

5. Специфический обмен глицина и серина. Их роль в биосинтезе биологически важных веществ.
Глицин является единственной из всех входящих в состав белков аминокислот, в молекуле которой отсутствует асимметричный атом углерода. Тем не менее метаболически он связан с химическими компонентами организма в большей степени, чем любая другая аминокислота.
Показано, что в реакции взаимопревращения глицина и серина участвует тетрагидрофолиевая кислота; эту реакцию катализирует пиридоксалевый фермент серин-оксиметилтрансфераза:

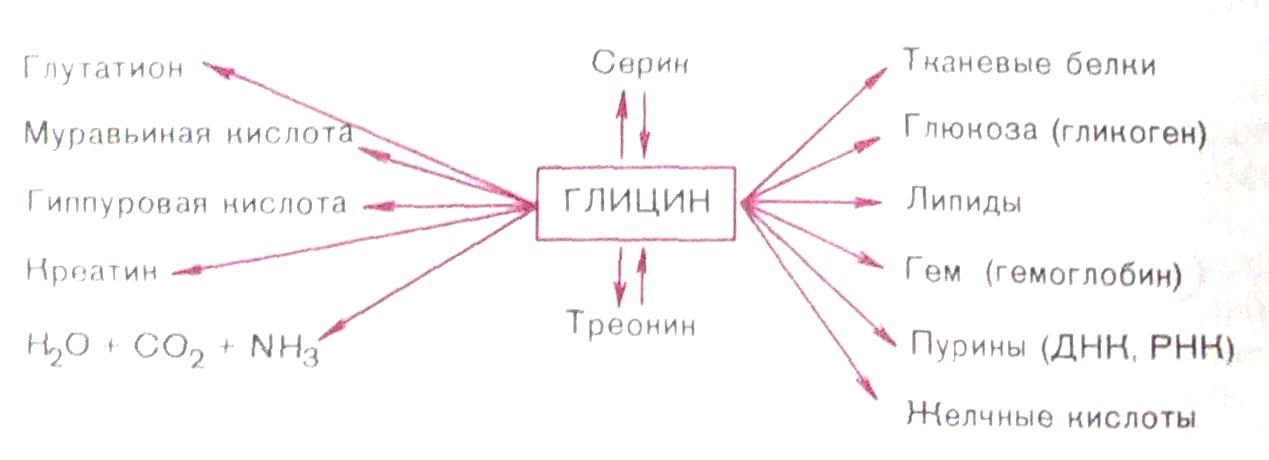
На схеме видно, что глицин в некоторых синтезах играет незаменимую роль, в частности в образовании белков, пуриновых нуклеотидов, гема гемоглобина, парных желчных кислот, креатина, глутатиона и др. Большинство этих реакций представлено в соответствующих разделах учебника. Здесь укажем на реакции, при помощи которых осуществляются взаимопревращения глицина, серина и треонина, а также на реакции катаболизма Имеются также доказательства взаимопревращения треонина и глицина в треонинальдолазной реакции:

Основным путем катаболизма глицина в животных тканях, однако, считается распад его на С02, NH3 и N5,-метилентетрагидрофолиевую кислоту по уравнению:
![]()
Механизм этой реакции, недавно раскрытый К. Тада, включает участие митохондриальной глицинрасщепляющей ферментной системы, отличной от глицинсинтазы и состоящей из 4 белков: Р-белка, содержащего пиридоксальфосфат (глициндекарбоксилаза); Н-белка, содержащего липое-вую кислоту; Т-белка, требующего присутствия ТГФК, и L-белка, названного липамиддегидрогеназой:

