

Вопросы и ответы экзамена ХОиВМС
Предмет органической химии [1. Стр. 9]
Предмет органической химии. Органическая химия изучает законы химических превращений, состав, свойства, способы получения и применение органических соединений.
Органическими соединениями называют соединения углерода – углеводороды и их производные. Кроме углерода и водорода в органические соединения практически могут входить почти все элементы периодической системы. Однако чаще всего в состав органических соединений входят кислород, галогены, азот, сера, фосфор и некоторые металлы.
Простейшие соединения углерода, такие, как оксиды, угольная кислота и ее соли, относят к неорганическим соединениям и их изучает неорганическая химия.
Соединения углерода выделяют в особую научную дисциплину по ряду причин.
Первая из них – многочисленность органических соединений. К настоящему времени их насчитывают около 5 млн., в то время как число известных соединений всех других элементов не превышает 700 000.
Вторая причина – сложность и своеобразие характера химического поведения органических соединений:
а) органические вещества отличаются известной инертностью и большинство химических реакций с их участием протекает гораздо медленнее, чем ионные реакции, характерные для неорганических веществ;
б) при нагревании даже до сравнительно невысокой температуры (100 0С) многие органические вещества разлагаются;
в) почти все органические соединения горючи, тогда как подавляющее большинство неорганических соединений не обладает горючестью.
Третья причина – большое практическое значение органических соединений: это пища, ткани, стройматериалы, краски, горючие и смазочные материалы, растворители, флотореагенты, сорбенты, экстрагирующие вещества, полупроводники, взрывчатые вещества, полимерные материалы, лекарства и т.д.
Теория химического строения органических соединений а. М. Бутлерова [1. Стр. 10]
Теория химического строения органических соединений А.М. Бутлерова. Вплоть до 20-х годов ХІХ столетия химики считали совершенно невозможным синтез органических веществ из неорганических соединений. В 1840 г. известный русский ученый И.И. Гесс высказал утверждение, что органические вещества образуются по тем же законам, что и неорганические. В 1845 г. Кольбе получил уксусную кислоту из древесного угля, серы, хлора и воды.
Постепенно химики научились получать и более сложные органические вещества. Так, в 1854 г. М. Бертло синтезировал вещества, относящиеся к классу жиров, а в 1861 г. А.М. Бутлеров получил из полиформальдегида и воды сахаристое вещество.
Со временем был накоплен огромный фактический материал, требовавший теоретического обобщения и объяснения возможных путей синтеза органических соединений и связи их свойств со строением.
Основополагающей теорией строения органических соединений явилась теория химического строения, созданная Александром Михайловичем Бутлеровым (1828-1886) и обнародованная им 19 сентября 1861 г. на 36-м съезде немецких естествоиспытателей в докладе «О химическом строении вещества».
Основные положения теории химического строения А. М. Бутлерова можно свести к следующему.
1. Атомы в молекуле органического соединения связаны друг с другом в определенной последовательности.
Свойства вещества зависят от их химического строения, т.е. от порядка соединения атомов в молекуле и характера их взаимного влияния. В результате каждое из веществ имеет свои особые физические и химические свойства.
2. Соединение атомов в молекуле происходит в соответствии с их валентностью. Валентности всех атомов в молекуле взаимно насыщены. Свободных валентностей у атомов в молекуле нет.
3. Атомы углерода способны соединяться между собой и образовывать углеродные цепи различного вида. Эти цепи могут быть открытыми или замкнутыми (кольца, циклы), прямыми или разветвленными.
В зависимости от числа связей, затрачиваемых атомами углерода на соединение друг с другом, цепи могут быть насыщенными (с одинарными связями) или ненасыщенными (с кратными – двойными или тройными связями).
4. Каждое органическое соединение имеет одну определенную формулу строения, отражающую порядок химической связи атомов в реально существующей молекуле. Строение молекулы как реально существующего объекта можно изучить экспериментально химическими и физическими методами.
