

Лекция № 5. Физико-химические основы гидрогенизационных процессов
• Гидрирование
• Гидроочистка
• Гидрокрекинг (алканов, алкенов, циклоалканов, аренов)
Процесс гидрогенизации углевородного сырья можно рассматривать как крекинг в присутствии избытка водорода, протекающий при высоких давлениях; свободный водород присоединяется к образовавшимся в результате крекинга продуктам, так что общее содержание в них водорода превышает содержание его в исходном сырье. В этом основное различие процессов гидрогенизации и собственно крекинга.
В нефтехимической промышленности с помощью реакций гидрирования получают циклогексан и его производные, многие амины, спирты и ряд других мономеров.
Гидрогенизационные процессы развиваются в двух направлениях: гидроочистка нефтяных фракций и деструктивное гидрирование тяжелых нефтяных дистиллятов и остатков (гидрокрекинг, гидродеалкилирование).
Гидрирование. Непредельные углеводороды легко гидрируются в присутствии катализаторов даже при комнатной температуре. В первую очередь гидрированию подвергаются алкадиены, затем алкены, имеющие двойную связь в конце цепи, и, наконец, алкены с двойной связью в середине молекулы.
Алкины так же как и алкены, легко вступают в реакции с водородом. Ацетилен в присутствии никелевого катализатора начинает реагировать с водородом уже при комнатной температуре, но лучше идет при 150оС:
СНСН
![]() СН2=СН2
СН3СН3
СН2=СН2
СН3СН3
Как и в случае алкенов, тройные связи на конце цепи гидрируются быстрее, чем внутренние.
Бензольные кольца гидрируются значительно труднее, чем непредельные алифатические соединения. Присоединение водорода к любой двойной связи экзотермично, гидрирование же бензола в 1,2-дигидробензол эндотермично. Далее дигидробензол гидрируется легче и экзотермически:
С6Н6 + Н2 С6Н8 - 24 кДж/моль
С6Н8 + Н2 С6Н10 +110 кДж/моль
С6Н10 + Н2 С6Н12 +119 кДж/моль
_________________________________
С6Н6 + 3Н2 С6Н12 + 206 кДж/моль
Гомологи бензола гидрируются труднее, чем бензол.
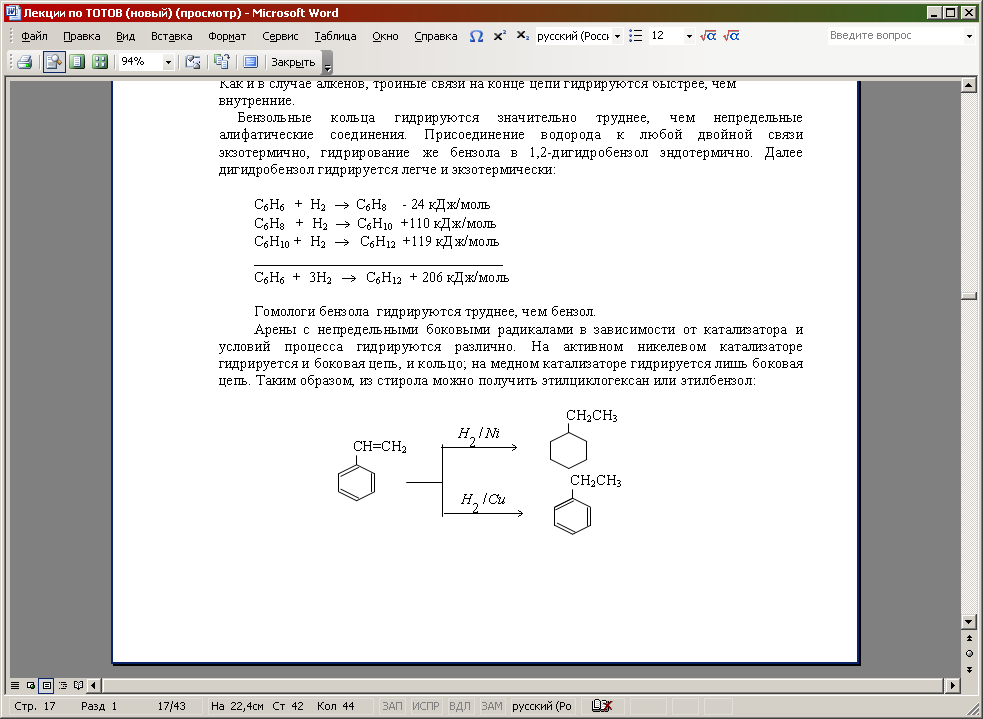
Арены с непредельными боковыми радикалами в зависимости от катализатора и условий процесса гидрируются различно. На активном никелевом катализаторе гидрируется и боковая цепь, и кольцо; на медном катализаторе гидрируется лишь боковая цепь. Таким образом, из стирола можно получить этилциклогексан или этилбензол:
Конденсированные арены гидрируются легче бензольных. Основной принцип при гидрирования конденсированных ароматических систем – последовательность насыщения бензольных колец водородом, причем по мере насыщения скорость реакции снижается:

Наряду с последовательным гидрированием ароматических колец возможно расщепление образовавщихся насыщенных колец и выделение алкилзамещенных аренов.
