

Список використаної літератури
Воюцкий С. С. Курс коллоидной химии [Текст] : учебник / С. С. Воюцкий. – 2-е изд., перераб. и доп. – М. : Химия, 1975. – 512 с. : ил.
Запольский А.К. Фізико-хімічні основи технології очищення стічних вод: [Текст]: Підручник. / А.К. Запольский, Н.А. Мішкова-Кліменко, І.М. Астрелін. – К.: Лібра, 2000. – 552 с.
Фролов Ю. Г. Курс коллоидной химии. Поверхностные явления и дисперсные системы [Текст] : учебник / Ю. Г. Фролов. – 2-е изд., перераб. и доп. – М. : Химия, 1989. – 464 с. : ил. – (Для высшей школы). – ISBN 5-7245-0244-5
Додаток б Методи дослідження каталізаторів і гетерогенно-каталітичних реакцій
При дослідженні гетерогенно-каталітичних процесів промислового значення здійснюють підбір каталізаторів за їх активністю і селективністю, визначають їхню структуру (розмір поверхні, пористість, розподіл об’єму пор за розміром пор) і механічну міцність, дістають дані про кінетику хімічних і фізичних стадій процесу, необхідні для переходу від лабораторного реактора до промислового і розрахункового вибору оптимального режиму роботи останнього.
Величина питомої площі поверхні каталізаторів, сумарний об’єм пop і розподіл їх за радіусами належать до найважливіших характеристик промислових контактних мас.
Визначення питомої площі поверхні каталізаторів. Для визначення питомої площі поверхні каталізаторів Sпит найширше використовується полімолекулярна фізична адсорбція газів за температур, близьких до температур їх кипіння (тобто за умов, коли гази фактично є парами). Теоретичні відомості відносно полімолекулярної та мономолекулярної адсорбції наведено в додатку А.
Рівняння взаємозв’язку об’єму газу Vr, адсорбованого за ізотермічних умов, з його рівноважним тиском Р описується рівнянням БЕТ:
|
(Б.1) |
де Р0 – тиск насиченої пари адсорбату;
а – стала рівняння БЕТ.
Параметр
![]() характеризує кількість
молекул пари, необхідних для покривання
адсорбенту щільним моношаром. Цей
параметр називають, також об’ємом
або місткістю
моношару (в останньому
випадку його виражають через кількість
грам адсорбату, що припадають на 1 г
адсорбента).
характеризує кількість
молекул пари, необхідних для покривання
адсорбенту щільним моношаром. Цей
параметр називають, також об’ємом
або місткістю
моношару (в останньому
випадку його виражають через кількість
грам адсорбату, що припадають на 1 г
адсорбента).
За експериментальними даними, ізотермами мономолекулярної ленгмюрівської адсорбції (рисунок Б.1, І) описується тільки незначна кількість процесів низькотемпературної фізичної адсорбції. Більшість, процесів характеризуються ізотермами полімолекулярної адсорбції типу ІІ, за формою яких, використовуючи теорію БЕТ, можна визначити питому площу поверхні адсорбенту. На таких експериментальних ізотермах є прямолінійний відрізок ВС. Точка В, у якій він починається, вказує на закінчення заповнення моношару (теорія БЕТ дозволяє знайти координати цієї точки аналітичним способом).
Пряма,
побудована згідно з рівнянням БЕТ у
координатах
![]() –
–
![]() (див. рисунок Б.1, ІІІ),
відтинає на осі ординат відрізок
(див. рисунок Б.1, ІІІ),
відтинає на осі ординат відрізок
![]() і
має кутовий коефіцієнт
і
має кутовий коефіцієнт
![]() .
За цими двома величинами можна обчислити
Vм
і а. Значення
сталої а залежить
від зміни енергії Гіббса, зумовленої
переходом пари, що перебуває у стані
рівноваги з рідиною, на поверхню
адсорбента (процес супроводжується
виділенням теплоти адсорбції першого
шару q1
і прихованої теплоти конденсації
адсорбату q2):
.
За цими двома величинами можна обчислити
Vм
і а. Значення
сталої а залежить
від зміни енергії Гіббса, зумовленої
переходом пари, що перебуває у стані
рівноваги з рідиною, на поверхню
адсорбента (процес супроводжується
виділенням теплоти адсорбції першого
шару q1
і прихованої теплоти конденсації
адсорбату q2):
![]() .
.

Рисунок Б.1– Ізотерми мономолекулярної (І) і полімолекулярної (ІІ, ІІІ) адсорбції
Для ізотерм типу ІІ (рисунок Б.1) значення а зазвичай перевищує 2.