А.М. Бутлеров провел ряд экспериментальных работ, подтвердив предсказания теории получением изобутана, третичного бутилового спирта и других веществ. Это позволило ему в 1864 г. заявить, что отныне имеется достаточно фактов, чтобы ручаться за возможность синтетического получения любого органического вещества.
3. Тетраэдрическая модель атома углерода [1. стр. 10] Тетраэдрическая модель атом углерода. Основные представления о химическом строении, заложенные А.М. Бутлеровым, были дополнены Вант-Гоффом и Ле Белем (1874), которые развили идею о пространственном расположении атомов в молекуле органического вещества и поставили вопрос о пространственной конфигурации и конформации молекул. Работа Вант-Гоффа «Химия в пространстве» положила начало плодотворному направлению органической химии – стереохимии, т.е. учению о пространственном строении.
4. Электронные представления в органической химии [1.стр. 10-12] Электронные представления в органической химии. Метод описания движения электронов в атомах и соответствующий математический аппарат был предложен в 1926 г. Шредингером на базе начинавшей формироваться к этому времени квантовой механики. По аналогии с уравнениями, описывающими упругие механические, звуковые и световые волны, уравнение движение электрона по орбитали получило название волнового уравнения Шредингера.
Согласно принципам квантовой механики можно определить лишь вероятность нахождения электрона в данной области пространства, окружающего точку с координатами (х, у, z), но не его точные координаты. Обычно функция вероятности обозначается через (х, у, z) и тогда электрон с максимальной вероятностью будет находиться в той области, где максимальна.
Если обозначить решение волнового уравнения Шредингера через (х, у, z) и назвать его волновой функцией, то 2 (х, у, z) оказывается пропорциональным (х, у, z). Подобрав соответствующий постоянный числовой множитель, можно получить рaвенство:
2 (х, у, z) = (х, у, z),
при этом волновая функция останется решением уравнения Шредингера.
Интерпретация как зарядового облака оказалась очень наглядной при изображениях атомных и молекулярных орбиталей. В этих случаях для каждой существует некая граничная поверхность, внутри которой сосредоточено, например, 90 или 99% заряда. Форма этих поверхностей являются важнейшими стереохимическими факторами и определяют ход многих химических реакций.
Вследствие того, что волновая функция прямо связана с распределением электронной плотности, можно утверждать (правда, с некоторой натяжкой), что волновая функция описывает орбиту движения электрона вокруг ядра. В теории химической связи такие волновые функции получили название атомных орбиталей, сокращенно АО.
Наиболее важной является классификация орбиталей по типам s, р, d и т.д. Все АО s–типа сферически симметричны, поэтому распределение заряда зависит только от радиуса. Орбитали всех других типов не имеют сферической симметрии. Например, имеются три АО р-типа, граничные поверхности которых похожи на гантели. Эти орбитали имеют ясно выраженную направленность, поэтому они могут быть обозначены как рx, рy, рd, где х, у и z сооветствуют трем осям координат, относительно которых симметрична соответствующая р-орбиталь. Все три р-орбитали совершенно эквивалентны, за исключением их направления, и все они линейно независимы.
Существует также пять АО d–типа с более сложной конфигурацией. Орбитали других типов в органических соединениях почти не встречаются.
Подразделение атомных (но не молекулярных) орбиталей на типы s, р, d, и т.д. довольно четкое; не существует промежуточных гибридизованных орбиталей между s– и р-орбиталями. По этой причине целесообразно ввести обазначение типов АО с помощью квантовых чисел. Набор стационарных состояний электронов определяется квантовыми числами: п, l, m, s. От различных значений этих чисел зависят симметрия и ориентация волновой функции и ее узловые свойства.
Главное квантовое число п определяет общий размер зарядового облака. Это означает, что число п определяет общий уровень энергии электрона.
