

Отклонения от закона Рауля.
Растворы, компоненты которых существенно различаются по физическим и химическим свойствам, подчиняются закону Рауля лишь в области очень малых концентраций; при больших концентрациях наблюдаются отклонения от закона Рауля. Случай, когда истинные парциальные давления паров над смесью больше, чем вычисленные по закону Рауля, называют положительными отклонениями. Противоположный случай, когда парциальные давления паров компонентов оказываются меньше вычисленных — отрицательные отклонения.
Причиной отклонений от закона Рауля является то обстоятельство, что однородные частицы взаимодействуют друг с другом иначе, чем разнородные (сильнее в случае положительных и слабее в случае отрицательных отклонений).
Реальные растворы с положительными отклонениями от закона Рауля образуются из чистых компонентов с поглощением теплоты (ΔНраств > 0); объём раствора оказывается больше, чем сумма исходных объёмов компонентов (ΔV > 0). Растворы с отрицательными отклонениями от закона Рауля образуются с выделением теплоты (ΔНраств < 0); объём раствора в этом случае будет меньше, чем сумма исходных объёмов компонентов (ΔV < 0).
2 Закон Рауля.
Температура кипения- это температура при которой давление насыщенного пара жидкости равно нормальному давлению. Δtкип=tкип.р-ра – tкип. раст-ля ; Δtкрист=tкрист.чистого р-ля- tкрист.р-ра
Δtкип= Кэ*Сm
Δtкрист=Кк*Сm
Эбуллиоскопическая константа – разница между температурой кипения раствора и температурой чистого растворителя.(Кэ)
Криоскопическая константа – разница между температурой замерзания раствора и температурой чистого растворителя.(Кк)
формулировка 2 закона: В разбавленном растворе неэлектролита повышение t кипения и понижение t кристаллизации прямопропорционально моляльной концентрации растворённого вещества.
![]() барометрическая формула
Больцмана.
барометрическая формула
Больцмана.
Следствия из закона Рауля:
1. Растворение нелетучего компонента в растворителе приводит к расширению температурной области существования жидкой фазы.
2. Понижение температуры замерзания и повышение температуры кипения прямо пропорциональны моляльной концентрации растворенного вещества.
3. Растворы, содержащие одинаковое число молей растворенных веществ в одинаковых молях растворителя, обнаруживают одно и то же понижение температуры замерзания и одно и то же повышение температуры кипения.
Билет 5
Термохимические законы
Одним из важнейших законов природы является закон сохранения энергии (первое начало термодинамики). Энергия не возникает из ничего и не исчезает, она может лишь переходить из одного вида в другой, поэтому в изолированной системе запас энергии постоянен, независимо от протекающих в ней процессов.
Каждая система обладает определенным запасом внутренней энергии, которая является функцией состояния системы, так как она не зависит только от состояния системы и не зависит от того, каким путем это состояние достигнуто.
Изменение внутренней энергии (ΔU) равно разности между количеством теплоты, полученной при протекании какого-либо процесса (Q), и количеством произведенной при этом работы (A): ΔU=Q-A.
Первый закон термохимии. Тепловой эффект прямой реакции равен по абсолютному значению и противоположен по знаку тепловому эффекту обратной реакции. Или, осуществив в системе какой-либо химический процесс, а затем – ему противоположный, мы возвращаем систему в первоначальное состояние с той же внутренней энергией, какую она имела.
Второй термохимический закон (закон Гесса). Тепловой эффект химической реакции не зависит от пути ее протекания и определяется только начальным и конечным состоянием системы.
Следствие из закона Гесса. Тепловой эффект реакции равен разности суммы теплот образования продуктов реакции и суммы теплот образования исходных веществ с учетом стехиометрических коэффициентов в уравнении реакции.
H0298=∑H0298 образ.продук.-∑Н0298обр.исх.вв.