Місткість
моношару
входить у вираз для обчислення
![]() (м2/г):
(м2/г):
![]()
де М – молярна маса адсорбату;
NA – число Авогадро. NA=6·1023;
SM – площа, яку займає адсорбована молекула на поверхні адсорбенту у заповненому моношарі (площа поперечного перерізу, ефективна молекулярна площа), м2, так для молекули бензолу SM=39·10-20.
При достатньо великій Sпит (до сотні квадратних метрів на один грам адсорбенту) для її визначення використовують адсорбцію азоту. При температурах 77–78 К азот дає високі значення сталої а (внаслідок великих значень теплоти адсорбції а = 220), що визначає крутий злам ізотерм II типу і досить точне визначення положення точки В навіть при короткому лінійному відрізку BС (рисунок Б.1). Це дає змогу перевірити правильність розрахунку величини Vм за рівнянням БЕТ, порівнюючи її з величиною Vм, знайденою за експериментальною ізотермою.
Для визначення малих Sпит (порядку кількох квадратних метрів на один грам адсорбенту) доцільніше використовувати аргон, криптон, ксенон, для пари яких Р0 достатньо малий, що дозволяє легко досягнути значень Р/Р0, необхідних при роботі з експериментальними вимірювальними установками.
Внаслідок непридатності методу БЕТ для роздільного визначення Sпит окремих компонентів каталізаторів і носіїв (для каталізаторів на носіях) рекомендується застосовувати методи, що базуються на високотемпературній (273 К і вище) хемосорбції водню, кисню, оксиду вуглтецю (II).
Для експериментального визначення кількості адсорбованої пари використовуються титрометричні (вимірюється зміна об’єму адсорбату), гравіметричні (визначається збільшення маси адсорбенту за рахунок адсорбції газу) і газохроматографічні (визначається за зміною складу газу в результаті адсорбції або десорбції) методи.
Визначення об’єму і розмірів пop. Знаючи розміри пop, можна визначити наявність (або відсутність і ступінь внутрішньо-дифузійного гальмування за конкретних умов каталітичного процесу, а також ступінь використання внутрішньої поверхні каталізатора, яка обернено пропорціональна радіусу пор. При визначенні характеристик пор найширше застосовується адсорбційний метод, що базується на зниженні тиску пари (порівняно з тиском насиченої пари адсорбату Ps) над циліндричним стовпчиком рідини у вузькій порі (капілярі) з радіусом rп, що властиве капілярній конденсації.

Рисунок Б.2 – Ізотерми адсорбції і десорбції пари бензолу на крупнопористому силікагелі при 293 К
Це зниження можна описати рівнянням Кельвіна:
![]()
де Vm – молярний об’єм рідини (конденсату);
rК – середній радіус Кельвіна (радіус циліндра, що відповідає даному значенню Ps/P);
σ – поверхневий натяг.
Експериментальні ізотерми адсорбції і десорбції пари на пористому каталізаторі, подані в координатах питома кількість адсорбованої речовини а – відносний тиск пари Р/Рs (рисунок Б.2), не співпадають і утворюють гістерезисну петлю. Це цілком можна пояснити, оскільки при адсорбції, що супроводжується капілярною конденсацією, складова поверхневого натягу в останньому рівнянні дорівнює σ∙cosθ3, а не σ, причому крайовий кут змочування θ3 між твердою речовиною і рідиною відрізняється від нуля. В результаті тиск рівноважної адсорбції Ра в області капілярної конденсації більший за відповідний тиск десорбції Рд (десорбція відбувається із цілком заповнених пop; θ3 = 0).
На етапі експериментального визначення розміру пор каталізатора необхідно провести адсорбцію (до Р/Рs ≈ 1) і десорбцію з наступним використанням для розрахунку десорбційної вітки, оскільки при цьому не виникає необхідність у поправці на кут змочування. Множенням значень G, знайдених експериментально, на Vm визначають об’єм пop, а підставлянням у рівняння (2) відповідного значення Р/Рs дістають rк.
Рівняння Кельвіна не дає
точного значення rп:
величина rК.
менша за rп
на товщину адсорбованого полімолекулярного
шару δ,
тобто
![]() .
.
У наближенні сталості δ в усій області Р/Рs, в якій відбувається капілярна конденсація, для моделі каталізатора з циліндричними або конічними порами можна записати
![]()
де Vm – об’єм 1 ммоль зрідженої пари за температури досліду, см3; Sпит – площа питомої поверхні каталізатора, обчислена за методом БЕТ, см2/г.

