

Тема 35.Системная красная волчанка. Склеродермия. Дерматомиозит
Системная красная волчанка, системная склеродермия и дерматомиозит относятся к диффузным болезням соединительной ткани, которые характеризуются системным типом воспаления различных органов и систем в сочетании с развитием аутоиммунных и иммунокомплексных процессов, а также избыточным фиброзообразованием. Практическим обоснованием для объединения этих заболеваний является схожесть клинических проявлений, особенно в ранней стадии, необходимость проведения дифференциальной диагностики, общие лабораторные показатели воспалительной активности, групповые и характерные для каждого заболевания иммунологические маркеры, близкие принципы противовоспалительной и иммуносупрессивной и терапии.
СИСТЕМНАЯ КРАСНАЯ ВОЛЧАНКА
Системная красная волчанка (СКВ) - системное аутоиммунное ревматическое заболевание неизвестной этиологии, характеризующееся гиперпродукцией широкого спектра органонеспецифических аутоантител к различным компонентам клеточного ядра с развитием иммуновоспалительного повреждения тканей и внутренних органов.
ЭПИДЕМИОЛОГИЯ.Заболеваемость СКВ составляет 4-250 случаев на 100 000 населения в год. Более 70% заболевают в возрасте 14-40 лет, пик заболеваемости приходится на 14-25 лет. Соотношение женщин и мужчин от 8:1 до 10:1, среди детей - 3:1. Наиболее часто развивается у женщин репродуктивного возраста. Риск возрастает во время беременности и в послеродовом периоде. Смертность при СКВ в 3 раза выше, чем в популяции.
ЭТИОЛОГИЯ СКВ до настоящего времени неизвестна. Обсуждается роль различных инфекционных агентов, токсических веществ, некоторых лекарственных препаратов. Косвенным подтверждением триггерной роли вирусной или бактериальной инфекции является более частое, чем в популяции, выявление серологических признаков инфекции вируса Эпштейн-Барр, способность бактериальной ДНК стимулировать синтез антиядерных аутоАТ, а также способность вирусных белков и «волчаночных» аутоАГ к «молекулярной мимикрии». Ультрафиолетовое облучение рассматривается как фактор, стимулирующий апоптоз клеток кожи, что приводит к появлению на мембране апоптозных клеток внутриклеточных АГ, индуцирующих развитие аутоиммунного воспаления у генетически предрасположенных индивидуумов. На наследственную предрасположенность указывает распространенность СКВ среди кровных родственников (5-12%). Отмечена связь с негенетически обусловленным дефицитом отдельных компонентов комплемента (С1q, С4, С2), полиморфизмом генов FcγRII рецепторов ФНО-α, увеличение частоты носительства HLA-B8 и HLA-DR3. Влияние нарушений гормональной регуляции обусловленоиммунорегуляторными возможностями гормональной системы. У женщин имеет значение избыточный синтез эстрогенов и пролактина, которые стумулируют иммунный ответ, и недостаток андрогенов, обладающих иммуносупрессивной активностью. У мужчин – гипоандрогенемия и гиперпродукция пролактина.Основной концепцией в настоящее время остается полиэтиологическая. Под влиянием неблагоприятных факторов начинает действовать любой из перечисленных факторов, что приводит к запуску патологической иммуновоспалительной реакции.
ПАТОГЕНЕЗ СКВ определяют два тесно взаимосвязанных процесса. На ранней стадии – преобладание поликлональной (В-клеточной) активации иммунитета, в дальнейшем – антигенспецифические (Т-клеточные) иммунные реакции. Фундаментальное иммунное нарушение – врожденные или индуцированные дефекты программированной гибели клеток. Характерна неконтролируемая продукция антител, образующих иммунные комплексы, которые откладываются в субэндотелиальном слое базальной мембраны сосудов многих органов, определяя различные признаки болезни. Схематически патогенез СКВ можно представить как комплексное воздействие генетических, гормональных и иммунорегуляторных факторов, направленных на продукцию антител В-лимфоцитами.
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ Начало болезни может быть представлено поражением одного или двух органов, однако для развернутой клинической картины характерна полиорганная вариабельная симптоматика. При обострении возможно вовлечение в патологических процесс ранее интактных органов и систем.
Конституциональные нарушения включают слабость, лихорадку, снижение массы тела и отражают активность патологического процесса.
Рис. 1 Волчаночная «бабочка»
Поражение кожи выявляют у 55-90% пациентов. Выделяют 28 вариантов кожных изменений при СКВ. В 40-70% случаев встречается фотосенсибилизация – повышенная чувствительность к инсоляции, которая в половине случаев п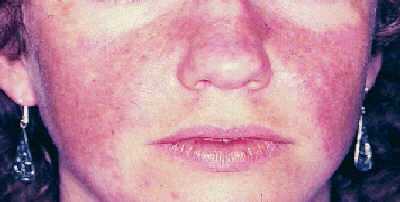 риводит
к обострению заболевания. Фотоиндуцируемые
кожные изменения развиваются на открытых
участках тела (лицо, зона «декольте»,
верхние конечности) и представлены
разнообразными макулярными, папулезными,
буллезными повреждениями, а также
классической эритемой. Прочие изменения
кожи могут быть представлены центробежной
эритемой Биетта, сосудистой (васкулитной)
«бабочкой»,
которая описывается как нестойкое
пульсирующее разлитое покраснение
цианотичного оттенка, усиливающаяся
при воздействии ветра, мороза, солнца,
психоэмоциональных нагрузок (рис. 1).
Могут быть поражения кожи в виде
дискоидной
красной волчанки,
характерными признаками которой являются
эритема, инфильтрация, геперкератоз и
атрофия, люпус-панникулита,
гипер- и депигментации, алопеции –
частого, но неспецифичного признака.
Диагностически значимыми являются
капилляриты
– отечная эритема с мелкоточечными
геморрагиями на подушечках пальцев
рук, ладонях и подошвах.
риводит
к обострению заболевания. Фотоиндуцируемые
кожные изменения развиваются на открытых
участках тела (лицо, зона «декольте»,
верхние конечности) и представлены
разнообразными макулярными, папулезными,
буллезными повреждениями, а также
классической эритемой. Прочие изменения
кожи могут быть представлены центробежной
эритемой Биетта, сосудистой (васкулитной)
«бабочкой»,
которая описывается как нестойкое
пульсирующее разлитое покраснение
цианотичного оттенка, усиливающаяся
при воздействии ветра, мороза, солнца,
психоэмоциональных нагрузок (рис. 1).
Могут быть поражения кожи в виде
дискоидной
красной волчанки,
характерными признаками которой являются
эритема, инфильтрация, геперкератоз и
атрофия, люпус-панникулита,
гипер- и депигментации, алопеции –
частого, но неспецифичного признака.
Диагностически значимыми являются
капилляриты
– отечная эритема с мелкоточечными
геморрагиями на подушечках пальцев
рук, ладонях и подошвах.
Поражение слизистыхдиагностируют у 7- 40% заболевших, чаще в виде энантемы (эритематозные участки с геморрагическими вкраплениями и эрозиями слизистой оболочки), афтозного стоматита (умеренно болезненные язвы), люпус-хейлита.