Квантовое число l характеризует свойства симметрии АО и связано с моментом импульса движущегося электрона. Квантовое число l может иметь значения от 0 до (n -1), т.е. l = 0 при n = 1; l = 0, 1 при n = 2; l = 0, 1, 2 при n = 3 и т.д., что соответствует типам орбиталей s, p, d и т.д. Для нас более важны геометрические характеристики орбиталей, указываемые символами s, p, d, …, чем цифровые значения l, поэтому всегда целесообразнее пользоваться первыми.
Магнитное квантовое число ml указывает на количество и направление в пространстве орбиталей данной формы. Оно имеет 2l + 1 значений: от –l до +l. Так, при l = 0 m = 0: возможна только одна s-орбиталь сферической симметрии. При l = 1 m = 1, 0, +1 и, следовательно, возможны три p-орбитали, направленные по трем осям координат (px, py, pz).
Спиновое
квантовое число
ms
соответствует
двум возможным ориентациям магнитного
момента электрона в магнитном поле:
вдоль силовых линий или против. Это
записывается как -½ и +½ или значками
 .
.
Согласно принципу Паули, в атоме не может быть двух электронов с одинаковым значениям всех четырех квантовых чисел, это значит, что одну и ту же АО могут занимать только два электрона с разными значениями спина (спаренные с антипараллельными спинами).
5. Химическая связь ( ионная, ковалентная, водородная) [1. стр. 12-13 ] Ионная связь. Электронная конфигурация инертного газа для любого атома может образоваться двумя различными способами. Один из них - перенос электронов: атомы одного из элементов отдают электроны, которые переходят к атомам другого элемента. В данном случае между этими атомами образуется так называемая ионная (электровалентная, гетерополярная) связь. Гетерополярная связь существует лишь в кристаллах (что, правда, в последнее время на основании спектроскопических данных тоже подвергается сомнению). Поэтому более правильным было бы говорить «ионное взаимодействие», а не ионная связь.
Отличительными чертами ионных соединений являются мгновенность протекания реакций, диссоциация (распад кристалла, молекулы, радикала или иона на фрагменты, имеющие меньшую молекулярную массу; термическая, фотохимическая и электролитическая) и сольватация (взаимодействие частиц растворённого вещества с молекулами растворителя, проводящее к образованию сольватов) ионов в водных растворах, высокие температуры плавления и кипения, растворимость в полярных растворителях, электрическая проводимость растворов расплавов.
Как правило, гетерополярные связи образуют между собой элементы начала и конца периодов таблицы Менделеева. Это объясняется тем, что различные атомы в разной степени способны принимать на свои орбиты электроны. Элементы седьмой группы, имеющие на внешней орбите семь электронов, легко принимают один электрон, недостающий до полного октета. Элементы же первой группы имеют на внешней орбите всего один электрон, который они легко отдают.
Таким образом, гетерополярная связь возникает между атомами, сильно отличающимися по электроотрицательности (Na-Cl, K -OH).
Ковалентная (гомеополярная) связь – химическая связь, образованная за счет взаимодействия орбиталей неспаренных электронов; наиболее распространенный в органической химии тип связи. Эта связь обладает максимальной прочностью.
Ковалентная связь и соответственно молекула могут быть неполярными, когда оба связанных атома обладают одинаковым сродством к электрону, например Н:Н. Такая связь обладает максимальной прочностью. Она может быть полярной, когда электронная пара вследствие большего сродства к электрону одного из атомов оттянута в его сторону:
Н→Cl
или
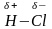
При таком способе обозначения символы δ- и δ+ (частичные заряды) означают, что на атоме со значком δ- избыточная электронная плотность, а на атоме со значком δ+ она несколько понижена.
Донорно-акцепторная или координационная (часто также используются термин диполярная) связь. Разновидностью донорно-акцепторной связи является связь азота с кислородом в нитросоединениях или N–оксидах. В этом случае азот связан с одним кислородным атомом обычной двойной ковалентной связью за счет обобщенных двух электронов азота и двух электронов кислорода, а оставшаяся неподеленная электронная пара (п-электроны) азота образует связь со вторым атомом кислорода, при этом, очевидно на азоте (донор) появляется положительный заряд, а на кислороде (акцептор) – отрицательный. Таким образом, связь между атомом азота и вторым атомом кислорода представляет собой сочетание ковалентной и ионной связей и носит название семиполярной связи.