Гидроочистка – процесс удаления из нефтепродуктов гетероатомов в результате гидрирования серу-, азот-, кислородсодержащих соединений. Одновременно гидрируются диены, алкены и отчасти полициклические арены и удаляются металлы, содержащиеся в виде металлорганических соединений. Гидроочистку проводят над гидрирующими серастойкими катализаторами.
Гидрокрекинг (деструктивное гидрирование, гидродеалкилирование) обычно осуществляется на бифункциональных катализаторах, активных как в реакциях гидрирования, так и в реакциях крекинга. Крекирующую функцию катализатора обеспечивают соединения кислотного характера, направляющие реакцию по карбкатионному механизму (оксид алюминия, алюмосиликаты, цеолиты), а гидрирующую – в основном металлы VIII группы (Fe, Co, Ni, Pt, Pd и др.).
Гидрокрекинг алкенов. Алкены в кислотных центрах катализатора превращаются в карбкатионы и вступают в реакции, характерные для этих частиц. Они изомеризуются и подвергаются распаду по -правилу. Одновременно на гидрирующих центрах катализатора происходит насыщение алкенов. На катализаторах с высокой кислотной активностью образуются низкомолекулярные разветвленные соединения, главным образом изобутан. На катализаторах с высокой гидрирующей активностью образуются малоразветвленные алканы с большой молекулярной массой.
Гидрокрекинг алканов. Алканы сначало подвергаются деструкции и часто изомеризации. Образующиеся осколки и двойные связи сразу же насыщаются водородом. Реакция деструктивного гидрирования алканов может быть описана уравнением:
CnH2n+2 + H2 CmH2m+2 + Cn-mH2(n-m)+2
В процессе гидрокрекинга возможен разрыв любой С–С-связи в молекуле алкана. Термодинамические расчеты показывают, что наибольшую константу равновесия имеет реакция отщепления метана. Однако в промышленных процессах основное значение имеют кинетические факторы, и скорость разрыва различных С–С-связей зависит от выбранного катализатора.
Тепловой эффект деструктивного гидрирования суммируется из эндотермических реакций крекинга и экзотермических реакций гидрирования и может колебаться в широких пределах.
Считают, что гидрокрекинг алканов на бифункциональных катализаторах протекает в несколько стадий. Вначале на активных центрах гидрирования – дегидрирования происходит дегидрирование углеводородов с образованием алкенов в очень малой концентрации. Алкены далее легко превращаются в карбкатионы и инициируют цепной карбкатионный процесс, аналогичный каталитическому крекингу.
Основные отличия гидрокрекинга от каталитического крекинга заключается в том, что общая конверсия алканов при гидрокрекинге выше, чем при каталитическом крекинге. Это обусловлено легкостью образования алкенов на гидрирующих – дегидрирующих центрах катализатора. При каталитическом крекинге алкены образуются значительно труднее – в основном за счет термического распада. Продукты гидрокрекинга имеют предельный характер. Катализаторы гидрокрекинга практически не закоксовываются, так как алкены подвергаются быстрому гидрированию и не успевают вступать в дальнейшие превращения по пути полимеризации и уплотнения.