Рисунок Б.3 – Інтегральний (а) і диференціальний (б) розподіл об’єму nop за радіусами для силікагелю
Значення середньостатистичної товщини адсорбційного шару відповідає початку гістерезису. На кожному етапі десорбції при сталості δ спостерігається така залежність:
д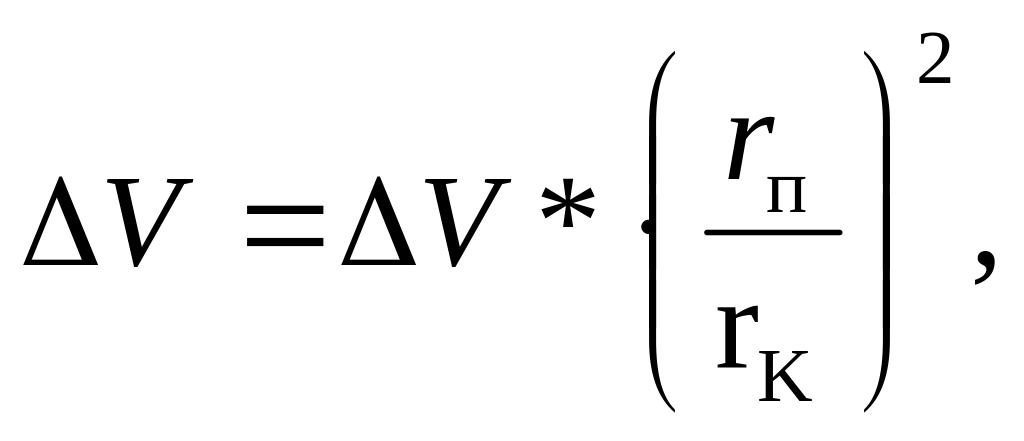 е
ΔV –
зміна сорбції, виражена об’ємом зрідженої
пари при температурі досліду; ΔV*
– приріст об’єму вільних пор.
е
ΔV –
зміна сорбції, виражена об’ємом зрідженої
пари при температурі досліду; ΔV*
– приріст об’єму вільних пор.
Розподіл об’єму пор за радіусами виражають відношенням ΔV*/ΔVrп (або dV*/drп). Цей розподіл можна подати інтегральними або диференціальними кривими (рисунок Б.3).
Об’єм пор Vпит та їхні еквівалентні радіуси визначають також способом ртутної порометрії, що базується на вимірюванні об’єму ртуті, що заповнює під тиском пори сорбента. Зовнішню поверхню ртуть не вкриває, оскільки вона не змочує більшість твердих тіл.
Кінетичні дослідження
Найважливішими характеристиками будь-якого каталізатора є його активність і селективність. Активність каталізатора вимірюється швидкістю перебігу реакції при його наявності, тобто кількістю основної вихідної речовини (реагенту) Gp, що реагує за одиницю часу на одиниці площі поверхні каталізатора S:
Я![]() кщо
швидкість реакції виражається через
концентрацію реагенту с,
або через його початкову
концентрацію с0
і ступінь перетворення
реагенту х, то
швидкість реакції становитиме
кщо
швидкість реакції виражається через
концентрацію реагенту с,
або через його початкову
концентрацію с0
і ступінь перетворення
реагенту х, то
швидкість реакції становитиме
![]()
Для визначення активності одиниці площі поверхні каталізатора необхідно виміряти всю його площу внутрішньої поверхні і повністю використати її в реакції, тобто проводити процес у кінетичній області. Для промислового процесу активність каталізатора найчастіше обчислюють відносно одиниці об’єму його шару (Vкат):
![]()
Внаслідок невизначеності об’єму киплячого шару каталізатора активність останнього обчислюють відносно одиниці його маси.
Селективність (вибірковість) каталізатора визначається як відношення швидкості утворення цільового продукту до швидкості реагування вихідної речовини (тобто до активності каталізатора). Можна визначити селективність через відношення швидкості цільової реакції до швидкості побічної реакції.
Активність і селективність характеризують не тільки сам каталізатор, але й умови реалізації гетерогенно-каталітичного процесу, оскільки залежать від області його перебігу і технологічних параметрів (температури, концентрації реагентів тощо).
Багаточисельні методи дослідження кінетики гетерогенно-каталітичних реакцій можна розділити на дві основні групи:
1) статичні, що здійснюються в замкнутих системах, тобто у реакторах періодичної дії;
2) динамічні, що здійснюються за стаціонарних умов у реакторах безперервної дії (проточних).