NH4Cl=NH3+HCl
H0(NH4Cl)=-314,8
H0(NH3)=-46,15
H0(HCl)=-91,54
H0298реакуии=(H0NH3+H0298HCl)-H0NH4Cl=177,01>0 эндотермическая
При термохимических расчетах энтальпию образования простых веществ в стандартных условиях принимают равной нулю.
Осмос- явление односторонней диффузии через полупроницаемую перегородку. Необходимым условием возникновения осмоса является наличие растворителя и раствора или двух растворов различной концентрации, разделенных полупроницаемой мембраной. С точки зрения термодинамики движущей силой осмоса является стремление системы к выравниванию концентраций, так как при этом энтропия системы возрастает, поскольку система переходит в менее упорядоченное состояние, энергия Гиббса системы соответственно уменьшается, химические потенциалы выравниваются. Поэтому осмос - самопроизвольный процесс.
Давление, которое создается при распределении веществ в среде растворителя называется осмотическим давлением.
Изотонический раствор – раствор с одинаковым осмотическим давлением.
Гипертанический раствор – раствор с высоким осмотическим давлением.
Гипотанический раствор-раствор с низким осмотическим давлением.
Законы осмотического давления.
Закон осмотического давления, открытый Ван-Гоффом, утверждает, что его величина в числовом выражении равна такому давлению, которое оказывало бы вещество данного раствора, будучи при этой же температуре идеальным газом, с условием, что его объем был бы равен объему раствора. Вант-Гофф предложил эмпирическое уравнение для расчета осмотического давления разбавленных растворов неэлектролитов: Р=СМ*R*Т
1) Термодинамика- наука о свойствах макросистем, изучающая превращения различных форм энергии, рассматривает результат тех сил, которые имеют место в макросистеме. Отдельные частицы не изучает. Химическая термодинамика- раздел термодинамики, который изучает применение законов термодинамики к физическим и химическим процессам. Конкретно изучает:
1)тепловые эффекты хим.рекций
2)химическое равновесие
3)фазовое равновесие.
Термодинамика включает в себя: термохимию, учения о хим.равновесии, жидких и твердых телах, фазовых переходах, возможность протекания процесса.
Хим.термодинамика рассматривает свойства системы, когда она находится в равновесном состоянии, те все параметры постоянны во всех точках и не измен.самопроизвольно.
В химической термодинамике рассматриваются системы: изолированные – не обменивающиеся с окружающей средой веществом и энергией; закрытые – обменивающиеся энергией с окружающей средой и не обменивающиеся веществом. Состояние системы можно охарактеризовать термодинамическими параметрами, к которым относятся: температура, давление, концентрация, плотность, объем.
Если состояние системы характеризуется постоянными и неизменными во времени значениями термодинамических параметров во всех точках системы, то она находится в состоянии равновесия. При изменении одного из параметров состояния система переходит в состояние нового равновесия. Химическая термодинамика рассматривает переходы из одного состояния в другое, при этом могут изменяться или оставаться постоянными некоторые параметры:
· изобарические – при постоянном давлении;
· изохорические – при постоянном объеме;
· изотермические – при постоянной температуре;
· изобарно - изотермические – при постоянном давлении и температуре и т.д.
Термодинамические свойства системы можно выразить с помощью нескольких функций состояния системы, называемых характеристическими функциями: внутренней энергии U, энтальпии H, энтропии S, энергии Гиббса G, энергии Гельмгольца F. Характеристические функции обладают одной особенностью: они зависят только от состояния системы и не зависят от способа (пути) достижения данного состояния системы. Их значение определяется параметрами системы (давлением, температурой и др.) и зависит от количества или массы вещества, поэтому принято относить их к одному молю вещества.