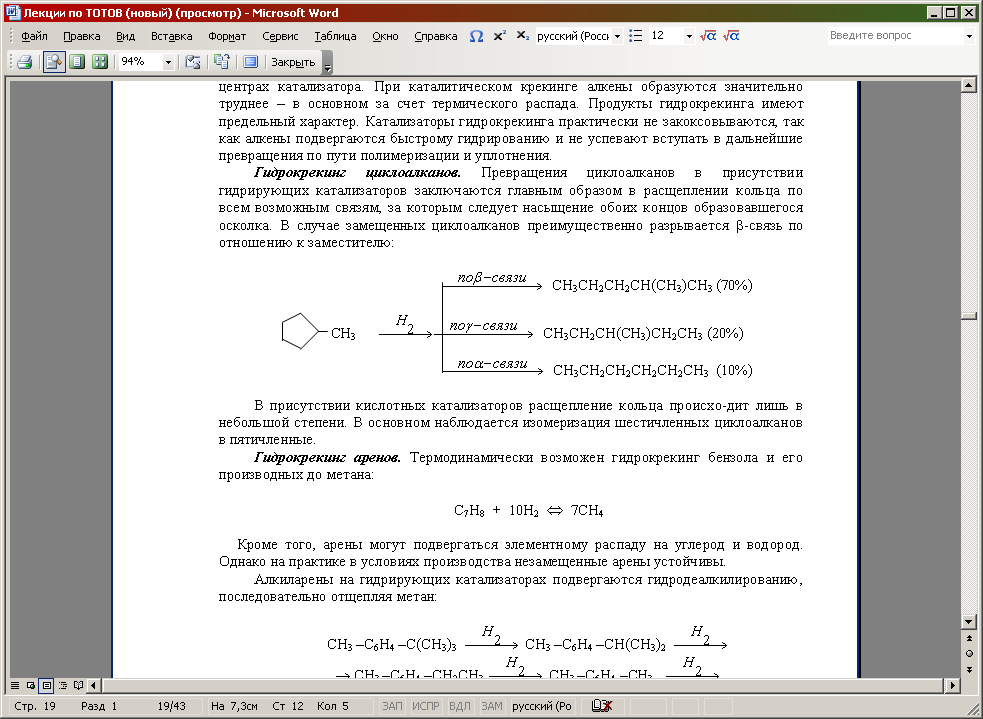
Гидрокрекинг циклоалканов. Превращения циклоалканов в присутствии гидрирующих катализаторов заключаются главным образом в расщеплении кольца по всем возможным связям, за которым следует насыщение обоих концов образовавшегося осколка. В случае замещенных циклоалканов преимущественно разрывается -связь по отношению к заместителю:
В присутствии кислотных катализаторов расщепление кольца происхо-дит лишь в небольшой степени. В основном наблюдается изомеризация шестичленных циклоалканов в пятичленные.
Гидрокрекинг аренов. Термодинамически возможен гидрокрекинг бензола и его производных до метана:
С7Н8 + 10Н2 7СН4
Кроме того, арены могут подвергаться элементному распаду на углерод и водород. Однако на практике в условиях производства незамещенные арены устойчивы.
Алкиларены на гидрирующих катализаторах подвергаются гидродеалкилированию, последовательно отщепляя метан:
СН3 –С6Н4 –С(СН3)3 СН3 –С6Н4 –СН(СН3)2
СН3 –С6Н4 –СН2СН3 СН3 –С6Н4 –СН3
С6Н5 –СН3 С6Н6 + 5СН4
На катализаторах с высокой кислотной активностью реакция аренов значительно многообразнее и сложнее, чем на гидрирующих катализаторах. Метил- и этилбензолы подвергаются в основном изомеризации по положению заместителей и диспропорционированию (см. лекция № 4).
Деалкилирование метил- и этилбензолов, в соответствии с ионным механизмом реакции, является сильно эндотермическим процессом и практически не идет.
Алкилбензолы, содержащие в боковой цепи 3 и более атома углерода, деалкилируются, как и при каталитическом крекинге и риформинге (см. лекции № 3, 4).
Полициклические арены на катализаторах с высокой кислотностью гидрируются до моноциклических с различными алкильными заместителями и далее расщепляются, как алкилбензолы. В результате гидрокрекинга поли-циклических аренов в значительном количестве образуются также производные тетралина и индана.
Сравнение скоростей гидрокрекинга углеводородов различных классов свидетельствует о том, что гидрирование полициклических структур до углеводородов, содержащих одно бензольное или алициклическое кольцо, происходит быстро. Гидрирование аренов и циклоалканов с разрушением кольца протекает сравнительно медленно. Относительно медленно проходит также гидрокрекинг алканов. Таким образом, в продуктах реакции накапливаются производные моноциклических аренов и циклоалканов, а также алканы, преимущественно разветвленные.
