

Впоросы похимии:
1.Основные типы органических реакций. Гомолитические и гетеролитические реакции. Нуклеофильные и электрофильные реакции.
2.Понятие об изомерии. Виды изомерии: структурная и пространственная. Цис-и-транс изомерия.
3.Электронное строение органических соединений. Гибридные орбитали. Образование и характеристика и связей.
4.Электроотрицательность атомов в органических молекулах.
Индуктивный эффект.
5.Сопряженные системы. Статический и динамический мезомерный эффекты. Сверхсопряжение.
6.Кето-енольная и кольчато-цепная таутомерия. Причины и проявление в различных классах органических соединений.
7.Гомологический ряд алканов. Номенклатура. Методы получения.
8.Химические свойства алканов.
9.Гомологический ряд алкенов. Номенклатура. Методы получения.
10.Химические свойства алкенов.
11.Реакции электрофильного присоединения в алкенах.
12.Химические свойства алкинов.
13.Гомологический ряд алкинов. Номенклатура. Методы получения.
14.Типы диеновых углеводородов. Различие в строении и основных свойствах.
15.Химические свойства сопряженных диеновых углеводородов.
16.Получение бутадиена, хлоропрена, изопрена. Полимеризация.
17.Механизм реакций электрофильного замещения в ароматическом ядре. Правила ориентации.
18.Химические свойства бензола и его производных: присоединение, замещение, окисление в ядре и боковой цепи.
19.Строение ароматических углеводородов. Правило Хюккеля. Конденсированные ароматические системы. Ароматические гетероциклы.
20.Гомологический ряд аренов. Номенклатура. Методы получения.
21.Галоидные алкилы. Номенклатура. Методы получения.
22.Химические свойства галогенпроизводных.
23.Сравнение реакционной способности предельных, непредельных и ароматических галогенпроизводных.
24.Механизмы реакций нуклеофильного замещения SN1 SN2.
25.Химические свойства алифатических аминов.
26.Химические свойства ароматических аминов (анилин).
27.Алифатические амины. Номенклатура. Получение.
28.Одноатомные предельные спирты. Номенклатура. Методы получения.
29.Химические свойства спиртов.
30.Непредельные и многоатомные спирты. Особенности строения и химических свойств.
31.Фенолы. Электрофильное и нуклеофильное (пикриновая кислота) замещение в ароматическом ядре.
32.Фенолы. Химические свойства. Гидрохинон и хинон.
33.Альдегиды и кетоны. Номенклатура. Методы получения.
34.Химические свойства альдегидов и кетонов.
35.Функциональные производные карбоновых кислот и их взаимопревращения.
36.Карбоновые кислоты. Номенклатура. Строение и методы получения.
37.Химические свойства карбоновых кислот. Механизм реакции этерификации и гидролиза.
38.Непредельные карбоновые кислоты. Дикарбоновые кислоты.
39.Химические свойства альдоз и кетоз.
40.Углеводы. Классификация. Строение.
41.Ди- и полисахариды. Строение, свойства, гидролиз.
Восстанавливающие и невосстанавливающие дисахариды: сахароза,
мальтоза, целлобиоза.
42.Аминокислоты. Методы получения и химические свойства.
43.Пятичленные гетероциклы. Их строение, свойства и взаимопревращения.
44.Шестичленные гетероциклы. Свойства пиридина, особенности его строения.
45.Основные понятия науки о полимерах. Основные отличия высокомолекулярных соединений от низкомолекулярных.
46.Классификация полимеров по составу, строению и методу синтеза. Молекулярно-массовые характеристики полимеров: средняя молекулярная масса.
47.Структура полимеров. Изомерия макромолекул. Физические и фазовое состояние полимеров: кристаллическое, стеклообразное, вязкотекучее и высокоэластичное.
48.Полиэтилен, полипропилен, каучуки, полиамиды, полиэфиры.
49.Растворение полимеров. Ограниченное и неограниченное набухание. Строение разбавленных растворов полимеров. Методы определения молекулярной массы полимеров. Растворы полимеров.
50.Полимеризация: катионная и анионная. Радикальная полимеризация: стадии процесса.
51.Поликонденсация.
52.Химические свойства полимеров.
53.Полимераналоговые превращения. Деструкция полимеров.
54.Фосфоросодержащие органические соединения. Строение ДНК и РНК.
Задачи по химии
Ответы на экзамен по химии:
1 Чаще всего органические реакции классифицируют по типу разрыва химических связей в реагирующих частицах. Из их числа можно выделить две большие группы реакций —радикальные и ионные.
Радикальные реакции — это процессы, идущие с гемолитическим разрывом ковалентной связи. При гемолитическом разрыве пара электронов, образующая связь, делится таким образом, что каждая из образующихся частиц получает по одному электрону. В результате гемолитического разрыва образуются свободные радикалы:
X:Y → X.+.Y
Нейтральный атом или частица с неспаренным электроном называется свободным радикалом.
Ионные реакции — это процессы, идущие с гетеролитическим разрывом ковалентных связей, когда оба электрона связи остаются с одной из ранее связанных частиц.
X:Y → X+ + :Y-
В результате гетеролитического разрыва связи получаются заряженные частицы: нуклеофильная и электрофильная.