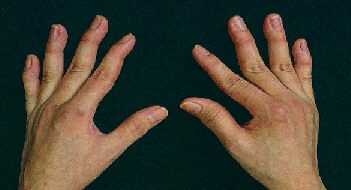
Рис. 2 Артропатия Жакку
 Поражение
опорно-двигательного аппарата
встречается у 80-90% пациентов на различных
этапах болезни, примерно в половине
случаев может быть первым признаком
болезни. Характерны артралгии,
скованность, симметричный артрит, чаще
мелких суставов кистей рук с поражением
периартикулярных
тканей
(тендиниты, теносиновиты) и развитием
артропатии
Жакку,
которая клинически выглядит как поражение
кисти при ревматоидном артрите (рис.
2). Поражение скелетной мускулатуры
(миалгии,
повышенная чувствительность мышц при
пальпации, мышечная слабость, атрофия)
в ряде случаев не отличаются от
классических идиопатических воспалительных
миопатий. Асептические
некрозы костей
наиболее часто развиваются в головке
бедренной кости и коленных суставах.
СКВ ассоциирована с высоким риском
остеопороза
(в 5 раз чаще).
Поражение
опорно-двигательного аппарата
встречается у 80-90% пациентов на различных
этапах болезни, примерно в половине
случаев может быть первым признаком
болезни. Характерны артралгии,
скованность, симметричный артрит, чаще
мелких суставов кистей рук с поражением
периартикулярных
тканей
(тендиниты, теносиновиты) и развитием
артропатии
Жакку,
которая клинически выглядит как поражение
кисти при ревматоидном артрите (рис.
2). Поражение скелетной мускулатуры
(миалгии,
повышенная чувствительность мышц при
пальпации, мышечная слабость, атрофия)
в ряде случаев не отличаются от
классических идиопатических воспалительных
миопатий. Асептические
некрозы костей
наиболее часто развиваются в головке
бедренной кости и коленных суставах.
СКВ ассоциирована с высоким риском
остеопороза
(в 5 раз чаще).
Поражение дыхательной системы может проявляться параличом или парезом голосовых связок гортани, развитием подглоточного стеноза, плевритом (40-60%), легочной гипертензией (5-14%) чаще вследствие ТЭЛА, острым волчаночным пневмонитом (1-4%), легочными (альвеолярными) геморрагиями (2%).
Поражение сердечно-сосудистой системы (52-89%)включает вовлечение перикарда (утолщением листков и/или наличие экссудата в сочетании с прочими серозитами), миокарда (миокардит, гипертрофия, миокардиодистрофия), эндокарда, коронарных артерий.Классический представитель клапанной патологии при СКВ - бородавчатый эндокардит Либмана-Сакса, который редко приводит к формированию стенозов и гемодинамически значимым нарушениям. Чаще вовлекается митральный клапан. Артериальная гипертензия наблюдается у 10-50%. Патология сосудов затрагивает в основном артерии среднего и мелкого калибра. Часто поражаются коронарные артерии. Развитие венозных тромбозов характерно для СКВ, ассоциированной с вторичным антифосфолипидным синдромом (АФС).
Поражение органов пищеварения проявляется анорексией, тошнотой, рвотой, диареей, асцитом (8-10%), васкулитом мезентериальных и внутриорганных артерий с развитием кишечных инфарктов и изъязвлений, панкреатита. Патология печени варьирует от незначительной гепатомегалии до тяжелого гепатита (10-31%).
Поражение почек (волчаночный нефрит) разнообразно и выявляется у 35-90% пациентов. Активные формы нефрита развиваются чаще в первые годы болезни. Выделяют быстропрогрессирующий нефрит с нефротическим синдромом, нефрит с выраженным нефритическим синдромом, нефрит с минимальным мочевым синдромом. В настоящее время используют морфологическую классификацию волчаночного нефрита.
Симптомы поражения нервной системы отмечаются у большинства пациентов, включают практически весь спектр неврологической симптоматики, могут быть первыми признаками болезни, возникающими задолго до появления развернутой картины СКВ. Наиболее частый вариант – транзиторные ишемические атаки, реже – инсульты,синдром псевдоопухоли мозга, эпилептические припадки (20-50%), хорея, церебральная атаксия, миелопатия / энцефаломиелопатия, периферические невропатии (2-21%), психические,нервно-психические и поведенческие проблемы (10-80%).
ЛАБОРАТОРНАЯ ДИАГНОСТИКА. Общий анализ крови. Увеличение СОЭ наблюдается часто, но, в отличие от лейкопении (чаще лимфопении), плохо коррелирует с клинической активностью заболевания. Может иметь место гипохромная анемия вследствие хронического воспаления, скрытого желудочно-кишечного кровотечения либо аутоиммунная гемолитическая анемия, тромбоцитопения. Общий анализ мочи– протеинурия, гематурия, лейкоцитурия, цилиндрурия. Изменения биохимического анализа крови неспецифичны, зависят от характера поражения внутренних органов.
Иммунологические нарушения.
Антинуклеарный фактор АНФ (ANA) - гетерогенная популяция аутоантител, реагирующая с различными компонентами клеточного ядра. Выявляется у 95% больных СКВ. Отсутствие АНФ ставит под сомнение диагноз СКВ.
Антитела к двуспиральной ДНК (20-60%). Повышение уровня коррелирует с активностью заболевания и развитием волчаночного нефрита.
Антитела к Sm (Smith) антигену – высокоспецифичны, но выявляют только в 10-30%.
Антитела к ядерному АГ пролиферирующих клеток (РСNA/cycline) - высокоспецифичны.
Антитела к Ro/SS-A антигену - ассоциируются с лимфопенией, тромбоцитопенией, фотодерматитом, легочным фиброзом, синдромом Шегрена.
АТ к рибосомальному белку Р встречаются у 10-20% пациентов, специфичны для СКВ, протекающей с волчаночным психозом.
Антитела к фосфолипидам, волчаночный антикоагулянт (ВА), ложноположительная реакция Вассермана – специфичны для АФС.
Определение LE клеток - лейкоцитов, фагоцитировавших ядерный материал. Для СКВ патогномонично определение большого числа LE-клеток.
Определение полного профиля аутоантител каждого пациента помогает предсказывать индивидуальные клинические проявления.
Инструментальные методы исследования
Опорно-двигательный аппарат (по показаниям): рентгенография костей и суставов, УЗИ мягких тканей и суставов, МРТ, денситометрия (определение минеральной плотности кости) – до начала лечения, затем – ежегодно. Дыхательная система: рентгенограмма ОГК (не реже 1 раза в год), по показаниям – функциональные тесты, фибробронхоскопия, КТ легких, ЭХО-КГ (для диагностики легочной гипертензии). Сердечно-сосудистая система: ЭКГ, ЭХО-КГ, УЗИ сосудов с измерением толщины интима-медиа, ХМ ЭКГ, ангиография по показаниям. Желудочно-кишечный тракт: ФГДС не реже 1 раза в год, УЗИ ОБП, по показаниям КТ, МРТ. Нервная система: МРТ, КТ, ЭЭГ по показаниям.
ДИАГНОСТИЧЕСКИЕ КРИТЕРИИ СКВ основаны на клинических и лабораторных признаках (табл. 35.1).
-
Таблица 35.1.
Диагностические критерии СКВ американской ассоциации ревматологов (1982 г.)
Критерий
Примечание
1. Сыпь на скулах
Фиксированная эритема на скуловых выступах, имеющая тенденцию к распространению к носогубной зоне.
