemical analogues. In this case the number of starting compounds would be more modest, but this does not fundamentally alter the process. The need for rigorous testing further along in the development pathway will remain the same. Studies performed subsequent to this selection contribute to the overall assessment of safety and efficacy of the candidate compound.
In the R&D stages downstream of discovery, the investigations are regulated by internationally accepted guidelines and quality requirements.
The different steps in classical drug development (drug life-cycle) are characterised by four well-defined stages, which are summarised in the diagram below.
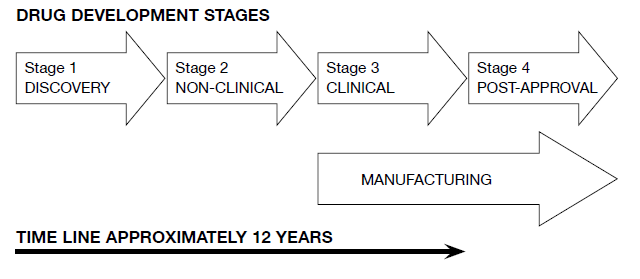
Stage 1
The first stage, the discovery of a potential NME, is not covered by a regulatory standard, nor are studies that demonstrate proof of concept. The WHO has recently published guidance on this early research phase : Quality Practices in Basic Biomedical Research – QPBR.
Stage 2
The position of GLP studies within the drug development process is specific to the second stage. These studies are termed “non-clinical” as they are not performed in humans. Their primary purpose is safety testing. Toxicology and safety pharmacology studies, with a potential extension to pharmacokinetics and bioavailability, are those studies where compliance with GLP is required. From the diagram above, the somewhat restricted scope of
GLP is evident.
Stage 3
The third stage, following on from safety studies of stage 2, encompasses clinical studies in human subjects. Here, GCP is the basic requirement for quality standards, ethical conduct and regulatory compliance. GCP must be instituted in all clinical trials from Phase I (to demonstrate tolerance of the test drug and to define human pharmacokinetics) through Phase II (where the dose-effect relationship is confirmed) to Phase III (full scale, often multi-centric, clinical efficacy trials in hundreds or thousands of subjects).
Stage 4
The fourth stage is post-approval. Here the drug has been registered and is available on the market. However, even after marketing approval, the use of the drug is monitored through formal pharmacovigilance procedures. Any subsequent clinical trials (Phase IV) must also comply with GCP.
Good Manufacturing Practice (GMP)
From stage 3 of development and continuing throughout the rest of the drug’s lifetime, GMP applies to all manufacturing of Active Pharmaceutical Ingredients (API – drug substance) and formulated medicines (drug product).
The scope of this handbook is restricted to the GLP-regulated area (stage 2 of the
above diagram) i.e. to the “ ... the non-clinical safety testing of test items contained in pharmaceutical products ... required by regulations for the purpose of registering or licensing ...The purpose of testing these test items is to obtain data on their properties and/or their safety with respect to human health and/or the environment.” (OECD Principles of GLP).