Нуклеофильная частица (нуклеофил) — это частица, имеющая пару электронов на внешнем электронном уровне. За счет пары электронов нуклеофил способен образовывать новую ковалентную связь.
Электрофильная частица (электрофил) - это частица, имеющая свободную орбиталь на внешнем электронном уровне. Электрофил представляет незаполненные, вакантные орбиталидля образования ковалентной связи за счет электронов той частицы, с которой он взаимодействует.
Частица с положительным зарядом на атоме углерода называется карбокатионом.
Согласно другой классификации, органические реакции делятся на термические, являющиеся результатом столкновений молекул при их тепловом движении, и фотохимические, при которых молекулы, поглощая квант света Av, переходят в более высокие энергетические состояния и далее подвергаются химическим превращениям. Для одних и тех же исходных соединений термические и фотохимические реакции обычно приводят к различным продуктам. Классическим примером здесь является термическое и фотохимическое хлорирование бензола — в первом случае образуется хлорбензол, во втором случае — гексахлорциклогексан.
Кроме того, в органической химии реакции часто классифицируются так же, как и в неорганической химии — по структурному признаку. В органической химии все структурные изменения рассматриваются относительно атома (или атомов) углерода, участвующих в реакции. Наиболее часто встречаются следующие типы превращений:
1) присоединение R-CH=CH2 + XY→ RCHX-CH2Y;
2) замещение R-CH2X + Y→ R-CH2Y + X;
3) отщепление R-CHX-CH2Y→ R-CH=CH2 + XY;
(элиминирование)
4) полимеризация n(СН2=СН2) → (—CH2—СН2—)n
В большинстве случаев элиминируемая молекула образуется при соединении двух частиц, отщепленных от соседних атомов углерода. Такой процесс называется 1,2-элиминированием.
Кроме приведенных четырех типов простейших механизмов, реакций на практике употребляются еще следующие обозначения некоторых классов реакций, приведенные ниже.
Окисление — реакция, при которой под действием окисляющего реагента вещество соединяется с кислородом (либо другим электроотрицательным элементом, например, галогеном) или теряет водород (в виде воды или молекулярного водорода).
Действие окисляющего реагента (окисление) обозначается в схеме реакции символом [О], а действие восстанавливающего реагента (восстановление) — символом [Н].
|
[O] |
|
CH3CHO |
→ |
CH3COOH |
|
[O] |
|
CH3OH |
→ |
CH2O + H2 |
|
кат |
|
CH3OH |
→ |
CH2O + H2 |
Отщепление водорода в последнем примере называется дегидрированием и проводится с помощью катализатора.
Восстановление - реакция, обратная окислению. Под действием восстанавливающего реагента соединение принимает атомы водорода или теряет атомы кислорода:
|
[H] |
|
CH3COCH3 |
→ |
CH3CH(OH)CH3 |
|
|
|
Гидрирование - реакция, представляющая собой частный случай восстановления. Водород присоединяется к кратной связи или ароматическому ядру в присутствии катализатора.
Конденсация — реакция, при которой происходит рост цепи. Сначала происходит присоединение, за которым обычно следует элиминирование.
Пиролиз — реакция, при которой соединение подвергается термическому разложению без доступа воздуха (и обычно при пониженном давлении) с образованием одного или нескольких продуктов. Примером пиролиза может служить термическое разложение каменного угля. Иногда вместо пиролиза употребляется термин "сухая перегонка" (в случае разложения каменного угля используется также термин "карбонизация").
Некоторые реакции получают свои названия по продуктам, к которым они приводят. Так, если в молекулу вводится метильная группа, то говорят о метилировании, если ацетил — то обацетилировании, если хлор — то о хлорировании и т.д.
ГОМОЛИТИЧЕСКИЕ РЕАКЦИИ (от греч. homos -равный, одинаковый, общий и lysis-разложение, распад), протекают с гемолизом хим. связи, т.е. с таким ее разрывом, при к-ром электронная пара, осуществляющая связь, разделяется между образующимися фрагментами (своб. радикалами):
![]()
Иногда к гомологическим относят все р-ции, протекающие с участием и образованием своб. радикалов или ион-радикалов (см. Радикалы свободные, Одноэлектроннып перенос).
Для осуществления гомолитических реакций обычно используют термохим., каталитич., фотохим., радиационно-хим. и электрохим. методы. Возможность реализации р-ции определяется энергией диссоциации связи. В газовой фазе гомолиз протекает значительно легче, чем гетеролиз (см.Гетеролитические реакции).
По гомолитич. механизму протекают мн. цепные р-ции, напр. фотогалогенирование предельных углеводородов (металепсия), радикальная полимеризация, нитрование алифатич. соединений.
ГЕТЕРОЛИТИЧЕСКИЕ РЕАКЦИИ (от
греч. heteros-иной, другой и lysis-разложение,
распад), протекают с гетеролизом хим.
связи, т.е. с таким ее разрывом, при к-ром
электронная пара,
осуществляющая связь, остается у одного
из атомов и,
как правило, образуются ионы:

Иногда к гетеролитическим относят также р-ции, в к-рых образование новой химической связи происходит в результате обобществления электроннойпары одного из реагентов.
Осуществлению гетеролитических реакций благоприятствует использование в кач-ве реагентов сильных к-т или оснований, высокая диэлектрич. проницаемость среды. Гетеролиз энергетически менее выгоден (на 300-650 кДж/моль), чем гомолиз (см. Гомолитические реакции), однако в р-рах благодаря сольватации образующихся ионов гетеролитические реакции протекают сравнительно легко.
В орг. химии гетеролитические реакции принято делить в зависимости от характера разрыва связи С—X на нуклеофильные реакции (заместитель X уходит с электронной парой и образуется карбкатион) и электрофильные реакции (электронная пара остается у атома С и образуется карбанион). Механизм гетеролитических реакций иногда может включать промежут. образование ионрадикалов, в этом случае говорят о наличии стадии одно-электронного переноса.
Типичные гетеролитические реакции-распад молекул на ионы, рекомбинация ионов, мн. процессы замещения, элиминирования и присоединения, в т.ч. такие промышленно важные, как нитрование и сульфироваиие ароматич. соединений, присоединение галогенов к олефинам в полярных р-рителях.
2 Изомерия (от др.-греч. ἴσος — «равный», и μέρος — «доля, часть») — явление, заключающееся в существовании химических соединений (изомеров), одинаковых по составу и молекулярной массе, но различающихся по строению или расположению атомов в пространстве и, вследствие этого, по свойствам. Структурная изомерия[править | править исходный текст]
Структурная изомерия — результат различий в химическом строении. К этому типу относят:
Изомерия углеродной цепи (углеродного скелета)[править | править исходный текст]
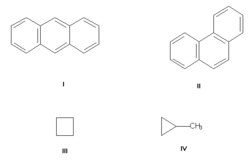
Изомерия углеродного скелета, обусловленная различным порядком связи атомов углерода. Простейший пример — бутан СН3—СН2—СН2—СН3 и изобутан (СН3)3СН. Другие примеры: антрацен и фенантрен (формулы I и II, соответственно), циклобутан и метилциклопропан (III и IV).
Валентная изомерия[править | править исходный текст]

