

8. Причины отклонения давления паров над раствором от з-на Рауля.
При образовании раствора, когда в чистый раст-ль вводится растворенное вещество,появляется новая структура с иным расположением частиц,чем в чистом растворителе,которая будет зависеть от состава раствора и будет специфична для каждого раствора.Изменяются и силы ММВ.В растворе кроме взаимодействия между однородными молекулами появляется взаимодействие и между разнородными частицами.Частицы растворенного вещества взаимодействуют друг с другом и с мол-ми раст-ля.Это является причиной отклонения давления паров над раствором от з-на Рауля.
«+» и «-» отклонения обусловлены разными факторами:
Если разнородные молекулы в растворе взаимно притягиваются с меньшей силой, чем однородные, т.е. FA-B < FA-A, то это облегчит переход из жидкой фазы в паровую (по сравнению с чистыми жидкостями) и будут наблюдаться положительные отклонения.
Усиление взаимного притяжения в разнородных молекулах (сольватация, водородная связь, образование химического соединения) затрудняет переход в газовую фазу и будут наблюдаться отрицательные отклонения.
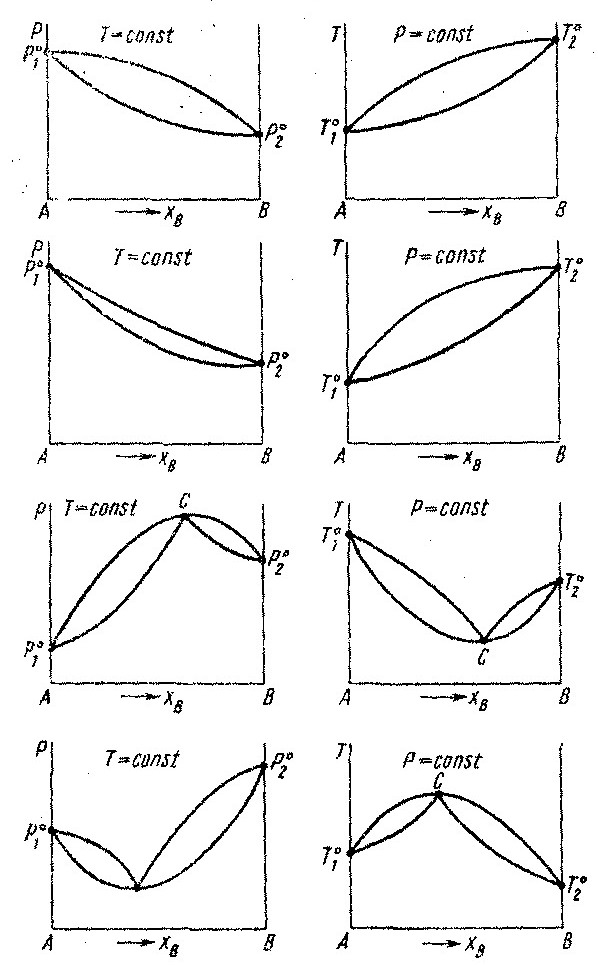
9.Диаграммы давление –состав и температура – состав для ревльных растворов
I)
II)
III)
IV)
I – Растворы с незначительными положительными отклонениями.
II – Растворы с незначительными отрицательными отклонениями.
III – Расворы со значительными положительными отклонениями.
IV – Растворы со значительными отрицательными отклоненями.
«+» и «-» отклонения обусловлены разными факторами:
Если разнородные молекулы в растворе взаимно притягиваются с меньшей силой, чем однородные, т.е. FA-B < FA-A, то это облегчит переход из жидкой фазы в паровую (по сравнению с чистыми жидкостями) и будут наблюдаться положительные отклонения.
Усиление взаимного притяжения в разнородных молекулах (сольватация, водородная связь, образование химического соединения) затрудняет переход в газовую фазу и будут наблюдаться отрицательные отклонения.