Контрольные вопросы
Сущность гидрогенизационных процессов.
Какие процессы протекают при гидроочистке?
Сущность процесса гидрокрекинга.
Катализаторы процесса гидрокрекинга.
Литература
Магарил Р.З. Теоретические основы химических процессов переработки нефти. Л.: Химия, 1985. -279 с.
Ахметов С.А. Технология глубокой переработки нефти и газа. – Уфа: «Гилем», 2002. – 672 с.
Технология переработки нефти Ч. 1. /Под редакцией О.Ф.Глаголевой, В.М.Капустина/. – М.: Химия, 2005, 400 с.
Лекция № 6. Физико-химические основы полимеризационных процессов
• Радикальная полимеризация
• Ионная полимеризация
Полимеризацией называется реакция соединения молекул мономера, протекающая за счет раскрытия кратных связей и не сопровождающаяся выделением побочных продуктов.
Полимеризация характерна для соединений с кратными связями, число и характер которых в молекуле мономера могут быть разными. Полимеризация всегда сопровождается понижением степени насыщенности реагирующих веществ, уменьшением общего числа молекул и увеличением их среднего молекулярного веса. Возможна также полимеризация насыщенных соединений циклического строения, содержащих в цикле гетероатом. В этих случаях при полимеризации происходит размыкание цикла и образование гетероцепного линейнего полимера.
Механизм полимеризации определяется химической природой растущих радикалов, являющихся промежуточными продуктами полимеризации. Если, эти частицы достаточно стабильны и характеризуются значительной продолжительностью жизни, полимеризация называется ступенчатой. Если же промежуточные продукты нестабильные – коротко живущие частицы, то полимеризация называется цепной.
В полимеризации могут одновременно участвовать два или несколько различных мономеров. Такую полимеризацию называют совместной, или сополимеризацией.
В зависимости от химической природы активных центров, участвующих в полимеризации, различают радикальную и ионную полимеризацию. Методы возбуждения и механизм этих видов цепной полимеризации различны. При радикальной полимеризации активными центрами реакции являются свободные радикалы. При ионной полимеризации активными центрами являются ионы – положительно или отрицательно заряженные частицы.
Радикальная полимеризация всегда протекает по цепному механизму и состоит из следующих реакций: инициирование (образование свободных радикалов), рост цепи, обрыв цепи.
В зависимости от способа образования свободных радикалов различают термическую, фотохимическую, радиационную и инициированную полимеризацию.
При термической полимеризации свободные радикалы возникают под действием тепла за счет раскрытия кратных связей мономера
 СН2
= СНR
нагревание
СН2
– CHR
СН2
= СНR
нагревание
СН2
– CHR
и последующего взаимодействия такого бирадикала с молекулой мономера
СН2 – CHR + СН2 = CHR СН2 – CHR – СН2 – CHR
Термическая полимеризация протекает крайне медленно, и скорость ее резко зависит от температуры. Впоследствии бирадикалы превращаются в полимерные монорадикалы.
При фотохимической полимеризации молекула мономера поглощает квант световой энергии и переходит в возбужденное состояние
СН2 = СНR + hv СН2 = СНR
В результате мономолекулярного превращения возбужденная молекула образует бирадикал
СН2 = СНR СН2 – CHR
Поскольку образование активных центров фотополимеризации протекает в результате прямого поглощения квантов энергии, реакция может проводиться при температурах, при которых полимеризация, инициируемые другими методами, не протекает. При фотохимической полимеризации бирадикалы также превращаются в полимерные монорадикалы.
При радиационной полимеризации образование свободных радикалов происходит при действии на мономер ионизирующих излучений (-лучей, рентгеновых лучей, ускоренных электронов, нейтронов, -частиц и т.д.).