Доказательства что наследственная некетогенная глициномии (повышение уровня глицина в крови) обусловлена недостаточностью Р- или Т-бедка глицин расщепляющей ферментной системы пенсии или мозга и что
каждый из этих белков контролируется отдельным геном. Серии легко превращается в пиру ват под действием сериндегндратазы. В связи с этим в тканях имеются условия для превращения глицина (через серии) в пиру ват. Этим путем осуществляется участие глицина в обмене углеводов. Важную роль играет серии в биосинтезе сложных белков-фосфоиротеинов, а также фоофоглицеридов. Помимо фоефатидилсерина, углеродный скелет и азот серина используются в биосинтезе фосфатидид-этаноламина и фосфатидилхолина.
6. Специфический обмен фенилаланина и тирозина. Образование биологически активных продуктов. Молекулярная патология (фенилкетонурия, алкаптонурия, альбинизм).
Фенилаланин-
незаменимая аминокислота, т.к. ткани
животных не обладают способностью
синтезировать его бензольное кольцо.
А тирозин полностью заменим при
достаточном поступлении фенилаланина
с пищей. Объясняется это тем, что основной
путь превращения фенилаланина начинается
с его окисления (точнее, гидроксилирования)
в тирозин Реакция гидроксилирования
катализируется специфической
фенилаланин-4-монооксигеназой, которая
в качестве кофермента содержит
тетрагидробиоптерин. Блокирование этой
реакции, наблюдаемое при нарушении
синтеза фенилаланин-4-монооксигеназы
в печени, приводит к развитию тяжелой
наследственной болезни – фенилкетонурии
(фенилпировиноградная олигофрения). В
процессе трансаминирования тирозин
превращается в n-оксифенилпировиноградную
кислоту, которая под действием
специфической оксидазы подвергается
окислению, декарбоксилированию,
гидроксилированию и внутримолекулярному
перемещению боковой цепи с образованием
гомогентизиновой кислоты; эта реакция
требует присутствия аскорбиновой
кислоты, роль которой пока не выяснена.
Дальнейшее превращение гомогентизиновой
кислоты в малеилацетоуксусную кислоту
катализируется оксидазой гомогентизиновой
кислоты. Малеилацетоуксусная кислота
под действием специфической изомеразы
в присутствии глутатиона превращается
в фумарилацетоуксусную кислоту,
подвергающуюся гидролизу с образованием
фумаровой и ацетоуксусной кислот,
дальнейшие превращения которых уже
известны.
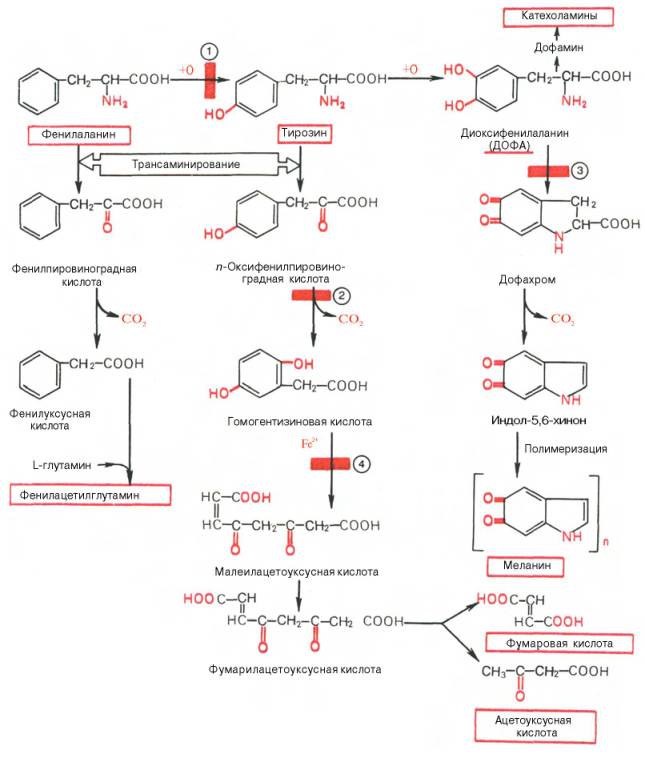
Цифры в кружках - участки блокирования реакций при фенилкетонурии (1), тирозинозе (2), альбинизме (3) и алкаптонурии (4).
Фенилаланин и тирозин являются предшественниками меланинов, обеспечивающих пигментацию кожи, глаз, волос, активное участие принимает фермент тирозиназа.
Фенилкетонурия (фенилпировиноградная олигофрения) развивается как результат потери способности организма синтезировать фенилаланин-4-монооксигеназу, катализирующую превращение фенилаланина в тирозин. Характерные особенности болезни – резкое замедление умственного развития ребенка, а также экскреция с мочой больших количеств фенил-пировиноградной кислоты (до 1–2 г/сут) и фенилацетилглутамина (до 2–3 г/сут). Решающим доказательством метаболического блока при фенилкетонурии являются данные о накоплении фенилаланина в тканях. Так, количество его в крови может достигать 600 мг/л (в норме 15 мг/л), в цереброспинальной жидкости – 80 мг/л (в норме 1,5 мг/л). Развитие болезни можно предотвратить, если значительно снизить прием фенилаланина с пищей с самого рождения ребенка.
Алкаптонурия характеризуется экскрецией с мочой больших количеств (до 0,5 г/сут) гомогентизиновой кислоты, окисление которой кислородом воздуха придает моче темную окраску. В далеко зашедших случаях развиваются охроноз, наблюдаются отложение пигмента в тканях и потемнение носа, ушей и склеры. Этот дефект связан с врожденным отсутствием в печени и почках оксидазы гомогентизиновой кислоты.
Альбинизм – врожденное отсутствие пигментов в коже, волосах и сетчатке. Метаболический дефект связан с потерей меланоцитами способности синтезировать тирозиназу – фермент, катализирующий окисление тирозина в диоксифенилаланин и диоксифенилаланинхинон, являющихся предшественниками меланина.