Статичні методи. Реакцію проводять у замкнутому об’ємі до досягнення термодинамічної рівноваги або до повного витрачання одного з вихідних реагентів. Кінетику реакції спостерігають за зміною концентрації компонентів реакційної суміші у часі. Особливо зручні статичні методи при дослідженні реакцій, що відбуваються із зміною об’єму, оскільки при цьому можна стежити за ходом реакції за зміною тиску.
Будь-який статичний метод має інтегральний характер, оскільки дає змогу вимірювати лише зміну концентрації речовини за який не будь проміжок часу, а ці дані є інтегралом швидкості реакції в часі або вздовж шару каталізатору. Для визначення ж швидкостей реакцій необхідно проводити диференціювання дослідних даних, що подаються зазвичай у вигляді кінетичних кривих у координатах с – τ.
При дослідженні кінетики гетерогенно-каталітичних реакцій, що відбуваються між газоподібними речовинами, статичний метод зазвичай використовують в циркуляційному варіанті, коли газова суміш безперервно прокачується через каталізатор, циркулюючи у замкнутій системі. Замкнутість системи дає змогу зводити баланс речовин за різницею, що особливо цінно при вивченні складних реакцій, де не всі компоненти вдається визначати аналітично. Примусова ж циркуляція реагентів дає змогу застосовувати дрібнозернистий каталізатор і цим самим усувати виникнення внутрішньо-дифузійних опорів. Варіюванням циркуляції можна досягти усунення зовнішньо-дифузійного гальмування процесу або хоча б виявити межі його дії.
Основним недоліком будь-яких статичних систем є обмеження можливості їх застосування для дослідження лише таких каталізаторів, стаціонарність стану яких або не залежить від реакційного середовища (цей випадок, згідно з теорією Г. К. Борєскова є рідкісним), або ж установлюється із швидкістю вищою, ніж швидкість реакції. При приблизній рівності часу досягнення стаціонарного стану і тривалості реакції результати досліджень будуть спотворені нестаціонарністю властивостей каталізатора, що спричинить невідповідність справжньої динаміки реакції і кінетичних рівнянь, отриманих на основі допущення про стаціонарність властивостей каталізатора.
Якщо ж час досягнення стаціонарного стану каталізатора значно перевищує тривалість експерименту, то приймається допущення про квазістаціонарність стану каталізатора і придатність отриманих кінетичних залежностей для адекватного описування процесу.
Обмежене застосування статичних методів при вивченні активності промислових каталізаторів зумовлено також неконтрольованими спотвореннями кінетичних даних можливими перепадами температур і концентрацій у реакційному просторі.
Динамічні (проточні) методи. Проточні методи дослідження кінетики гетерогенно-каталітичних реакцій реалізуються за стаціонарних умов процесу в реакторах безперервної дії. Реакційну суміш пропускають з певною швидкістю через шар каталізатора при сталості вихідних концентрацій, температур, тисків, умов перемішування в кожному досліді й аналізують склад суміші на вході в реактор або по його довжині. При практичному здійсненні проточних методів використовують інтегральні і диференціальні реактори.
Проточні інтегральні реактори найчастіше являють собою заповнені каталізатором трубки, аналогічні тим, що застосовуються в промислових апаратах і досить близькі до них за умовами роботи. Стадія модельної проточної установки необхідна при розробці промислових гетерогенно-каталітичних процесів, оскільки дає змогу крім хімічних і кінетичних даних вивчити технологічні особливості процесу, напрацювати зразки цільового продукту, дістати відомості про тривалість стійкої роботи каталізатора тощо.
Застосування проточного інтегрального реактора для кінетичних досліджень базується на дещо спрощеному допущенні про рух газу в нерухомому шарі каталізатора у режимі ідеального витіснення (при нехтуванні радіальними градієнтами концентрації, тиску і температури) і при квазістаціонарному стані системи.
Хід кінетичних досліджень у
проточних інтегральних реакторах
зводиться до вимірювання концентрації
реагентів (або продуктів) сг
і температури реагуючої суміші Т
у послідовних точках
реакційного шару з координатами l,
що вимірюються часом τ, за який потік з
лінійною швидкістю ω проходить відстань
від входу до даного перерізу: τ = l/ω.
Варіювання часу
досягається зміною швидкості потоку.
Експериментальна кінетична залежність
має вигляд
![]() .
Графічним або більш точним чисельним
диференціюванням із застосуванням ЕОМ
визначають залежність швидкості реакції
.
Графічним або більш точним чисельним
диференціюванням із застосуванням ЕОМ
визначають залежність швидкості реакції
![]() від концентрації реагентів
(або продуктів) і температури:
від концентрації реагентів
(або продуктів) і температури:
![]() .
.