Валентная — особый вид структурной изомерии, при которой изомеры можно перевести друг в друга лишь за счёт перераспределения связей. Например, валентными изомерами бензола (V) являются бицикло[2.2.0]гекса-2,5-диен (VI, «бензол Дьюара»), призман (VII, «бензол Ладенбурга»), бензвален (VIII).
Изомерия функциональной группы (межклассовая изомерия)[править | править исходный текст]
Различается характером функциональной группы; например, этанол (CH3—CH2—OH) и диметиловый эфир (CH3—O—CH3).
Изомерия положения[править | править исходный текст]
Тип структурной изомерии, характеризующийся различием положения одинаковых функциональных групп или кратных связей при одинаковом углеродном скелете. Пример: 2-хлорбутановая кислота и 4-хлорбутановая кислота.
Метамерия[править | править исходный текст]
Метамерия — вид структурной изомерии, для которого характерно различное распределение углеродных атомов между несколькими углеводородными радикалами, разделенными в молекуле гетероатомом. Метамерия известна в рядах алифатических простых эфиров, сложных эфиров, тиоспиртов и аминов. В настоящее время термин используется редко.
На данный вид изомерии ещё указывал А. М. Бутлеров, называя его «изомерия нецельных структур».
Пример: CH3CH2OCH2CH3 — диэтиловый эфир и CH3OCH2CH2CH3 — метилпропиловый эфир
Пространственная изомерия (стереоизомерия)[править | править исходный текст]
Основная статья: Стереоизомеры
Пространственная изомерия (стереоизомерия) возникает в результате различий в пространственной конфигурации молекул, имеющих одинаковое химическое строение. Для обозначения пространственных изомеров разных типов разработана стереохимическая номенклатура, собранная в разделе E номенклатурных правил ИЮПАК по химии[источник не указан 1106 дней].
Этот тип изомерии подразделяют на энантиомерию (оптическую изомерию) и диастереомерию.
Энантиомерия (оптическая изомерия)[править | править исходный текст]
Основная статья: Оптическая изомерия

Энантиомерами (оптическими изомерами, зеркальными изомерами) являются пары оптических антиподов — веществ, характеризующихся противоположными по знаку и одинаковыми по величине вращениями плоскости поляризации света при идентичности всех других физических и химических свойств (за исключением реакций с другими оптически активными веществами и физических свойств в хиральной среде). Необходимая и достаточная причина возникновения оптических антиподов — принадлежность молекулы к одной из следующих точечных групп симметрии: Cn, Dn, T, O или I (хиральность). Чаще всего речь идет об асимметрическом атоме углерода, то есть об атоме, связанном с четырьмя разными заместителями.
Асимметрическими могут быть и другие атомы, например атомы кремния, азота, фосфора, серы. Наличие асимметрического атома — не единственная причина энантиомерии. Так, имеют оптические антиподы производные адамантана (IX), ферроцена (X), 1,3-дифенилаллена (XI), 6,6'-динитро-2,2'-дифеновой кислоты (XII). Причина оптической активности последнего соединения —атропоизомерия, то есть пространственная изомерия, вызванная отсутствием вращения вокруг простой связи. Энантиомерия также проявляется в спиральных конформациях белков, нуклеиновых кислот, в гексагелицене (XIII).
Диастереомерия[править | править исходный текст]
Диастереомерными считают любые комбинации пространственных изомеров, не составляющие пару оптических антиподов. Различают σ- и π-диастереомеры.
σ—диастереомерия[править | править исходный текст]
σ-диастереомеры отличаются друг от друга конфигурацией части имеющихся в них элементов хиральности. Так, диастереомерами являются (+)-винная кислота и мезо-винная кислота, D-глюкоза и D-манноза, например:

π—диастереомерия (геометрическая изомерия)[править | править исходный текст]
π-диастереомеры, называемые также геометрическими изомерами, отличаются друг от друга различным пространственным расположением заместителей относительно плоскостидвойной связи (чаще всего С=С и С=N) или цикла. К ним относятся, например, малеиновая и фумаровая кислоты (формулы XIV и XV соответственно), (Е)- и (Z)-бензальдоксимы (XVI и XVII), цис- и транс-1,2-диметилциклопентаны (XVIII и XIX).

3 ?
4 Молекула органического соединения представляет собой совокупность атомов, связанных в определенном порядке, как правило, ковалентными связями. При этом связанные атомы могут различаться по величине электроотрицательности. Величины электроотрицательностей в значительной степени определяют такие важнейшие характеристики связи, как полярность и прочность (энергия образования). В свою очередь, полярность и прочность связей в молекуле, в значительной степени, определяют возможности молекулы вступать в те или иные химические реакции.
Электроотрицательность атома углерода зависит от состояния его гибридизации. Это связано с долей s-орбитали в гибридной орбитали: она меньше у sp3- и больше у sp2- и sp-гибридных атомов.
Все составляющие молекулу атомы находятся во взаимосвязи и испытывают взаимное влияние. Это влияние передается, в основном, через систему ковалентных связей, с помощью так называемых электронных эффектов.
Электронными эффектами называют смещение электронной плотности в молекуле под влиянием заместителей.
Атомы, связанные полярной связью, несут частичные заряды, обозначаемые греческой буквой "дельта" (d). Атом, "оттягивающий" электронную плотность s-связи в свою сторону, приобретает отрицательный заряд d-. При рассмотрении пары атомов, связанных ковалентной связью, более электроотрицательный атом называют электроноакцептором. Его партнер по s-связи соответственно будет иметь равный по величине дефицит электронной плотности, т.е. частичный положительный заряд d+, будет называться электронодонором.
Смещение электронной плотности по цепи s-связей называется индуктивным эффектом и обозначается I.
Индуктивный эффект передается по цепи с затуханием. Направление смещения электронной плотности всех s -связей обозначается прямыми стрелками.
В зависимости от того, удаляется ли электронная плотность от рассматриваемого атома углерода или приближается к нему, индуктивный эффект называют отрицательным (-I) или положительным (+I). Знак и величина индуктивного эффекта определяются различиями в электроотрицательности между рассматриваемым атомом углерода и группой, его вызывающей.
Электроноакцепторные заместители, т.е. атом или группа атомов, смещающие электронную плотность s-связи от атома углерода к себе, проявляют отрицательный индуктивный эффект(-I-эффект).
Электродонорные заместители, т.е. атом или группа атомов, смещающие электронную плотность к атому углерода от себя, проявляют положительный индуктивный эффект (+I-эффект).
+I-эффект проявляют алифатические углеводородные радикалы, т.е. алкильные радикалы (метил, этил и т.д.). Большинство функциональных групп проявляют -I-эффект: галогены, аминогруппа, гидроксильная, карбонильная, карбоксильная группы.
Индуктивный эффект проявляется и в случае, когда связанные атомы углерода различны по состоянию гибридизации.
При передаче индуктивного эффекта метальной группы на двойную связь в первую очередь ее влияние испытывает подвижная p-связь.
Влияние заместителя на распределение электронной плотности, передаваемое по p-связям, называют мезомерным эффектом (М). Мезомерный эффект также может быть отрицательным и положительным. В структурных формулах его изображают изогнутой стрелкой, начинающейся у центра электронной плотности и завершающейся в том месте, куда смещается электронная плотность.
Наличие электронных эффектов ведет к перераспределению электронной плотности в молекуле и появлению частичных зарядов на отдельных атомах. Это определяет реакционную способность молекулы.
Индукти́вный эффе́кт (полярный эффект) — смещение электронной плотности химической связи по σ-связям. Является разновидностью эффекта поля.
Понятие об индуктивном эффекте было введено К. Ингольдом[1], им же были введены обозначения:
+I-эффект в случае повышения заместителем электронной плотности;
–I-эффект в случае понижения заместителем электронной плотности
В качестве вещества сравнения берут незамещённое соединение, то есть нулевой индуктивный эффект принимается для атома водорода.
Характерной особенностью индуктивного эффекта по сравнению с мезомерным эффектом является его быстрое затухание по цепочке связей.
Среди наиболее характерных +I-групп можно выделить: алкильные группы, металлы, металлоидные группы (силильные, борные, фосфорные и пр.); среди наиболее характерных групп с –I-эффектом выделяются заряженные группы (из-за эффекта поля), такие как триалкиламмониевые, диалкилсульфониевые и прочие ониевые соли, нитрогруппа, гидроксигруппа, алкоксигруппа, аминогруппа, галогены и т. п.
Фактически эффект обусловлен в первую очередь атомом, с которым непосредственно связан исходный атом углерода и определяется, таким образом, разницейэлектроотрицательностей атомов.
Количественная оценка индуктивного эффекта может быть произведена при помощи уравнения Тафта[
5 В простейшем случае сопряженные системы —
это системы с чередующимися двойными и одинарными связями. Они могут быть открытыми и закрытыми. Открытая система имеется в диеновых углеводородах (УВ).
Все атомы С находятся в состоянии sp-гибридиза-ции. Четыре негибридные р-орбитами, перекрываясь между собой, образуют единую электронную систему. Этот вид сопряжения называется p, p-сопряжением.
Происходит сопряжение р-электронов с S-электро-нами. Этот вид сопряжения называется р, р-сопряже-нием. Закрытая система имеется в ароматических УВ.
Сопряжение – процесс энергетически выгодный, энергия (Е) при этом выделяется. Энергия сопряжения бутадиена – 1,3 составляет 15 кДж/моль, энергия сопряжения бензола – 228 кДж/моль.
2. Ароматичность
Это понятие, включающее различные свойства ароматических соединений. Условия ароматичности:
1) плоский замкнутый цикл;
2) все атомы С находятся в sp2-гибридизации;
3) образуется единая сопряженная система всех атомов цикла;
4) выполняется правило Хюккеля: в сопряжении участвуют 4n + 2 р-электронов, где n = 1, 2, 3...
Простейший представитель ароматических углеводородов – бензол. Он соответствует всем четырем условиям ароматичности. Правило Хюккеля: 4n + 2 = 6, n = 1.
Нафталин – ароматическое соединение 4n + 2 = 10, n = 2.
Пиридин – ароматическое гетероциклическое соединение. Взаимное влияние атомов в молекуле
В 1861 г. русский ученый A. M. Бутлеров выдвинул положение: «Атомы в молекулах взаимно влияют друг на друга». В настоящее время это влияние передается двумя путями: индуктивным и мезомерным эффектами.
Индуктивный эффект – это передача электронного влияния по цепи р-связи. Известно, что связь между атомами с различной электроотрицательностью (ЭО) поляризована, смещена к более электроотрицательному атому. Это приводит к появлению на атомах эффективных (реальных) зарядов (d). Такое электронное смещение называется индуктивным и обозначается буквой «I» и стрелкой «?».
? + ? –
СН3 – СН2 ? X, Х = Hal-, НО-, HS-, NH2– и др.
Индуктивный эффект может быть положительным или отрицательным. Если заместитель X притягивает электроны химической связи сильнее, чем атом Н, то он проявляет – I.I (H) = 0. В нашем примере X проявляет – I.
Если заместитель X притягивает электроны связи слабее, чем атом Н, то он проявляет +I. Все алкилы (R = СН3-, C2H5– и т. д.), Меп+ проявляют +I.
Мезомерный эффект (эффект сопряжения, резонансный эффект), вид взаимного влияния атомов в молекуле илиионе. заключающийся в статической поляризации сопряженной системы связей. Обусловлен смещением π-электронов сопряженных связей или неподеленных пар в сторону атомов с недостроенной до октета электронной оболочкой. Электронное смещение (обозначается изогнутой стрелкой) приводит к появлению частичных электрических зарядов на концах сопряженной цепи. Положительным мезомерным эффектом (+ М) обладают электронодонорные группы, способные к частичной или полной передаче пары электронов в общую сопряженную систему. Электроноакцепторные группы, поляризующие сопряженную систему в противоположном направлении, характеризуются отрицательным мезомерным эффектом (—М): + М-Эффект уменьшается:
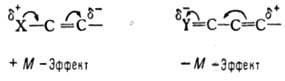
при переходе от отрицательно заряженных заместителей к положительно заряженным, с возрастанием электроотрицательности атома, с увеличением номера периода в периодической системе. Увеличению — М-эффекта способствуют: наличие положительного заряда, более высокий номер группы и периода в периодической системе:
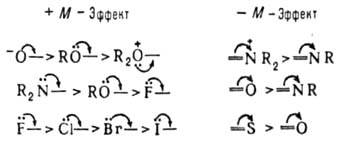
Важная особенность мезомерного эффекта - альтернирующий характер обусловливаемой им поляризации. Так, + М-эффект группы ОН приводит к увеличению электронной плотности в орто-и пара-положениях бензольного кольца (формула I), чем и объясняется электрофильное замещение только по этим положениям. Заместители с -М-эффектом понижают электронную плотность в орто- и пара-положениях бензольного кольца (II); этим объясняется электрофильная атака в мета-положения таких молекул.