2. Дискоидная сыпь
Эритематозные приподнимающиеся бляшки с прилипающими кожными чешуйками и фолликулярными пробками; на старых очагах могут быть атрофические рубцы.
3.Фотосенсибилизация
Кожная сыпь, возникающая в результате необычной реакции на солнечный свет.
4. Язвы в ротовойполости
Изъязвление полости рта или носоглотки; обычно безболезненные.
5. Артрит
Неэрозивный артрит, поражающий 2 или более периферических сустава, проявляющийся болезненностью, отёком и выпотом.
6. Серозит
Плеврит (плевральные боли, или шум трения плевры, или наличие плеврального выпота) или перикардит (подтверждённый с помощью эхокардиографии или выслушиванием шума трения перикарда).
7. Поражение почек
Персистирующая протеинурия >0,5 г/сут или цилиндрурия (эритроцитарная, гемоглобиновая, зернистая или смешанная).
8. Поражение ЦНС
Судороги или психоз (в отсутствие приёма ЛС или метаболических нарушений).
9. Гематологические
нарушения
Гемолитическая анемия с ретикулоцитозом, или лейкопения <4,0х109/л (зарегистрированная 2 и более раза), или тромбоцитопения <100х109/л (в отсутствие приёма ЛС).
10. Иммунологические
нарушения
Анти-ДНК или анти-Sm или аФЛ – увеличение уровня IgG или IgM (АТ кардиолипину) или положительный тест на ВА при использовании стандартных методов, или ложноположительная реакция Вассермана в течение как минимум 6 месяцев при подтверждённом отсутствии сифилиса с помощью реакции иммобилизации бледной трепонемы и теста флюоресцентной адсорбции трепонемных АТ.
11. Антинуклеарные антитела
Повышение уровня АНА методом ИФА или другим методом (при отсутствии приема лекарственных препаратов, вызывающих волчаночноподобный синдром).
Диагноз СКВ устанавливают при выявлении 4 или более из 11 вышеперечисленных критериев.
ФОРМУЛИРОВКА ДИАГНОЗА. При формулировке диагноза необходимо указывать вариант течения СКВ (острое, подострое, хроническое), степень активности, перечень клинических симптомов и лабораторных нарушений на момент обследования.
Острое течение – быстрое развитие мультиорганных проявлений, включая поражение почек и ЦНС, высокая иммунологическая активность.
Подострое – в дебюте наблюдаются конституциональные симптомы, неспецифическое поражение кожи и суставов. Заболевание протекает волнообразно, с периодическим возникновением обострений и развитием полиорганной симптоматики в течение 2-3 лет с момента появления первых симптомов.
Первично-хронический вариант течения – свойственно длительное превалирование одного или нескольких симптомов (дискоидные высыпания, синдром Рейно, артрит, судорожный синдром, гематологические нарушения, синдром Шегрена). Множественные органные поражения появляются к 5-10-му годам болезни. Первично-хронический вариант течения наиболее часто наблюдается при сочетании СКВ и вторичного АФС.
Выделяют также отдельные иммунологические вариантыСКВ: с дебютом в детском и подростковом возрасте, у пожилых, у мужчин, подострая кожная красная волчанка, вторичный антифосфолипидный синдром, синдром неонатальной волчанки.
Активность СКВ определяют при помощи индексов с балльной системой оценки: индекс SLAM (SystemLupusActivityMeasurement), SLEDAI (SystemicLupusErythematosusDiseaseActivityIndex), ECLAM (EuropeanConsensusLupusActivityMeasurement) - I минимальная, II умеренная, III высокая.
ЛЕЧЕНИЕ. Терапия СКВ - пожизненный процесс. Немедикаментозная терапия подразумевает исключение психоэмоциональных нагрузок, ограничение пребывания на солнце, активное лечение сопутствующих инфекционных заболеваний, эффективную контрацепцию в период обострения заболевания и на фоне лечения цитотоксическими препаратами.
Медикаментозная терапия.Наиболее значимые лекарственные средства для лечения СКВ – глюкокортикоиды (ГК), цитотоксические препараты, гидроксихлорохин, НПВП.
Доза ГКС зависит от активности заболевания.Пациентам с низкой активностью заболевания назначают преднизолон в дозе <10 мг/сут., при умеренной активности - 20–40 мг/сут. в течение 2–4 недель с постепенным снижением до поддерживающей дозы. При лечении тяжёлых клинических проявлений со стороны ЦНС, почек (гломерулонефрит), тромбоцитопении, гемолитической анемии назначают высокие дозы ГК и цитотоксических препаратов. Абсолютное показание для назначения высоких доз ГК (1 мг/кг/сут и более) – высокая активность СКВ, на фоне которой в отсутствие лечения очень быстро развивается необратимое поражение жизненно важных органов. Длительность приема максимальной дозы варьирует от 4 до 12 недель. Снижение дозы проводится постепенно, под тщательным клинико-лабораторным контролем. Поддерживающая терапия – 5-10 мг/сутки (пожизненно). Пульс-терапия (500-1000 мг метилпреднизолона внутривенно капельно в течение как минимум 30 минут 3 дня подряд) проводится при высокой активности СКВ для достижения быстрого эффекта, а также снижения дозы пероральных ГК.
Цитотоксические препараты. Показаны при активном волчаночном нефрите, генерализованном васкулите, высокой общей активностиь болезни, резистентности к ГК, при появлении побочных реакций ГК на первых этапах лечения и необходимости уменьшения поддерживающей дозы преднизолона.
Циклофосфамид (ЦФ) (внутривенно) – препарат выбора при волчаночном нефрите и тяжелом поражении ЦНС. Подавляющая терапия – 500 мг 1 раз в неделю 4 недели или 1000 мг 1 – 2 раза при комбинированной пульс терапии или 200 мг через день 10 раз (до суммарной дозы 2000 мг в месяц); поддерживающая терапия 1000 мг 1 раз в месяц - 6 месяцев, затем 200 мг 1 раз в неделю с постепенным увеличением интервалов между инъекциями (до 5 лет).
Азатиоприн. Используют для поддержки ремиссии волчаночного нефрита, индуцированной ЦФ, лечения резистентных к ГК форм аутоиммунной гемолитической анемии, тромбоцитопении, торпидных поражений кожи. Подавляющая терапия – 100 – 150 мг в сутки; поддерживающая терапия – 50 – 100 мг в сутки (до 5 лет).
Микофенолата мофетил (селсепт). Показан при диффузном пролиферативном волчаночным нефрите, рефрактерном к ЦФ. Препарат оказывает выраженный антипролиферативный эффект в отношении лимфоцитов и проявляет цитостатическую, а не цитотоксическую активность. Терапевтическая доза 2-3 г/сут. в 2 приема, поддерживающая – 1 г/сут.
Метотрексат назначается при рефрактерных к монотерапии ГК волчаночном артрите и поражениях кожи.
Циклоспорин А в дозе <5 мг/(кг х сут.) - альтернативный препарат второго ряда при непереносимости и неэффективности ГК и цитостатиков с положительным влиянием на активность заболевания, тяжелую органную патологию.
НПВП – применяют в стандартных дозах для лечения мышечно-скелетных проявлений, лихорадки, умеренно выраженного серозита.