Наиболее распространенной является инициированная полимеризация, при которой свободные радикалы образуются в результате термического гомолитического распада нестойких веществ (инициаторов), введенных в среду мономера. К таким веществам относятся органические перекиси и гидроперекиси, неорганические перекиси, озониды, некоторые азо- и диазосоединения и др. Количество применяемого при полимеризации инициатора невелико и колеблется в пределах от 0,1 до 1 % от веса мономера. Энергия активации, характеризующая инициирование, обычно близка к энергии связи, разрывающейся при распаде инициатора. Для большинства инициаторов эти величины лежат в пределах от 25 до 35 ккал/моль. Поэтому достаточно высокие скорости инициирования могут быть достигнуты при температуре выше 50оС. При высоких температурах инициированная полимеризация может происходить без введения инициаторов, за счет разложения небольших количеств перекисных примесей, образующихся при взаимодействии мономера с кислородом воздуха, или других случайных примесей.
Для радикальной полимеризации при нормальных и пониженных температурах используют окислительно-восстановительное инициирование, происходящее за счет окислительно-восстановительной реакции в среде, содержащей мономер. Полимеризацию в этом случае вызывают свободные радикалы, являющиеся промежуточными продуктами при окислительно- восстановительных реакциях. Типичный пример окислительно-восстановительной реакции – взаимодействие перекиси водорода с ионами двухвалентного железа
Fe2+ + H2O2 Fe3+ + OH + OH
Радикал OH, присоединяясь к молекуле мономера, инициирует радикальную полимеризацию.
Особенностью окислительно-восстановительного инициирования является очень низкая энергия активации (12-20 ккал/моль).
Реакция роста цепи при радикальной полимеризации состоит из последовательного ряда элементарных актов взаимодействия свободного радикала с молекулами мономера, причем растущая цепь сама является свободным радикалом с возрастающим в процессе реакции молекулярным весом.
В результате реакции роста цепи -связь превращается в -связь, сопровождающую-ся выделением тепла за счет разности энергий - и -связей, т.е.
R + CH2 = CHX RCH2CHX + СН2СНХ
RCH2CHXСН2СНХ + … + CH2 = CHX
R(CH2CHX)nСН2СНХ
Энергия активации реакции роста цепи лежит в пределах 3-10 ккал/моль.
Реакция обрыва цепи приводит к исчезновению в системе активных радикалов и может происходить при взаимодействии двух растущих радикалов в результате их рекомбинации
R(CH2CHX)nСН2СНХ +
+ ХНССН2(CH2CHX)mR R(CH2CHX)nСН2СНХ
ХНССН2(CH2CHX)mR;
или диспропорционирования



R(CH2CHX)nСН2СНХ + ХНССН2(CH2CHX)mR
R(CH2CHX)nСН=СНХ + ХН2ССН2(CH2CHX)mR
При диспропорционировании молекула полимера имеет на одном конце двойные связи. Энергия активации обрыва цепи обычно не превышает 1,5 ккал/моль.
Для радикальной полимеризации весьма характерны реакции передачи цепи, сущность которых состоит в обрыве растущего радикального атома или группы атомов от какой-либо молекулы (передатчика цепи). В результате этого радикал превращается в валентнонасыщенную молекулу и образуется новый радикал, способный к продолжению кинетической цепи. Передача цепи может осуществляться через молекулы мономера, растворителя, полимерные молекулы, уже успевшие образоваться в реакционной системе. В последнем случае образуются разветвленные макромолекулы.
Путем введения в полимеризующуюся систему веществ, через которых легко осуществляется передача цепи, можно регулировать средний молекулярный вес полимера. Такие вещества называются регуляторами. В качестве регуляторов применяют хлорированные углеводороды (СС14, СС12СС12 и др.), меркаптаны, тиогликолевую кислоту и другие (в количестве 2-6 % от веса мономера).
Ионная полимеризация протекает в присутствии катализаторов, которые в отличие от инициаторов не расходуются в процессе полимеризации и не входят в состав полимера. В отличие от радикальной полимеризации, протекающей путем передачи по цепи непарного электрона, ионная полимеризация идет с образованием либо иона карбония, либо карбаниона с последующей передачей по цепи положительного или отрицательного заряда. Поэтому различают катионную и анионную полимеризации.
Катионная полимеризация. Возникновение активного центра при катионной полимеризации связано с потерей одним атомом углерода электрона и образованием карбониевого иона и соответствующего аниона (противоиона), которые в средах с невысокой диэлектрической проницаемостью остаются в непосредственной близости от катиона, образуя с ним ионную пару. При катионной полимеризации реакционно-способный конец растущей цепи заряжен положительно.