К донорно-акцепторному типу связей относятся и связи в комплексных соединениях. От описанных выше они отличаются лишь меньшей прочностью. Так как передача электронов осуществляется не полностью.
Водородная связь. Присутствию у ряда атомов неподеленной пары электронов обязана своим существованием и так называемая водородная связь. Она образуется между молекулами, содержащими электроотрицательные атомы (О, N, F, реже Сl, Вr, S ), которые имеют неподеленную электронную пару, и молекулами с активными атомами водорода. Активными называются атомы водорода, связанные с другим атомом сильно полярной связью. Например,
I→O;
H→S;
H→N и т.д.
и т.д.
Атом водорода имеет гораздо меньшие размеры, чем все другие атомы. Он очень слабо экранирован и может вследствие этого подходить очень близко к неподеленным парам электронов других атомов.
Водородная связь (обозначается тремя точками) по своему характеру в основном является электростатической. Энергия водородной связи значительно ниже энергии ковалентной связи (примерно 4-33 кДж/моль), тем не менее она играет значительную роль в определении как химических, так и физических свойств соединений. Водородная связь может оказаться достаточно прочной для существования независимых частиц в растворе например катиона оксония (Н3О)+. Более слабые водородные связи приводят к образованию ассоциированных систем. Наличием водородных связей в таких соединениях объясняется уменьшение летучести, увеличение вязкости и изменение других физических свойств. Эти явления наблюдаются во многих чистых жидкостях, например, в аммиаке, воде, фтористом водороде, первичных и вторичных аминах, спиртах, фенолах, минеральных и органических кислотах.
Известны примеры особо прочных внутримолекулярных водородных связей. Такого типа связи возникают, если структура самой молекулы способствует их образованию.
Благодаря водородным связям фиксируются вторичные и третичные структуры белков, образуются связи в двойных спиралях ДНК и т.д. Решающую роль водородных связей часто объясняет малые энергии, необходимые для многих биохимических превращений.
6. Классификация органических соединений [1. стр. 15] Классификация органических соединений. В зависимости от структуры углеродного скелета (углеводородная часть и особенно углеродная часть сложного органического вещества) все органические соединения классифицируются следующим образом.
1. Ациклические (алифатические) соединения. Скелет составлен из непосредственно связанных атомов углерода в виде неразветвленной (нормальной)

или разветвленной цепи

2. Карбоциклические соединения. В их молекулах углеродные цепи замкнуты в цикл:

3. Гетероциклические соединения. В циклы молекул этих соединений кроме атомов углерода входят атомы других элементов:

В каждом из этих рядов все соединения распределяются по классам в зависимости от имеющихся в них функциональных групп. Например, класс ациклических спиртов (группа - ОН), класс ациклических аминов (группа - NН2), класс карбоциклических карбоновых кислот (группа - СООН) и т.д.
7. Основы номенклатуры в органической химии [1. стр. 16] Основы номенклатуры в органической химии. На первых этапах развития химии вновь открытым соединениям присваивались, как правило, тривиальные названия, чаще связанные с источником получения, чем со структурой и до сих пор являются общепринятыми: муравьиная кислота, уксусный альдегид, ацетон, хлороформ и т.д. В химии природных соединений это часто практикуется и в наши дни. Количество новых соединений росло очень быстро и возникла необходимость связывать названия со строением вещества. В связи с этим появилась рациональные названия: метилэтилкетон, диметиламин, триметилуксусная кислота, триметилкарбинол и т. п. В этих случаях, как правило, соединение рассматривают как замещенное теми или иными радикалами простейшее соединение данного ряда.
На международных съездах химиков была разработана и затем предложена (1957 и 1965 гг.) в качестве официальной научной номенклатуры так называемая номенклатура ИЮПАК (IUPAC – International Union of Pure and Applied Chemistry - Международный союз теоретической и прикладной химии).