Статический мезомерный эффект проявляется в значениях длин и силовых постоянных связей молекул, их дипольных моментов, констант равновесий (но не скоростей) реакций. Количественной мерой мезомерного эффекта групп, связанных с бензольным кольцом, служат sс (или sR)-константы заместителей, полученные вычитанием индукционных sI-констант из s-констант Гаммета. Мезомерный эффект поддается полуколичественной оценке при помощи теории возмущений.
Близок к мезомерному эффекту электромерный эффект ( + E, - E), заключающийся в поляризации сопряженной системы связей реагирующей молекулы или иона в переходном состоянии под влиянием электрических полей реагентов. В отличие от статического мезомерного эффекта этот эффект носит динамический характер. Его величина определяется поляризуемостью сопряженной системы, а направление поляризации всегда способствует понижению энергии переходного состояния. Поэтому, несмотря на сходство механизмов, возможны различные направления мезомерной и электромерной поляризации связей.
Гиперконъюгация — лишь один из видов сопряжения простых и кратных связей. Более широкие представления о сопряжении простой связи с двойной (σ,π-сопряжение) и двух простых связей (σ,σ-сопряжение) развиты А. Н. Несмеяновым, указавшим на ряд аналогий химического поведения π,π- σ,π- и σ,σ-сопряженных систем, например в реакциях 1,4-присоединения.
Простейшим примером молекулы, где связь Н—С сопряжена со связью С=С, может служить молекула пропилена. В этой молекуле благодаря влиянию метильной группы возникает электрическая асимметрия двойной связи

которая выражается, например, в дипольном моменте молекулы пропилена (0,35 D). Смещение π-электронной пары, очевидно, связано с некоторым смещением σ-электронов связей С—Н метильной группы в том же направлении. Обычно это изображают следующим образом:

В этом и заключается статический эффект сопряжения простых и двойных связей (σ,π-сопряжение).
6 Таутомерия — равновесная динамическая изомерия. Сущность её заключается во взаимном превращении изомеров с переносом какой-либо подвижной группы соответствующим перераспределением электронной плотности.
Кето-енольная таутомерия. Определенная протонная подвижность атома водорода у альфа-атома углерода в монокарбонильных соединениях (слабого СН-кислотного центра) проявляется в их способности к реакциям конденсации. Представим, что по каким-либо причинам подвижность такого атома водорода возросла настолько, что он оказался способным отщепиться в виде протона Н. Это должно привести к образованию мезомерного иона, отрицательный заряд которого рассредоточен между атомами углерода и кислорода. Обратное присоединение протона к этому иону в соответствии с его двумя граничными структурами может приводить либо к исходному карбонильному соединению, либо к енолу.

Поэтому, в принципе, карбонильное соединение в кетонной форме может существовать в равновесии с изомером — енольной формой. Такой вид изомерии называют кето-енольной таутомерией, а изомеры, находящиеся в состоянии подвижного равновесия, — таутомерами. В рассматриваемом случае между кетонной и енольной формами осуществляется перенос протона, поэтому такое равновесие называется прототропной таутомерией. В монокарбонильных соединениях (альдегидах, кетонах, сложных эфирах) равновесие практически полностью смещено в сторону кетонной формы. Например, содержание енольной формы в ацетоне составляет всего 0,00025%. При наличии второй электроноакцепторной группы у альфа-атома углерода (например, нитрогруппы —NО2, карбонильной группы =С=О) содержание енольной формы резко возрастает. Так, в 1,3-дикарбонильном соединении ацетилацетоне енольная форма преобладает.
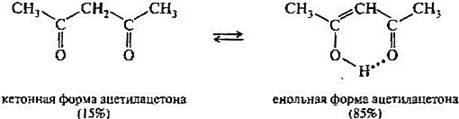
Многие реакции, включающие образование и превращения карбонильных соединен in vivo, как будет показано в дальнейшем, протекают через промежуточные енольные формы или производные этих форм.
Химическая сущность мутаротации состоит в способности моносахаридов (или моносахаридных звеньев в составе олиго- и полисахаридов) к существованию в виде равновесной смеси таутомеров — открытой и циклических форм. Такой вид таутомерии называется цикло-оксо-таутомерией (ранее еще называли кольчато-цепной). В растворах установление равновесия между четырьмя циклическими таутомерами моносахаридов протекает через открытую форму — оксоформу. Взаимопревращение альфа- и бета-аномеров друг в друга через промежуточную оксоформу называется аномеризацией. Таким образом, в растворе D-глюкоза существует в виде пяти таутомеров альфа- и бета-аномеров пиранозных и фуранозных циклических форм и оксоформы.