Аминохинолиновые производные. Гидроксихлорохинназначают пациентам без тяжелых висцеральных проявлений, в период снижения доз ГК и цитостатиков для поддержания ремиссии. Подавляющая терапия – 600 мг в сутки; поддерживающая терапия – 200 – 400 мг в сутки (длительно, нередко пожизненно).
Экстракорпоральные методы лечения. Плазмаферез показан при цитопении, криоглобулинемии, васкулите, поражении ЦНС, тромботической тромбоцитопенической пурпуре, для лечения наиболее тяжёлых пациентов с быстро нарастающим нарушением функций жизненно важных органов (пневмонит, поражение ЦНС, быстропрогрессирующий люпус-нефрит с почечной недостаточностью) в сочетании с интенсивной терапией циклофосфамидом и ГКС. Наиболее эффективный метод лечения критических состояний и прогностически неблагоприятных вариантов СКВ – синхронная интенсивная терапия.
Биологические агенты.При лечении СКВ используются препараты, блокирующие пролиферацию В-клеток – рекомбинантные химерные (мышь-человек) моноклональные антитела к поверхностному антигену В лимфоцитов CD20 (ритуксимаб «МабТера»). Возможно развитие ремиссии продолжительностью до 12 месяцев.
АНТИФОСФОЛИПИДНЫЙ СИНДРОМ.
Антифосфолипидный синдром (АФС)– клинико-лабораторный симптомокомплекс, включающий рецидивирующие венозные и/или артериальные тромбозы, различные формы акушерской патологии (в первую очередь привычное невынашивание беременности), тромбоцитопению и связанный с синтезом антифосфлипидных антител (аФЛ).
Антифосфолипидные антитела - гетерогенная популяция аутоантител, реагирующих с широким спектром антигенных детерминант фосфолипидов или фосфолипидсвязывающих белков (β-2 – гликопротеин I и др.). К основным, диагностически значимым, аФЛ относят:
антитела к кардиолипину (классов IgG и IgM)
волчаночный антикоагулянт (ВА)
антитела к β-2 – гликопротеину-1.
Показания к определению антител к фосфолипидам: СКВ, венозный тромбоз в возрасте до 40 лет, артериальный тромбоз до 40 лет, необъясненный неонатальный тромбоз, идиопатическая тромбоцитопения, кожный некроз на фоне приема непрямых антикоагулянтов, необъяснимое удлинение АЧТВ, наличие родственников с тромботическими нарушениями. При диагностике АФС необходимо учитывать наличие у родственников ревматических заболеваний, рецидивирующих инсультов в возрасте до 40 лет, рецидивирующих инфарктов в возрасте до 40 лет, рецидивирующего тромбофлебита, спонтанных абортов, преэклампсии, эклампсии. Имеют значение перенесенные инфекции, прием лекарственных препаратов (гормональные контрацептивы, новокаинамид, гидралазин), которые могут провоцировать дебют АФС.
Классификация антифосфолипидного синдрома
Первичный АФС (выставляется при исключении вторичного АФС при длительном наблюдении за пациентом);
Вторичный АФС при: ревматических и аутоиммунных заболеваниях, злокачественных новообразованиях, применении лекарственных препаратов, инфекционных заболеваниях, наличии иных причин;
Катастрофический АФЛ синдром – распространенный тромбоз с полиорганной недостаточностью, гибелью пациента, несмотря на лечение;
Синдром Снеддона – сочетание сосудисто-мозговых нарушений и ливедо при выявлении в крови аФЛ.
Клинические и лабораторные критерии АФС представлены в табл. 35.2.
-
Таблица 35.2.
Критерии антифосфолипидного синдрома
Клинический
критерий
Характеристика
Сосудистый
тромбоз
Один или более эпизодов артериального, венозного тромбоза или тромбоза мелких сосудовлюбой ткани или органе. Тромбоз должен быть объективизирован с помощью получения изображения или гистопатологического исследования, при котором не выявляются признаки воспаления в стенке сосуда.
Патология
беременности
А) Один или более случай необъяснимой внутриутробной смерти морфологически нормального плода после 10 недель беременности, подтвержденный при ультразвуковом исследовании или морфологически
Б) Один или более эпизод преждевременных родов морфологически нормального ребенка до 34 недель беременности из-за тяжелой преэклампсии, или эклампсии или плацентарной недостаточности
В) Три или более последовательных необъяснимых самопроизвольных аборта при исключении гинекологической патологии или гормональной недостаточности, или хромосомных аномалий у обоих родителей.
Лабораторные критерии
Антитела к кардиолипину и β2-ГП1 IgG и/или IgM изотипов в среднем или высоком титре (40 GPL или выше), подтвержденные дважды с промежутком не менее 12 недель.
ВА - найденный в плазме по крайней мере 2 раза с интервалом не менее 12 недель.
Определенный АФС диагностируется при наличии одного клинического и одного серологического критерия. АФС исключается, если менее 12 недель или более 5 лет выявляются аФЛ без клинических проявлений или клинические проявления без аФЛ.
В основе сосудистой патологии при АФС – невоспалительная тромботическая васкулопатия, затрагивающая сосуды любого калибра и локализации (от капилляров до крупных сосудов, включая аорту). В связи с этим спектр клинических проявлений АФС чрезвычайно разнообразен. Самые частые и характерные проявления АФС – венозные и/или артериальные тромбозы, которые развиваются примерно у трети пациентов, в сыворотке которых выявлен аФЛ, и акушерская патология.
СИСТЕМНАЯ СКЛЕРОДЕРМИЯ
Системная склеродермия(ССД)(системный склероз) - аутоиммунное заболевание соединительной ткани с характерным поражением кожи, сосудов, опорно-двигательного аппарата и внутренних органов (легкие, сердце, пищеварительный тракт, почки) в основе которого лежат нарушения микроциркуляции, воспаление и генерализованный фиброз.
ССД относят к группе склеродермических болезней, которая также включает ограниченную (очаговую) склеродермию, диффузный эозинофильный фасциит, склеродерму Бушке, мультифокальный фиброз, индуцированные формы склеродермии и псевдосклеродермические синдромы.
РАСПРОСТРАНЕННОСТЬ. Первичная заболеваемость колеблется от 3,7 до 20,0 на 1 млн. населения в год. Регистрируют во всех странах мира, но лица негроидной расы подвержены заболеванию в большей степени, чем представители европеоидной расы. Соотношение мужчин к женщинам - 1:7. Заболевают преимущественно лица в возрасте 30-50 лет.
ЭТИОЛОГИЯ.Предполагается многофакторный генез ССД, сочетающий наследственную предрасположенность с воздействием неблагоприятных экзо- и эндогенных влияний. Выявлены ассоциации ССД с аллелями системы гистосовместимости HLA класса II (HLA-DR5, HLA-DR3, HLA-DR2). Принципиально важна корреляция иммуногенетических маркеров с иммунологическими (специфические аутоАТ) и клиническими проявлениями ССД. Среди инфекционных агентов обсуждают возможную роль ретровирусов и цитомегаловируса, активацию латентных вирусов, наличие механизмов молекулярной мимикрии и др. В последние годы особое внимание обращено на триггерное воздействие химагентов (промышленных, бытовых, алиментарных). Наряду с кремнием (шахтеры), токсичным рапсовым маслом (Испания) и содержащих L-триптофан пищевых добавок, выявлена и доказана роль отдельных органических растворителей, продуктов нефти, хлорвинила, пестицитов, отдельных лекарств (блеомицин), силиконовых имплантантов.