В основу названия соединения по этой номенклатуре положена углеродная цепь молекулы, содержащая максимально число функциональных групп (гидроксил, карбоксил, аминогруппа и т. д.). Начало нумерации цепи определяет наиболее старшая функциональная группа. Порядок старшинства основных функций следующий:
COOH › C ≡ N › CHO › C = O › OH › NH2 › NO › Hal
Главная функция обозначается в названия суффиксом: гидроксильная группа –ол; альдегидная –ал; кетонная –он и т.д., остальные – префиксами. Например:

5-окси-4,5 –диметил-3- 2-хлор-4-амино-3-метокси-
изопропилгексан-3-он-2 2-бутеновая кислота
8. Предельные углеводороды (формула, строение, изомерия, способы получения, превращение и применение)[1. стр.17 ] Предельные, или насыщенные, углеводороды (алканы, парафины). К предельным углеводородам – парафинам, или алканам, - относят соединения с открытой цепью, атомы углерода в которых соединены друг с другом простыми (одинарными) связями. Они до предела насыщены водородом и в обычных условиях мало реакционноспособны (отсюда и их название «парафины» - от лат. parum affinis - малое сродство).
Гомологический ряд. Ряд соединений, обладающих сходными свойствами и строением, называют гомологическим рядом. Для алканов этот ряд начинается с метана как простейшего представителя предельных углеводородов:
CH4 - метан C5H12 - пентан
C2H6 - этан C6H14 - гексан
C3H8 - пропан C7H16 - гептан
C4H10 - бутан С8H18 – октан и т. д.
Как видно, каждый последующий член ряда отличаются от предыдущего по составу на группу СН2. Эту группу СН2 называют гомологической разностью, а члены ряда называют гомологами. Отсюда можно вывести и общую формулу ряда предельных углеводородов: CnH2n+2.
Строение. В молекулах насыщенных углеводородов атомы углерода находится в состоянии sр3-гибридизации и каждый из них образуют четыре -связи с атомами водорода или углерода.
Первым и простейшим по строению членом гомологического ряда предельных углеводородов является СН4.
Атом углерода в молекуле метана расположен в центре тетраэдра, а атомы водорода – в его вершинах. Все валентные углы между направлениями связей в молекуле метана равны между собой и составляют угол 109028’.
Если от молекулы углеводорода отнять один атом водорода, то получится одновалентный углеводородный остаток, или радикал, алкил. Названия радикалов производят от названий соответствующих углеводородов, заменяя суффикс –ан на –ил. Например: метил –СН3, этил –С2Н5 и т. д. Радикал часто обозначают Аlk- (алкил) или R.
При соединении друг с другом двух метильных радикалов образуется следующий гомолог ряда этан - С2Н6. Структурная формула его имеет вид СН3-СН3.
При отнятии от молекулы этана С2 Н6 любого из атомов водорода (они все структурно равноценны) образуется одновалентный радикал С2 Н5-, или СН3-СН2-, называемый этилом.
Изомерия. Четвертый член гомологического ряда предельных углеводородов бутан отличается от трех предыдущих гомологов тем, что имеет два изомера: нормальный бутан с неразветвленной углеродной цепью и изобутан с разветвленной углеродной цепью.
Вид изомерии, когда вещества отличаются друг от друга порядком связи атомов в молекуле, называют структурной изомерией или изомерией углеродного скелета.
Радикалами называются первичными, вторичными и третичными в зависимости от того, у какого атома углерода находится свободная валентность. Если атом углерода связан с одним радикалом, он называется первичным, с двумя радикалами – вторичными, с тремя – третичным.
Первые члены гомологического ряда алканов – метан, этан, пропан – существуют только в одной форме. Они не имеют изомеров. У бутана существуют два изомера. У углеводорода состава C5H12 - пентана известны три изомера: пентан, изопентан и неопентан.