В смеси таутомеров преобладают пиранозные формы. Оксоформа, а также таутомеры с фуранозными циклами содержатся в малых количествах. Важно, однако, не абсолютное содержание того или иного таутомера, а возможность их перехода друг в друга, что приводит к пополнению количества нужной формы по мере ее расходования в каком-либо процессе. Например, несмотря на незначительное содержание оксоформы, глюкоза вступает в реакции, характерные для альдегидной группы. Это обусловлено сдвигом равновесия в сторону открытой формы по мере ее расходования в соответствующей реакции. Таутомерия лежит в основе множественности химических свойств моносахаридов.
? причины таутомерии
7 Гомологичный ряд
? номенклатура алканов
Получение алканов
8 Химические свойства алканов ?
9 Гомологический ряд алкенов. Получение алкенов.
10 Алкены химически активны. Их химические свойства во многом определяются наличием двойной связи. Для алкенов наиболее характерны реакции электрофильного присоединения иреакции радикального присоединения. Реакции нуклеофильного присоединения обычно требуют наличие сильного нуклеофила и для алкенов не типичны.
Особенностью алкенов являются также реакции циклоприсоединения и метатезиса.
Алкены легко вступают в реакции окисления, гидрируются сильными восстановителями или водородом под действием катализаторов до алканов, а также способны к аллильному радикальному замещению.
11. Реакции электрофильного присоединения по двойной связи алкенов
Граничными
орбиталями ВЗМО и НСМО алкенов являются
занятая ![]() -
и пустая
*-орбитали.
Следовательно, в реакциях с электрофилами
(Е+)
будет участвовать
-орбиталь,
а в реакциях с нуклеофилами (Nu-)
-
*-орбиталь
связи С=С (см. рис. 3). В большинстве
случаев простые алкены легко вступают
в реакции с электрофилами, а с нуклеофилами
реагируют с большим трудом. Это объясняется
тем, что обычно НСМО большинства
электрофилов по энергии близки к
энергии
-ВЗМО
алкенов, тогда как ВЗМО большинства
нуклеофилов лежат значительно
ниже
*-НСМО.
-
и пустая
*-орбитали.
Следовательно, в реакциях с электрофилами
(Е+)
будет участвовать
-орбиталь,
а в реакциях с нуклеофилами (Nu-)
-
*-орбиталь
связи С=С (см. рис. 3). В большинстве
случаев простые алкены легко вступают
в реакции с электрофилами, а с нуклеофилами
реагируют с большим трудом. Это объясняется
тем, что обычно НСМО большинства
электрофилов по энергии близки к
энергии
-ВЗМО
алкенов, тогда как ВЗМО большинства
нуклеофилов лежат значительно
ниже
*-НСМО.

Простые алкены реагируют лишь с очень сильными нуклеофильными агентами (карбанионы) в жестких условиях, однако введение электроноакцепторных групп в алкены, например, NO2, COR и др., приводит к понижению *-уровня, благодаря чему алкен приобретает способность реагировать с нуклеофилами средней силы (аммиак, RO-, Nє C-, енолят-анион и т. д.).
В результате взаимодействия электрофильного агента Е+ с алкеном образуется карбокатион, обладающий высокой реакционной способностью. Карбокатион далее стабилизируется за счет быстрого присоединения нуклеофильного агента Nu-:


Поскольку
медленной стадией является присоединение
электрофила, то процесс присоединения
любого полярного агента Е+![]() Nu-
следует
рассматривать именно как электрофильное
присоединение к кратной связи алкена.
Известно большое число реакций этого
типа, где роль электрофильного агента
выполняют галогены, галогеноводороды,
вода, соли двухвалентной ртути и другие
полярные реагенты. Электрофильное
присоединение к двойной связи в
классификации механизмов органических
реакций имеет символ АdE(Addition
Electrophilic) и в
зависимости от числа реагирующих молекул
обозначается как АdE2
(бимолекулярная реакция) или АdE3
(тримолекулярная реакция).
Nu-
следует
рассматривать именно как электрофильное
присоединение к кратной связи алкена.
Известно большое число реакций этого
типа, где роль электрофильного агента
выполняют галогены, галогеноводороды,
вода, соли двухвалентной ртути и другие
полярные реагенты. Электрофильное
присоединение к двойной связи в
классификации механизмов органических
реакций имеет символ АdE(Addition
Electrophilic) и в
зависимости от числа реагирующих молекул
обозначается как АdE2
(бимолекулярная реакция) или АdE3
(тримолекулярная реакция).
12. Химические свойства алкинов.
1.Присоединение галогенов |
|
2.Присоединение водорода |
|
3.Присоединение галогенводородов |
|
4.Присоединение воды |
|
5.Присоединение спиртов |
|
6.Присоединение кислот |
|
7.Присоединение синильной кислоты |
|
8.Реакция димеризации |
|
Гомологический ряд алкинов:
Этин: C2H2
Пропин: C3H4
Бутин: C4H6
Пентин: C5H8
Гексин: C6H10
Гептин: C7H12
Октин: C8H14
Нонин: C9H16
Децин: C10H18
Номенклатура алкинов
Молучение алкинов
14 ДИЕНОВЫЕ УГЛЕВОДОРОДЫ (АЛКАДИЕНЫ)
Диеновые углеводороды или алкадиены – это непредельные углеводороды, содержащие две двойные углерод - углеродные связи. Общая формула алкадиенов CnH2n-2. В зависимости от взаимного расположения двойных связей диены подразделяются на три типа:
1) углеводороды с кумулированными двойными связями, т.е. примыкающими к одному атому углерода. Например, пропадиен или аллен CH2=C=CH2;
2) углеводороды с изолированными двойными связями, т.е разделенными двумя и более простыми связями. Например, пентадиен -1,4 CH2=CH–CH2–CH=CH2;
3) углеводороды с сопряженными двойными связями, т.е. разделенными одной простой связью. Например, бутадиен -1,3 или дивинил CH2=CH–CH=CH2, 2-метилбутадиен -1,3 или изопрен
CH2=С–CH=CH2. I CH3 |
Наибольший интерес представляют углеводороды с сопряженными двойными связями.
Получение
Углеводороды с сопряженными двойными связями получают:
1) дегидрированием алканов, содержащихся в природном газе и газах нефтепереработки, при пропускании их над нагретым катализатором
CH3–CH2–CH2–CH3 ––~600°С;Cr2O3,Al2O3® CH2=CH–CH=CH2 + 2H2
CH3– |
CH–CH2–CH3 ––~600°С;Cr2O3,Al2O3® CH2= I CH3 |
C–CH=CH2 + 2H2 I CH3 |
2) дегидрированием и дегидратацией этилового спирта при пропускании паров спирта над нагретыми катализаторами (метод акад. С.В.Лебедева)
2CH3CH2OH ––~450°С;ZnO,Al2O3® CH2=CH–CH=CH2 + 2H2O + H2
Физические свойства
Бутадиен -1,3 – легко сжижающийся газ с неприятным запахом, t°пл.= -108,9°C, t°кип.= -4,5°C; растворяется в эфире, бензоле, не растворяется в воде. 2- Метилбутадиен -1,3 – летучая жидкость, t°пл.= -146°C, t°кип.= 34,1°C; растворяется в большинстве углеводородных растворителях, эфире, спирте, не растворяется в воде.
Химические свойства
Атомы углерода в молекуле бутадиена-1,3 находятся в sp2 - гибридном состоянии, что означает расположение этих атомов в одной плоскости и наличие у каждого из них одной p- орбитали, занятой одним электроном и расположенной перпендикулярно к упомянутой плоскости.
|
|
Схематическое изображение строения молекул дидивинила (а) и вид модели сверху (b). Перекрывание электронных облаков между С1–С2 и С3–С4 больше, чем между С2–С3. |
|
 a)
a) b)
b)
p- Орбитали всех атомов углерода перекрываются друг с другом, т.е. не только между первым и вторым, третьим и четвертым атомами, но и также между вторым и третьим. Отсюда видно, что связь между вторым и третьим атомами углерода не является простой s- связью, а обладает некоторой плотностью p- электронов, т.е. слабым характером двойной связи. Это означает, что s- электроны не принадлежат строго определенным парам атомов углерода. В молекуле отсутствуют в классическом понимании одинарные и двойные связи, а наблюдается делокализация p- электронов, т.е. равномерное распределение p- электронной плотности по всей молекуле с образованием единого p- электронного облака. Взаимодействие двух или нескольких соседних p- связей с образованием единого p- электронного облака, в результате чего происходит передача взаимовлияния атомов в этой системе, называется эффектом сопряжения. Таким образом, молекула бутадиена -1,3 характеризуется системой сопряженных двойных связей. Такая особенность в строении диеновых углеводородов делает их способными присоединять различные реагенты не только к соседним углеродным атомам (1,2- присоединение), но и к двум концам сопряженной системы (1,4- присоединение) с образованием двойной связи между вторым и третьим углеродными атомами. Отметим, что очень часто продукт 1,4- присоединения является основным. Рассмотрим реакции галогенирования и гидрогалогенирования сопряженных диенов.