Кінетика процесу стає відомою,
коли знайдено такі функції
![]() ,
інтегральні криві
(розв’язки) яких мало відрізняються
від експериментальних кінетичних кривих
,
інтегральні криві
(розв’язки) яких мало відрізняються
від експериментальних кінетичних кривих
![]() .
.
Зазвичай проведення дослідів на проточних інтегральних реакторах дає великий об’єм інформації, що дозволяє скласти математичний опис гетерогенно-каталітичних процесів з високим ступенем надійності і тим самим успішно розв’язати задачу переходу від лабораторного або дослідного реактора до промислового реактора будь-якої конструкції та оптимізувати режим роботи останнього.
У проточних диференціальних реакторах безпосередньо визначають швидкість реакції. Для створення диференціального реактора при дослідженні гетерогенно-каталітичних реакцій за участю газоподібних реагентів застосовують тонкий шар каталізатора (Δl) при великих швидкостях прокачування через нього реакційної суміші, завдяки чому зміна ступеня перетворення у шарі невелика при будь-яких змінах в експерименті початкових концентрацій реагенту с0. Тоді кількість (або зміна концентрації) перетвореної речовини може слугувати мірою швидкості реакції:
![]()
Слід зазначити, що використання для розрахунку швидкості реакції в диференціальних реакторах описаного типу малої концентраційної різниці реакційної суміші на вході в шар і на виході з шару каталізатора не забезпечує достатньої точності кінетичних даних.
Значно точніше каталітичну активність можна виміряти в безградієнтних проточно-циркуляційних реакторах. У таких реакторах забезпечується інтенсивна циркуляція реакційної суміші через каталізатор у замкнутому об’ємі при безперервному введенні і виведенні газового потоку і значному перевищенні витрати циркулюючого газу порівняно з витратою вихідного газу, що вводиться у систему.
Високі лінійні швидкості газів у циклі і малі ступені перетворення в диференціальному шарі каталізатора зумовлюють практичну відсутність або ж мінімальні градієнти концентрацій і температур у шарі. Реактор у цьому випадку працює в режимі, аналогічному повному змішуванню (тобто без зовнішньо дифузійних гальмувань). Пряме вимірювання швидкості реакції можна проводити у кожному досліді тривалістю τ за значною різницею концентрацій суміші, що надходить у циркуляційний контур і виходить із нього, що різко зменшує вплив похибок аналізу на точність визначення швидкості реакції порівняно з проточними диференціальними реакторами. Кінетичні залежності з урахуванням характеристичного рівняння реактора ідеального змішування безперервної дії (РІЗ-Б), що працює в установленому режимі, матимуть вигляд:
![]()
де τ – час досліду;
k – константа швидкості реакції;
х– ступінь перетворення;
п – порядок реакції,
с0 –початкова концентрація.
Крім розглянутих аспектів, активність і селективність каталізаторів можна оцінити, застосовуючи інші методи та реактори (наприклад, імпульсний метод з використанням газового хроматографа, реактор із суспендованим шаром каталізатора тощо). Вибір методу або типу реактора для проведення кінетичних досліджень конкретної гетерогенно-каталітичної реакції диктується, в основному, прагненням забезпечити адекватне моделювання виробничих умов.
Завдання перенесення результатів лабораторних досліджень гетерогенно-каталітичних реакцій у заводську практику найнадійніше вирішуються розробкою математичних моделей хімічних реакторів, що включають ідентифікований опис структури потоку (його гідродинаміки) в реакторі і кінетичні залежності реакції. Ідентифікація гідродинамічної обстановки полягає в установленні відповідності реальної структури потоку в реакторі якій-небудь з відомих моделей: ідеального витіснення, ідеального змішування, дифузійної тощо. Розв’язок цієї задачі дозволяє скласти математичний опис хімічного реактора у вигляді системи рівнянь матеріального і теплового балансів.
Визначення кінетичної складової математичних моделей здійснюється за експериментальними даними, одержаними за допомогою описаних вище диференціальних або інтегральних реакторів, розв’язуванням так званої оберненої задачі хімічної кінетики. Процедура розв’язування такої задачі включає: вибір можливих варіантів кінетичної схеми (із урахуванням області перебігу процесу); статистичну оцінку параметрів математичного опису кінетики за отриманими експериментальними даними; оцінку прийнятих гіпотез про механізм реакції і планування додаткових експериментів для зменшення довірчих інтервалів кінетичних параметрів і вибору схеми, що адекватно описує кінетику процесу у досліджуваній області режимних параметрів. Ітераційний процес вибракування неадекватних моделей проводиться до одержання математичної моделі, яка б не мала протиріччя всій сукупності експериментальних даних.