В каждом теле, в каждом веществе в скрытом виде заключена внутренняя энергия, которая включает в себя энергию поступательно, колебательного, вращательного движения молекул; энергию размещения электронов на различных уровнях и подуровнях в атомных и молекулярных орбиталях; энергию связи ядерных частиц в ядре. Она представляет собой способность системы к совершению работы или передаче теплоты. Однако можно определить ее изменение U при переходе из одного состояния в другое:
ΔU = U2 - U1 ,
где U2 и U1- внутренняя энергия системы в конечном и начальном состояниях. Если ΔU > 0 –внутренняя энергия системы возрастает, еслиΔU < 0 – внутренняя энергия системы убывает. U – термодинамическая функция состояния, так как ее количество не будет зависеть от пути и способа перехода системы, а будет определяться лишь разностью в этих состояниях. При переходе из одного состояния в другое система может обмениваться с окружающей средой веществом или энергией в форме теплоты и работы.
А- работа- мера энергии, переданной от одного тела к другому за счёт перемещения масс под действием каких-либо сил.
Q- мера энергии переданной от одного тела другому за счёт разницы температур этих тел, перенос вещества при этом не происходит.
А и Q зависят от способа проведения процесса.
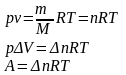
где Δn- изменение кол-ва вещ-ва, R- унив.газовая постоянная.
Δn=1 моль, ΔТ= 1К. А=R, где А- работа. которую совершает 1 моль газа при повыш.Т на 1 К.
1)А=0, Rv>0 => ΔU>0- эндотермич.процесс
2)Qv=0, A>0 => ΔU<0- экзотермич.процесс
Энтальпия, также тепловая функция и теплосодержание — термодинамический потенциал, характеризующий состояние системы в термодинамическом равновесии при выборе в качестве независимых переменных давления, энтропии и числа частиц.
Проще говоря, энтальпия — это та энергия, которая доступна для преобразования в теплоту при определенных температуре и давлении. Все химические реакции сопровождаются выделением (экзотермические) или поглощением (эндотермические) тепла. Мерой теплоты реакции служит изменение энтальпии ΔН, которая соответствует теплообмену при постоянном давлении. В случае экзотермических реакций система теряет тепло и ΔН — величина отрицательная. В случае эндотермических реакций система поглощает тепло и ΔН — величина положительная. Количественное соотношение между изменением внутренней энергии, теплотой и работой устанавливает первый закон термодинамики:
ΔН=ΔU+pΔV= ΔU + А.
Он устанавливает связь между кол-вом теплоты, выделенным или полученным в процессе реакций; кол-вом работы, произведенной системой и изменением внутр.энергии. Иными словами, если к системе подводится теплота Q, то она расходуется на изменение внутренней энергии системы ΔU и на совершение системой работы А над окружающей средой.
2) ХИМИЧЕСКОЕ РАВНОВЕСИЕ –
состояние химической системы, при
котором возможны реакции, идущие с
равными скоростями в противоположных
направлениях. При химическом равновесии
концентрации реагентов, температура и
другие параметры системы не изменяются
со временем. Количественная оценка
процесса эл-ой диссоциации характеризуется
двумя величинами: степенью диссоциации
а, и Кд. Степенью диссоциации наз.долю
диссоциированных на ионы молекул, те
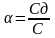
Степень
эл-ой диссоциации зависит как от природы
растворённого вещества, так и от
концентрации раствора, увеличиваясь с
его разбавлением. По значению а электролиты
условно делят на 3 типа: сильные (а>=30%),
средней силы (a=3-30%),
слабые (a<30%).