ПАТОГЕНЕЗ ССД чрезвычайно сложен. В его основе - нарушение иммунитета, микроциркуляции и фиброзообразования, взаимодействующие на уровне основных клеточных и рецепторно-лигандных систем. Характерен широкий спектр разнообразных нарушений клеточного и гуморального иммунитета: Т-клеточная активация, дизрегуляция в системе Th1- Th2-клеток, повышение уровня отдельных иммунорегуляторных цитокинов, наличие специфических антинуклеарных и антинуклеолярных АТ. Поражение сосудов и нарушение микроциркуляции – важнейшее звено патогенеза и морфогенеза ССД. Характерны признаки активации и деструкция эндотелия, пролиферация гладкомышечных клеток, утолщение интимы, сужение просвета сосудов микроциркуляторного русла, тромбозы. Повышенное коллагено- и фиброзообразование обусловлены гиперактивностью фибробластов, что ведет к избыточной продукции компонентов межклеточного матрикса, увеличению неофибриллогенеза и генерализованному фиброзу, свойственному ССД.
КЛАССИФИКАЦИЯ
Клинические формы: диффузная, лимитированная, перекрестная (оverlap-синдром), висцеральная форма, ювенильная склеродермия, индуцированная склеродермия, пресклеродермия.
Варианты течения:
Острое быстро прогрессирующее – диффузная форма поражения кожи и внутренних органов (сердце, легкие, почки) в первые 2 года от начала заболевания.
Подострое умеренно прогрессирующее – преобладание клинических и лабораторных признаков иммунного воспаления (плотный отек кожи, артрит, миозит), нередко overlаp-синдромы.
Хроническое медленно прогрессирующее течение – характерно преобладание сосудистой патологии (многолетний синдром Рейно с постепенным нарастанием сосудистых ишемических расстройств, висцеральной патологии).
Стадии ССД: I – начальная, выявляют 1-3 локализации болезни; II – генерализации, отражающая системный, полисиндромный характер процесса; III – поздняя, терминальная, когда имеется недостаточность одного или нескольких органов.
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ.
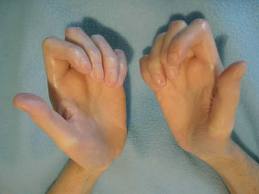
Рис.3 Склеродактлия
 Поражение
кожи
(склеродерма)
происходит у превалирующего большинства
пациентов, что характерным образом
меняет их облик.
Кожные
изменения имеют определенную стадийность,
включая стадиюплотного
отека, индурации и атрофии с
преимущественной локализацией на лице
и кистях.Нередко
кожные изменения сочетаются с сосудистой
патологией итрофическими нарушениями
(изъязвления,
гнойники, деформация ногтей, облысение).
При ограниченной склеродермии хронического
течения нередки телеангиэктазии
с локализацией на лице, слизистой губ,
реже – языке, твердом небе, груди, спине,
конечностях. Характерный признак
заболевания - склеродактилия
– плотный отек и индурация кожи кистей
с нарастающим ограничением движений и
развитием контрактур (рис. 3). Симптом
«кисета»
- уменьшение ротовой апертуры, истончение
красной каймы губ, вокруг которых
формируются радиальные складки. Прочие
симптомы: гиперпигментация
«соль с перцем»; атрофия
волосяных фолликулов, потовых и сальных
желез.
Поражение
кожи
(склеродерма)
происходит у превалирующего большинства
пациентов, что характерным образом
меняет их облик.
Кожные
изменения имеют определенную стадийность,
включая стадиюплотного
отека, индурации и атрофии с
преимущественной локализацией на лице
и кистях.Нередко
кожные изменения сочетаются с сосудистой
патологией итрофическими нарушениями
(изъязвления,
гнойники, деформация ногтей, облысение).
При ограниченной склеродермии хронического
течения нередки телеангиэктазии
с локализацией на лице, слизистой губ,
реже – языке, твердом небе, груди, спине,
конечностях. Характерный признак
заболевания - склеродактилия
– плотный отек и индурация кожи кистей
с нарастающим ограничением движений и
развитием контрактур (рис. 3). Симптом
«кисета»
- уменьшение ротовой апертуры, истончение
красной каймы губ, вокруг которых
формируются радиальные складки. Прочие
симптомы: гиперпигментация
«соль с перцем»; атрофия
волосяных фолликулов, потовых и сальных
желез.
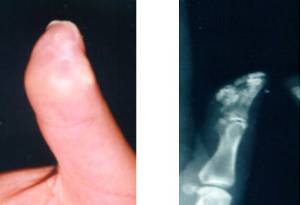
Рис. 4. Кальцинаты мягких тканей
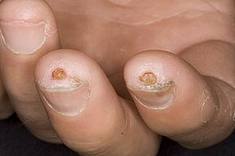
Рис. 5. Дигитальные язвочки и рубчики («крысиные укусы»)
Поражение опорно-двигательного аппарата. Наиболее частым проявлением являются полиартралгии, утренняя скованность, реже – полиартрит, в 20% - эрозивная артропатия. Типичное поражение мышц - фиброзная миопатия, реже - воспалительная миопатия. Акроостеолиз проявляется укорочением и деформацией пальцев вследствие резорбции концевых отделов дистальных фаланг, а в некоторых случаях – лучевых костей и отростков нижней челюсти. Кальциноз мягких тканей (рис. 4), преимущественно в области пальцев рук и периартикулярно (синдром Тибьержа-Вейсенбаха) – часть CREST-синдрома (сочетание кальциноза, синдрома Рейно, эзофагита, склеродактилии, телеангиэктазии) или лимитированной ССД хронического течения. Одним из наиболее ярких сосудистых нарушений являетсясиндром Рейно – симметричные вазоспастические кризы с побледнением и / или цианозом и чувством онемения пальцев рук, реже ног, с распространением на кисти и стопы, иногда - на подбородок, кончик носа и языка, что проявляется нарушением артикуляции, цианозом подобно синдрому «перемежающейся хромоты». Провоцируется холодом или эмоциональным стрессом. Следствием сосудистых нарушений являются дигитальные язвы с локализацией на дистальных фалангах пальцев кистей рук, резко болезненные, торпидные к лечению, язвенные поражения кожи над локтевыми, коленными суставами, в области лодыжек и пяток, ушной раковины и др.), сухая гангрена, дигитальные рубчики – точечные участки атрофии кожи дистальных фаланг пальцев рук («крысиные укусы») (рис. 5).Поражение желудочно-кишечного тракта. Склеродермический эзофагит – наиболее частое поражение пищевода и ЖКТ в целом. Проявляется дисфагией, чувством кома за грудиной, стойкой изжогой. Поражение кишечника – абдоминальные боли, диарея, синдром нарушенного всасывания, обстипационный синдром вплоть до кишечной непроходимости (псевдообструкция). Лимитированная форма хронического течения часто сочетается с первичным биллиарным циррозом.
Поражение легких.Вовлечение легких наблюдается в 70% больных с ССД и по частоте уступает только поражению пищевода. Основные клинико-морфологические виды поражения легких - интерстициальное заболевание и легочная гипертензия.
Интерстициальное заболевание легких – фиброзирующий альвеолит и диффузный пневмосклероз, преимущественно базальных отделов. Легочная гипертензия (10-40%) – изолированная или в сочетании с базальным пневмофиброзом.