Как видно, реакции бромирования и гидрохлорирования приводят к продуктам 1,2- и 1,4- присоединения, причем количество последних зависит, в частности, от природы реагента и условий проведения реакции. Важной особенностью сопряженных диеновых углеводородов является, кроме того, их способность вступать в реакцию полимеризации. Полимеризация, как и у олефинов, осуществляется под влиянием катализаторов или инициаторов. Она может протекать по схемам 1,2- и 1,4- присоединения.

Полимеризация диеновых соединений
В упрощенном виде реакцию полимеризации бутадиена -1,3 по схеме 1,4 присоединения можно представить следующим образом:
|
––––® |
|

 .
.
В полимеризации участвуют обе двойные связи диена. В процессе реакции они разрываются, пары электронов, образующие s- связи разобщаются, после чего каждый неспаренный электрон участвует в образовании новых связей: электроны второго и третьего углеродных атомов в результате обобщения дают двойную связь, а электроны крайних в цепи углеродных атомов при обобщении с электронами соответствующих атомов другой молекулы мономера связывают мономеры в полимерную цепочку.
Элементная ячейка полибутадиена представляется следующим образом :
![]() .
.
Как видно, образующийся полимер характеризуется транс- конфигурацией элементной ячейки полимера. Однако наиболее ценные в практическом отношении продукты получаются при стереорегулярной (иными словами, пространственно упорядоченной) полимеризации диеновых углеводородов по схеме 1,4- присоединения с образованием цис- конфигурации полимерной цепи. Например, цис- полибутадиен
![]() .
.
Натуральный и синтетический каучуки
Натуральный каучук получают из млечного сока (латекса) каучуконосного дерева гевеи, растущего в тропических лесах Бразилии.
При нагревании без доступа воздуха каучук распадается с образованием диенового углеводорода – 2- метилбутадиена-1,3 или изопрена. Каучук – это стереорегулярный полимер, в котором молекулы изопрена соединены друг с другом по схеме 1,4- присоединения с цис- конфигурацией полимерной цепи :

Молекулярная масса натурального каучука колеблется в пределах от 7.104 до 2,5.106.
транс- Полимер изопрена также встречается в природе в виде гуттаперчи.

Натуральный каучук обладает уникальным комплексом свойств: высокой текучестью, устойчивостью к износу, клейкостью, водо- и газонепроницаемостью. Для придания каучуку необходимых физико-механических свойств: прочности, эластичности, стойкости к действию растворителей и агрессивных химических сред – каучук подвергают вулканизации нагреванием до 130-140°С с серой. В упрощенном виде процесс вулканизации каучука можно представить следующим образом :

Свойства сопряженных диеновых углеводородов