Растворы со значительными положительными или отрицательными отклонениями от идеальности способны образовывать азеотропные смеси, которые на диаграммах состояния изображены точкой с. Азеотропные смеси – это растворы при испарении которых получается пар того же состава, что и исходная жидкая смесь. В реальных растворах азеотропная смесь имеет самую низкую или самую высокую температуру кипения.При изменении внешнего давления изменяется не только Ткип,но и состав азеотропного раствора, т.е. азеотропная смесь не является хим. соединением. Азеотропные смеси образуются не только в системах со значительными отклонениями от з-на Рауля,но и в системах с незначительными отклонениями,если компоненты раствора имеют близкие Ткип, т.е. почти одинаковые давления пара над чистыми компонентами. В этом случае на диаграмме состояния появляется экстремум, лежащий в средней части диаграммы.Чем больше различие между Р01 и Р02 тем больше положение экстремума сдвинутов сторону одного из компонентов:при максимуме – в сторону более летучего;при минимуме – в сторону менее летучего.
10.Первый з-н Канавалова
Пар над смесью 2х летучих жидкостей относительно богаче тем из компонентов, прибавление которого к смеси повышает общее давление пара при данной температуре или понижает температуру кипения.
Графическое доказательство
На основе рис. Можно показать, что в паровой фазе содержится больше НКК, чем в жидкой фазе, находящейся в равновесии с паровой фазой. Для доказательства этого нагреваем изобарически жидкость состава х1 до температуры кипения Тк. При этой температуре жид-ть испаряется и образуется пар состава у1, который при конденсации образует жидкость состава х2. Из рисунка следует, что у2 > у1, то есть в паровой фазе НКК содержится больше, чем в жидкой фазе. Следовательно, в паре содержится больше того компонента, который кипит при более низкой температуре.
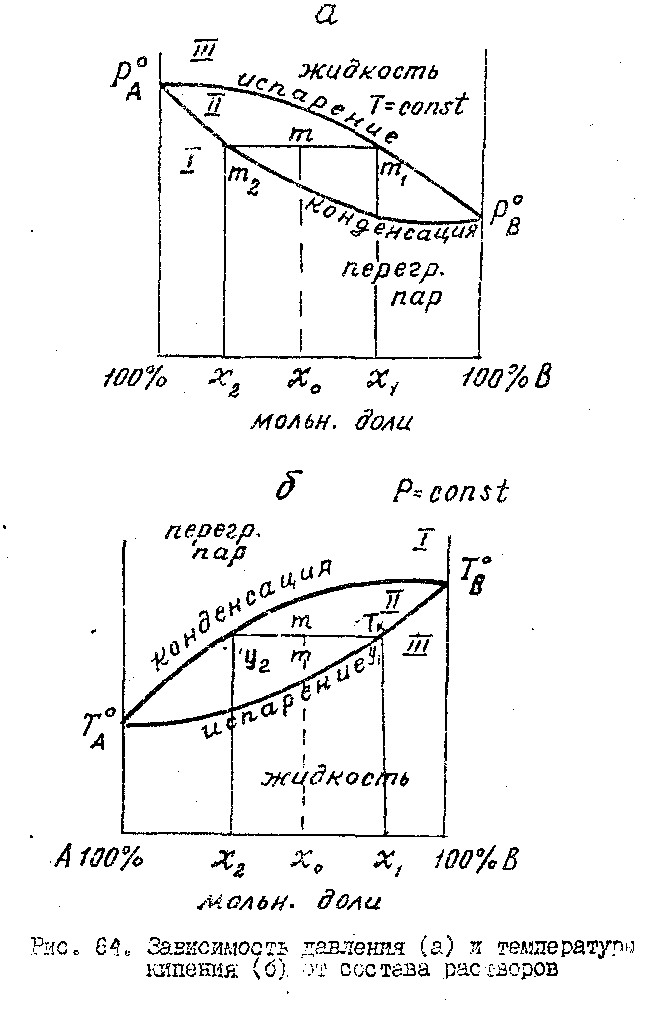
I – область пара, II – область жидкости, III – область равновесия пара и ж-ти.
Термодинамический вывод: исходя из уравнения Гиббса - Дюгема, которое для бинарной смеси жидкостей на небольшом удалении от экстремума можно записать в таком виде.
![]()
Подставляем в это выражение
Х1=1-Х2 :
![]()
Выделяем из dp1 :
![]()
Давление p1 и р2 по з-ну Дальтона можно связать с составом паровой фазы:
р1=(1-у2)Р и р2=у2Р
Подставляем в предыдущее уравнение:
![]()
Полное давление по з-ну Дальтона равно сумме парциальных давлений:
Р=р1+ р2
Дифференцируем dP=dp1 +dp2
![]()
Поделим на dx2
![]()