Поражение сердца. Патофизиологические изменения сердца при ССД включаютфиброз, поражение мелких сосудов, нарушение микроциркуляции с развитием ишемии миокарда при интактных коронарных сосудах. Характерно развитие систолической и диастолической дисфункции. В 70% случаев диагностируются нарушения ритма и проводимости, удлинение QT. Поражение эндокарда протекает с формированием относительно доброкачественного склеродермического порока (чаще митрального). Перикардит чаще имеет бессимптомое течение. При эхокардиографическом исследовании выявляется в 70-80% случает, чаще адгезивный, реже - экссудативный.
Поражение почек. Те или иные признаки почечной дисфункции при ССД диагностируется в 50% случаев. Основные клинические формы: острые фатальные и хронические латентные. К острым фатальным нарушениям относят острую нефропатию (склеродермический почечный криз): быстропрогрессирующая почечная недостаточность, злокачественная (гиперрениновая) АГ, нарастающая протеинурия, олиго- и анурия, микроангиопатическая гемолитическая анемия, тромбоцитопения, энцефало- и ретинопатия с острым началом без предвестников.
Хронические латентные формы - малосимптомная патология, в основе которой лежит снижение почечного кровотока с последующим уменьшением СКФ. Сочетается с минимальной или умеренной протеинурией, часто отмечаются артериальная гипертензия и начальные признаки ХПН.
КЛАССИФИКАЦИОННЫЕ КРИТЕРИИ. (Американская ассоциация ревматологов.)
«Большой критерий». Проксимальная склеродермия: симметричное утолщение и индурация кожи пальцев, распространяющиеся проксимально от пястно-фаланговых и плюсне-фаланговых суставов. Изменения могут затрагивать лицо, шею, грудную клетку, живот.
«Малые критерии». Склеродактилия: перечисленные выше кожные изменения, ограниченные пальцами; дигитальные рубчики – участки западения кожи на кончиках пальцев или потеря вещества подушечек пальцев; двусторонний базальный пневмофиброз: сетчатые или линейно-узловые тени, наиболее выраженные в базальных участках легких при стандартном рентгенологическом исследовании; могут быть проявления по типу «сотового легкого».
Постановка диагноза – большой критерий или 2 малых критерия.Чувствительность 97%, специфичность 98%. Недостаток: не охватывают всех клинических форм заболевания.
ЛАБОРАТОРНЫЕ ИССЛЕДОВАНИЯ. Общий анализ крови: гипохромная анемия, умеренное повышение СОЭ, снижение гематокрита. Общий анализ мочи: гипостенурия, микрогематурия, протеинурия, цилиндрурия, лейкоцитурия. Биохимический анализ крови: нет характерных изменений. Иммунологические исследования: АНФ у 95% в умеренном титре, склеродермоспецифические аутоантитела: АТ к топоизомеразе (анти-Scl-70), антицентромерные АТ (20%), АТ к РНК-полимеразе III (20-25%) и др. антинуклеарные АТ; РФ (у 45%) при сочетании с с-мом Шёгрена и ревматоидным артритом.
ИНСТРУМЕНТАЛЬНЫЕ ИССЛЕДОВАНИЯ
Капилляроскопия ногтевого ложа: дилатация и редукция капилляров.
Исследования висцеральных органов: пищевод – манометрия, эндоскопия / pH-метрия, рентгенография/эндоскопия; желудок – сцинтиграфия, эндоскопия; тонкий кишечник – рентгеноконтрастное исследование, дыхательный водородный тест, обзорная рентгенография; аноректальный отдел – манометрия; легкие – рентгенография, ФВД, КТГ, БАЛ, сцинтиграфия, торакоскопическая биопсия легких, допплер-эхокардиография, катетеризация правого желудочка и др.; сердце – ЭКГ, ЭХО-КГ, сцинтиграфия, рентгенография; почки – мониторинг АД, креатинин крови, уровень ренина в плазме, ОАК, офтальмоскопия, биопсия почки.
ЛЕЧЕНИЕ.
Немедикаментозное лечение. Общие рекомендации - избегать психоэмоциональных нагрузок, длительного воздействия холода и вибрации, уменьшить пребывание на солнце; ношение теплой одежды, прекращение курения, отказ от потребления кофе, избегать применение симпатомиметиков, бета-адреноблокаторов.
Медикаментозное лечение. Включает применение антифиброзных, сосудистых, противовоспалительных, иммуносупрессивных средств, экстракорпоральные методы (плазмаферез), реабилитационную и симптоматическую терапию.
Выбор препарата и показание к противовоспалительной терапии. НПВП – мышечно-суставные проявления, стойкий субфебрилитет. Глюкокортикостероиды – прогрессирующее диффузное поражение кожи, артрит, теносиновит, миозит, серозит (15-20 мг/сут), фиброзирующий альвеолит (40-50 мг/сут.). Циклофосфамид – фиброзирующий альвеолит, ранняя ССД, быстропрогрессирующее течение. Метотрексат – выраженное поражение суставов и мышц, при перекрестной форме ССД.Аминохинолиновые препараты включают в комплексную терапию, особенно при хроническом течении ССД. Трансплантация аутологичных стволовых клеток иногда с успехом применяется при тяжелой, прогностически неблагоприятной ССД.
Антифиброзная терапия. D-пеницилламин (купренил, бианодин и др.) - средство выбора при быстропрогрессирующей склеродермии, диффузной индурации кожи и висцерофиброзах. Применение длительное (не менее 6-12 мес.) по схеме (250- 500-750-1000 мг/день с последующим снижением) и использованием поддерживающих доз (250- 300 мг/день) в течение 2-5 лет. Клинический эффект проявляется положительной динамикой кожного синдрома (уменьшение индурации и др.), суставно-мышечного (с увеличением объема движений) и сосудистого (уменьшение синдрома Рейно, улучшение трофики).
Сосудистая терапия.Показания: синдром Рейно и его осложнения (ишемия, некрозы), легочная гипертензия, почечная АГ. Группы препаратов: 1. Блокаторы кальциевых каналов (нифедипин 30-60 мг/сут., амлодипин 5-20 мг/сут., нормодипин 5-10 мг/сут., верапамил 120-360 мг/сут., диалтиазем 90-240 мг/сут.); 2. Ингибиторы АПФ (каптоприл до 300 мг в сутки, лизиноприл («Диротон») 20 мг/сут. и др.) – при истинной склеродермической почке, АГ; 3. Простагландин Е1(вазапростан, алпростадил) - препарат выбора при прогрессирующем синдроме Рейно, тяжелых сосудистых поражениях с ишемическими некрозами пальцев рук и ног. Вазодилататоры целесообразно сочетать с антиагрегантами (трентал 400-800 мг/сут., вазонит 600-1200 мг/сут. и др. внутрь). Рекомендуется проведение 2-3 курсов в год внутривенного введения, в интервалах – пероральный прием. При наличии признаков гиперкоагуляции, микротромбозов рекомендовано включение в терапевтический комплекс антикоагулянтов.
Прогноз.5-летняя выживаемость 34-73 %. Риск смерти в 4,7 раза выше, чем в популяции.