В катионную полимеризации легко вступают мономеры винилового и дивинилового рядов, содержащие электродонорные заместители у двойной связи, например, изобутилен, пропилен, -метилстирол, винилакриловые эфиры, изопрен и др. С увеличением электроположительности заместителя способность виниловых мономеров к катионной полимеризации возрастает. Примером катионной полимеризации может служить полимеризация стирола в присутствии SnCl4:
1) образование активного центра – иона карбония
S nCl4 + 2СН2=СН [SnCl4]C-H2–CH–CH2–C+H
C6H5 C6H5 C6H5
2) рост цепи
[ SnCl4]C-H2–CH–CH2–C+H + nСН2=СН
C6H5 C6H5 C6H5
[SnCl4]C-H2–CH–[–CH2–CH–]n–СН2–С+Н
C6H5 C6H5 C6H5
образование макромолекулы (передача цепи)

 [SnCl4]C-H2–CH–[–CH2–CH–]n–СН2–С+Н
[SnCl4]C-H2–CH–[–CH2–CH–]n–СН2–С+Н
 C6H5
C6H5
C6H5
C6H5
C6H5
C6H5
CH3–CH–[–CH2–CH–]n–СН=СН + SnCl4
C6H5 C6H5 C6H5
Цепь растет последовательным присоединением молекул мономера к катиону. Обрыв цепи при катионной полимеризации – явление весьма редкое. Ограничение длины образующихся макромолекул происходит главным образом в результате передачи цепи, которая обычно осуществляется переносом противоиона от растущего макроиона к какой-либо другой частице. Кроме того, передача цепи может происходить переносом ионов по схеме:
R1+ + RH R1H + R+
В качестве катализаторов при катионной полимеризации применяют электроноакцепторные соединения (BF3, SnCl4, TiCl4, AlBr3, FeCl3 и др.). Большую роль при катионной полимеризации играют сокатализаторы: вода, протонные кислоты, спирты и другие, образующие комплексы с катализаторами.
Анионная полимеризация. При анионной полимеризации возникновение активного центра связано с образованием карбаниона. Конец растущей цепи заряжен отрицательно.
В анионную полимеризацию легко вступают мономеры винилового и дивинилового рядов, содержащие электроноакцепторные заместители у двойной связи, например, акрилонитрил, акриловые и метакриловые эфиры и др. Способность виниловых и дивиниловых мономеров к анионной полимеризации возрастает с увеличением электроотрицательности заместителя. Механизм анионной полимеризации, инициируемой, например, амидом натрия в жидком аммиаке, можно представить следующей схемой:
1) инициирование – образование карбаниона
N
 aNH2
Na+
+ NH2-
+ CH2=CHX
NH2
–CH2
–CHX-
Na+
aNH2
Na+
+ NH2-
+ CH2=CHX
NH2
–CH2
–CHX-
Na+
Карбанион, вероятно, остается в непосредственной близости от положительно заряженного иона натрия, образуя с ним ионную пару;
2) рост цепи
NH2 –CH2 –CHX- Na+ + nCH2=CHX
NH2 –CH2 –[–CHX–CH2–]n –CHX- Na+
3) передача цепи (например, через растворитель)
N

 H2
–CH2
–[–CHX–CH2–]n
–CHX-
Na+
+ NH3
H2
–CH2
–[–CHX–CH2–]n
–CHX-
Na+
+ NH3
NH2 –CH2 –[–CHX–CH2–]n –CH2X + NaNH2
Рост цепи и образование макромолекулы при анионной полимеризации происходит, очевидно, аналогично катионной полимеризации.
Катализаторами анионной полимеризации служат электронодонорные соединения, к которым амиды щелочных металлов, щелочные металлы и их растворы в жидком аммиаке, металлоорганические соединения щелочных металлов и др.
Скорость анионной полимеризации увеличивается с повышением концентрации мономера и катализатора, а молекулярный вес образующегося полимера прямо пропорционален концентрации мономера и не зависит от концентрации катализатора.
Контрольные вопросы
Сущность полимеризационных процессов.
Какие реакции протекают при полимеризационных процессах.
Основные продукты полимеризационных процессов углеводородного сырья.
Литература
1. Магарил Р.З. Теоретические основы химических процессов переработки нефти. Л.: Химия, 1985. -279 с.
2. Сагинаев А.Т. Теоретические основы химических процессов переработки нефти, газа и угля. – Атырау, «РИО АИНиГ», 2007. – 171 с.