Степень диссоциации слабого электролита
уменьшается, если в р-р вводят один из
ионов, образующихся при его диссоциации-
одноимённый ион. Степень ЭД связана с
изотоническим коэффициентом i:
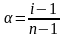 ,
где n-
число ионов, на которые распадается
молекула электролита. На основе принципа
Ле Шателье установили, что разбавление
раствора должно приводить к увеличению
степени диссоциации α. Но эту взаимосвязь
можно выразить и количественно. Рассмотрим
электролит, молекулы которого XY
распадаются в растворе на два иона –
катион и анион. Фактически, степень
диссоциации α представляет собой долю
распавшихся молекул. Если общую
концентрацию электролита (С) умножить
на эту долю, то получим концентрацию
любого из ионов. Иными словами, αС = [X+]
моль/л, и αC = [Y–] моль/л. Если электролит
сильный (α = 1), концентрация каждого из
ионов просто равна исходной концентрации
электролита. Например, в 1 литре 0,1 М
раствора NaCl содержится 0,1 моль ионов
Na+ и 0,1 моль ионов Cl–. К физическому
смыслу произведения αС можно прийти и
другим путем. Допустим, имеется раствор
циановодорода HCN (синильной кислоты) с
концентрацией С моль/л. В растворе
циановодород слабо диссоциирует на
ионы: HCN = H+ + CN–
,
где n-
число ионов, на которые распадается
молекула электролита. На основе принципа
Ле Шателье установили, что разбавление
раствора должно приводить к увеличению
степени диссоциации α. Но эту взаимосвязь
можно выразить и количественно. Рассмотрим
электролит, молекулы которого XY
распадаются в растворе на два иона –
катион и анион. Фактически, степень
диссоциации α представляет собой долю
распавшихся молекул. Если общую
концентрацию электролита (С) умножить
на эту долю, то получим концентрацию
любого из ионов. Иными словами, αС = [X+]
моль/л, и αC = [Y–] моль/л. Если электролит
сильный (α = 1), концентрация каждого из
ионов просто равна исходной концентрации
электролита. Например, в 1 литре 0,1 М
раствора NaCl содержится 0,1 моль ионов
Na+ и 0,1 моль ионов Cl–. К физическому
смыслу произведения αС можно прийти и
другим путем. Допустим, имеется раствор
циановодорода HCN (синильной кислоты) с
концентрацией С моль/л. В растворе
циановодород слабо диссоциирует на
ионы: HCN = H+ + CN–
Напомним,
что в выражении для степени диссоциации
α величина n0 отражает количество молекул
(или молей) HCN, изначально попавших в
раствор. В этом случае:
 ,
иными словами:
,
иными словами:
Отсюда следует, что
[H+] = Cα, [CN–] = Cα.
Итак, мы приходим к тому же выводу: чтобы получить концентрацию любого из ионов в растворе, достаточно общую концентрацию вещества С умножить на коэффициент α – степень его диссоциации.
Если теперь записать выражение для константы диссоциации HCN, то можно выразить Кд через Сα:

Концентрация недиссоциированных молекул [HCN] в равновесии должна быть меньше исходной концентрации С как раз на величину Сα – отсюда выражение (С – Сα) в знаменателе дроби. Если вынести концентрацию С за скобки и сократить, то получим следующее выражение:
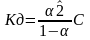 ,
Это не что иное, как математическая
формулировка закона разбавления
Оствальда: Закон разбавления Оствальда
— соотношение, выражающее зависимость
эквивалентной электропроводности
разбавленного раствора бинарного
слабого электролита от концентрации
раствора. Закон Оствальда справедлив
для бинарных электролитов, т.е. веществ,
молекулы которых в растворе распадаются
на два иона – катион и анион. В случае
сильных электролитов, когда α близка к
1, знаменатель дроби стремится к нулю,
а константа диссоциации Кд – к
бесконечности.
,
Это не что иное, как математическая
формулировка закона разбавления
Оствальда: Закон разбавления Оствальда
— соотношение, выражающее зависимость
эквивалентной электропроводности
разбавленного раствора бинарного
слабого электролита от концентрации
раствора. Закон Оствальда справедлив
для бинарных электролитов, т.е. веществ,
молекулы которых в растворе распадаются
на два иона – катион и анион. В случае
сильных электролитов, когда α близка к
1, знаменатель дроби стремится к нулю,
а константа диссоциации Кд – к
бесконечности.