ДЕРМАТОМИОЗИТ - ПОЛИМИОЗИТ
Полимиозит и дерматомиозит (ДМ, ПМ) являются наиболее частыми формамиидиопатических воспалительных миопатии (ИВМ). ИВМ - хронические диффузные заболевания поперечнополосатой мускулатуры, основным проявлением которых является мышечная слабость.
ЭПИДЕМИОЛОГИЯ. Частота ПМ/ДМ в популяции составляет 2 -10 случаев на 1 млн. населения в год. ДМ, реже ПМ, ассоциированный с опухолью, составляет около 20% от всех случаев воспалительных миопатий. Половой диморфизм не выявлен.
КЛАССИФИКАЦИЯ ИВМ
Первичный идиопатический полимиозит.
Первичный идиопатический дерматомиозит.
Миозит, ассоциированный с системными заболеваниями соединительной ткани.
Ювенильный полимиозит / дерматомиозит.
Миозит, сочетающийся со злокачественными опухолями.
Миозит с «включениями».
Другие формы воспалительных миопатий: гранулематозный миозит, эозинофильный миозит, миозит при васкулитах, орбитальный миозит (глазных мышц), фокальный (узелковый) миозит, оссифицирующий миозит.
ЭТИОЛОГИЯ. Несмотря на то, что причина ПМ/ДМ неизвестна, существует ряд доказательств влияния факторов внешней среды на развитие иммунных нарушений у предрасположенных индувидуумов с последующим развитием аутоиммунного миозита. На роль генетической предрасположенности указывает связь с HLA-B8/DR3, HLA-B14, HLA-B40, ассоциированных с определенными иммунными нарушениями. О возможной этиологической роли инфекционных факторов косвенно свидетельствует более частое начало заболевания зимой и ранней весной (особенно у пациентов с ювенильными формами), что по времени совпадает с эпидемиями инфекций.
ПАТОГЕНЕЗ. ПМ/ДМ рассматривают как аутоиммунные заболевания, связанные с развитием воспалительной клеточной инфильтрации скелетной мышцы и иммунологических нарушений (в частности, продукция аутоАТ). При ДМ наиболее существенное значение имеют гуморальные иммунные нарушения, связанные с отложением в мелких сосудах иммунных комплексов, активацией комплемента и развитием васкулопатии, сопровождающейся воспалительной инфильтрацией скелетной мышцы.В клеточном инфильтрате преобладают Т-лимфоциты, макрофаги и В-лимфоциты. При ПМ в инфильтрате преобладают CD8 Т-лимфоциты, при этом отсутствуют признаки васкулопатии или иммунокомплексного поражения сосудов.
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ. Началу заболевания может предшествовать инсоляция, работа с химикатами и другие факторы.
Поражение мышц. Мышечный синдром – решающий в клинической диагностике ДМ. Характерно развитие тяжелого, нередко некротического миозита скелетной мускулатуры проксимальных отделов конечностей. Развитие мышечной слабости обычно имеет медленный прогрессирующий характер (в течение нескольких недель или месяцев). Пациенты испытывают трудность при подъеме с низкой поверхности, по лестнице, вхождении в транспорт («симптом автобуса»), при одевании («симптом рубашки»), причесывании («симптом расчески»), возможны неожиданные падения, затруднения при вставании с постели и поворотах в ней. Движения в кистях, пальцах, стопах могут не нарушаться. При поражении мышц гортани и глотки развивается дисфагия, гнусавость, иногда афония, охриплость, «поперхивание» пищей, выливание жидкой пищи через нос. Развивающаяся псевдобульбарная симптоматика имитирует неврологическое заболевание. Возможно вовлечение в воспалительный процесс мимической мускулатуры (маскообразность лица), глазодвигательных мышц (диплопия, птоз век), мышц сфинктеров (недержание кала и мочи). Мышцы болезненны при пальпации, отечны, тестоваты на ощупь, в дальнейшем они уплотняются.
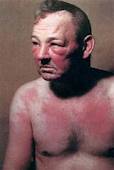 Поражение
кожи.
Наиболее
специфичный признак
«гелиотропная» сыпь
- эритема с фиолетовым оттенком,
локализующаяся на верхних веках и
нередко сочетающаяся с отеком (рис. 6).
Поражение
кожи.
Наиболее
специфичный признак
«гелиотропная» сыпь
- эритема с фиолетовым оттенком,
локализующаяся на верхних веках и
нередко сочетающаяся с отеком (рис. 6).
Папулы Готтрона – чешуйчатые эритематозные высыпания над пястно-фаланговыми суставами или над разгибательными поверхностями локтевых и коленных суставов.
Рис. 6. Гелиотропная эритема
 Симптом
Готтрона
– эритема без папул аналогичной
локализации. Прочие
изменения включают: шелушение
и трещины кожи на кончиках пальцев
(«рука механика»), околоногтевую эритему,
гипертрофию кутикулы, фотодерматит,
инфаркты околоногтевого ложа, петехии,
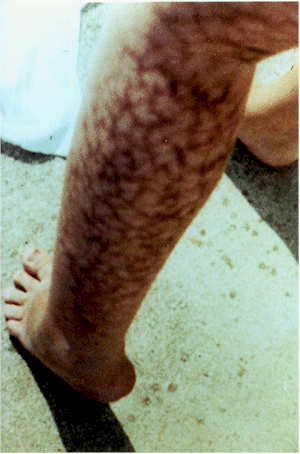
сетчатое ливедо (рис. 7), телеангиэктазии,
кожный зуд, панникулит (редко). Феномен
Рейно–
чаще встречается при ДМ, антисинтетазном
синдроме и у больных с перекрестным
синдромом ПМ/ ДМ/ ССД.
Симптом
Готтрона
– эритема без папул аналогичной
локализации. Прочие
изменения включают: шелушение
и трещины кожи на кончиках пальцев
(«рука механика»), околоногтевую эритему,
гипертрофию кутикулы, фотодерматит,
инфаркты околоногтевого ложа, петехии,
сетчатое ливедо (рис. 7), телеангиэктазии,
кожный зуд, панникулит (редко). Феномен
Рейно–
чаще встречается при ДМ, антисинтетазном
синдроме и у больных с перекрестным
синдромом ПМ/ ДМ/ ССД.
Кальциноз мягких тканей характерен для ювенильного ДМ или поздних стадий ДМ/ПМ. Кальцинаты располагаются подкожно в соединительной ткани, вокруг мышечных волокон, в зонах микротравматизации над локтевыми и коленными суставами, на сгибательных поверхностях пальцев рук и ягодиц.
Рис. 7Сетчатое ливедо
Суставы – преходящее симметричное поражение мелких суставов кистей, лучезапястных, редко – артропатия Жакку (хронический деформирующий артрит с подвывихами суставов кистей при отсутствии эрозивных изменений).Органы дыхания. Наиболее частое органное поражение, которое отмечают 80% случаев. При ДМ/ПМ могут возникать поражения межребрных мышц и диафрагмы, пневмония, фиброзирующий альвеолит. Поражение межребных мышц и диафрагмы возникает у подавляющего большинства пациентов, что приводит к эксираторной одышке и развитию дыхательной недостаточности по рестриктивному типу (40% случаев). Снижение кашлевого клиренса легких, легочного кровотока создает «благоприятные» условия для развития гиповентиляционой пневмонии и легочной гипертензии. Пневмонии – наиболее частая форма поражения легких (30-55%). Главную роль играет аспирация пищи, связанная с поражением мышц глотки и верхней трети пищевода. Частота аспирационного синдрома достигает 30%. Фиброзирующий альвеолит – наиболее тяжелый вариант поражения легких. Его частота составляет 10-60%.
Поражение сердца в большинстве протекает бессимптомно. Иногда при специальных исследованиях выявляют нарушения ритма и проводимости. Крайне редко могут возникать миокардит, перикардит, ХСН.
Поражение ЖКТ: дисфагия, назальная регургитация жидкой пищи (вследствие вовлечения мышц глотки), гипотония верхней трети пищевода, реже – рефлюкс-эзофагит, запор или диарея, ульцерация, особенно при ювенильном дерматополимиозите.
Поражение почек возникает крайне редко, как правило, при перекрестных синдромах, в основном, с ССД.
Миозит при злокачественных новообразованиях. Чаще ДМ, а не ПМ. Соотношение мужчин к женщинам = 1:1. Опухоли могут возникать до появления признаков ИВМ, одновременно с ними или после. Частота онкопатологии в 12 раз больше, чем в популяции. Васкулит, некроз кожи, амиотрофический синдром ассоциирован с онкопатологией при ИВМ в большей степени, чем легочный фиброз, миозитспецифические АТ и др. системные проявления. Наиболее часто – рак яичников и носоглоточный рак.
ДИАГНОСТИКА основывается на данных анамнеза, результатах клинического обследования, морфологического исследования биоптатов скелетной мышцы и электромиографии (ЭМГ). Должны учитываться жалобы и обстоятельства возникновения заболевания - боли или слабость в мышцах, дистальное или проксимальное поражение, эффект от отдыха, как быстро нарастает мышечная слабость, другие симптомы (утомляемость, сыпь, нарушение глотания, артралгии), прием миотоксических препаратов, эффект от ГКС и др.
Изменения общего анализа крови носят неспецифический характер: в ряде случаев отмечают увеличение СОЭ (преимущественно при развитии системных проявлений). Биохимический анализ. Наиболее важным биохимическим маркером повреждения скелетной мускулатуры является повышение КФК, которое обладает при ДМ/ПМ высокой чувствительность и специфичностью. Увеличение активности КФК в разные периоды болезни отмечают у 95% пациентов. Активность КФК не коррелирует с клинической активностью мышечного поражения и в ряде случаев может быть нормальной у больных с классическим ДМ/ПМ, несмотря на тяжелое поражение мышц по данным морфологического исследования. В поздних стадиях, при тяжелой мышечной атрофии, в дебюте и при опухолевом миозите КФК также может быть в норме. У большинства пациентов происходит увеличение миоглобина, АЛТ, АСТ, ЛДГ. Иммунологические исследования. АНФ выявляют у 50-90% пациентов. Виды антител, выявляемые при миозите подразделяют на миозит-специфические и миозит-ассоциированные. Их клиническое значение определяется наличием клинико-иммунологических ассоциаций. Так, Анти-Jо-1 ассоциируются с острым тяжелым ПМ, интерстициальными заболеваниями легких, Анти-ЕJ – с «рукой механика» и т.д.
Золотой стандарт диагностики ИВМ - мышечная биопсия. Для ПМ характерна инфильтрация мононуклеарными клетками (в основном лимфоцитами) с локализацией в эндомизии, некроз и регенерация мышечных волокон. Для ДМ – мононуклеарная инфильтрация вокруг фасций и кровеносных сосудов, признаки васкулопатии (тромбоз капилляров, отек, гиперплазия, вакуолизация и дегенерация клеток эндотелия). В поздних стадиях ДМ/ПМ определяется атрофия мышечных фибрилл, замещение жировой тканью, фиброзом. Для морфологического исследования легких характерно неравномерное распределение участков поражения между участками здоровой ткани.
Инструментальные методы.
Игольчатая ЭМГ – характерно уменьшение амплитуды и длительности потенциалов действия мышечных волокон, полифазия, псевдополифазия, спонтанная электрическая активность мышечных волокон.
Капилляроскопия позволяет выявить уменьшение числа капилляров, увеличение их размера, образование новых капилляров, «кустовидные» капилляры.
Рентгенологическое исследование легких показано всем пациентам для оценки поражения легких. К более чувствительным методам относят КТ высокого разрешения.
МРТ мышечной ткани позволяет выявить ранние изменения - отек мышечной ткани, который является индикатором активности заболевания, а также проводить дифференциальную диагностику между воспалительным отеком мышцы и атрофией с жировым замещением. Обязательным является диагностика онкологических заболеваний, включая определение онкомаркеров: РSA, СА-125, СА-15.3 (с-r яичников и молочной железы) и др.
Диагностические критерии
Наиболее информативными признаками для диагностики ДМ и ПМ (A. Bohan и J.B. Petter (1975) являются:
Симметричная проксимальная мышечная слабость мышц плечевого и тазового пояса.
Признаки миозита в биоптате скелетной мышцы (некроз мышечных волокон 1 –го и 2 –го типа, фагоцитоз, дегенерация и регенерация миофибрилл с разным диаметром мышечных волокон, эндомизиальные, перимизиальные, периваскулярные или интерстициальные мононуклеарные клетки).
Повышение КФК, альдолазы, ЛДГ, АСТ, АЛТ.
Первично-мышечные изменения на игольчатой ЭМГ (триада признаков).
Типичные кожные изменения.
При наличии первого признака и по крайней мере четырех из последующих чувствительность для диагностики дерматомиозита составляет 94%, специфичность 99%.Для диагностики полимиозита чувствительность составляет 99%, специфичность 95%.
ЛЕЧЕНИЕ. Основой лечения ИВМявляется назначение глюкокортикоидов. Начало терапии в течение первых трех месяцев от начала симптомов обусловливает более благоприятный прогноз. Принципы лечения ГК: как можно более раннее назначение; адекватная начальная доза (1,0-2,0 мг/кг в сутки); длительность приема начальной дозы не менее 2-3 месяцев; медленный темп снижения дозы (1/4 дозы от суммарной 1 раз в месяц); адекватная поддерживающая доза; при ювенильном ДМ, тяжелом поражении пищевода, развитии системных проявлений показана пульс-терапия.
Препараты «второго» ряда: метотрексат 7,5-25 мг/неделю, азатиоприн 2-3 мг/сутки, хлорамбуцил, циклофосфамид, циклоспорин, аминохинолиновые препараты, ФНО-α (инфликсимаб), блокатор пролиферации В-лимфоцитов (ритуксимаб). Показания: при факторах риска неблагоприятного прогноза (позднее назначение ГКС, тяжелая мышечная слабость, дисфагия), недостаточная эффективность ГКС; невозможность назначить адекватную дозу ГКС из-за побочных эффектов.
Прогноз: до эры ГКС смертность составляла 30-50%, сейчас 5-летняя выживаемость (исключая миозит на фоне злокачественных образований) 90%.
ВОЕННО-ВРАЧЕБНАЯ ЭКСПЕРТИЗА. Освидетельствование лиц с системными болезнями соединительной ткани проводится по статье 64а Постановления МО и МЗ РБ № 51/70 от 20.12.2010г. Вне зависимости от выраженности изменений со стороны органов и систем, частоты обострений и степени функциональных нарушений лица всех категорий годности признаются негодными к военной службе с исключением с воинского учета (НГИ).