С увеличением числа углеродных атомов в молекуле углеводорода возрастает и число возможных изомеров. Для углеводородов C1-С9 изомеры получены, для высших членов ряда получены лишь некоторые.
Различные геометрические формы молекул, переходящие друг в друга путем поворота вокруг простых связей называют конформациями или поворотными изомерами (конформерами).
Номенклатура. Первые четыре предельных углеводорода имеют эмпирические названия: метан, этан, пропан, бутан. Начиная с пентана названия углеводородов образуют из греческих и латинских (нонан) числительных добавлением суффикса –ан: пентан, гексан, гептан, октан, нонан, декан, ундекан и т.д.
Сложнее обстоит дело с номенклатурой углеводородов изостроения, для этого используют рациональную и заместительную номенклатуры. В последнее время применяют международную номенклатуру, или номенклатуру ИЮПАК.
Способы получения. Главными источниками получения алканов являются нефть, природные и попутные газы и каменные угли.
1. Предельные углеводороды с небольшим числом углеродных атомов от C1 до С11 можно выделить фракционной перегонкой нефти, газа или смесей углеводородов, получаемых гидрированием угля.
2. Гидрирование непредельных углеводородов в присутствии катализаторов – платины, палладия, никеля или хромита меди.
3. Алканы получают реакцией восстановления галогенопроизводных водородом в момент его выделения или водородом, возбужденным катализатором, а также иодоводородом.
4. Синтез алканов можно осуществить по реакции Вюрца (1855) действием металлического натрия на моногалогенопроизводные. Вместо натрия могут быть использованы другие металлы, например литий, магний, цинк.
5. Предельные углеводороды могут быть получены из галогенопроизводных действием металлического лития и солей меди (реакция Кори - Хауса).
Физические свойства алканов определяются их составом и строением. Первые четыре члена гомологического ряда при обычных условиях – газы, С5-С15 – жидкости, начиная с С16 – твердые тела.
С увеличением молекулярной массы повышается температура плавления и кипения алканов, возрастает плотность. Алканы с нормальной цепью углеродных атомов кипят при более высокой температуре, чем алканы с разветвленной цепью.
Все углеводороды парафинового ряда легче воды и нерастворимы в ней, но хорошо растворимы друг в друге и сами являются хорошими растворителями жиров, масел и других органических соединений.
Закономерность в изменении свойств алканов по мере усложнения состава была открыта немецким ученым К. Шорлеммером.
Химические свойства. Предельные углеводороды при обычных условиях не взаимодействуют ни с концентрированными кислотами, ни со щелочами, ни даже с такими активными радикалами, как металлический натрий и перманганат калия. Им свойственны реакции замещения водородных атомов, расщепления, окисления и изомеризации.
Реакции замещения могут протекать по радикальному или ионному механизму. Их условно обозначают буквой S (от лат. subctitutio - замещение). Радикальные и ионные цепные реакции можно затормозить или остановить введением в реакционную систему веществ, легко реагирующих с радикалами или ионами. Такие вещества называют ингибиторами.
Рассмотрим некоторые реакции замещения с участием предельных углеводородов.
1. Галогенирование. Наибольшее практическое значение имеют хлорирование и бромирование алканов. В обычных условиях молекулярный хлор и бром практически не реагируют с насыщенными углеводородами. Только в атомарном состоянии они способны вырывать атом водорода из молекулы алкана. Поэтому предварительно необходим разрыв молекулы галогена до свободных радикалов, которые зарождают цепную реакцию.
Стадии зарождения, или инициирования цепи, ее роста и обрыва характерны для всех цепных реакций. Большой вклад в изучение цепных реакций внес Н. Н. Семенов. Его исследования отмечены Нобелевской премии.
В промышленности используют термическое хлорирование алканов при температуре порядка 3000С. Процесс хлорирования алканов в присутствии катализаторов может быть иметь цепной ионный механизм.
Реакция со свободным фтором идет со взрывом. Фторирование алканов проводят фтором (разбавленным азотом или растворителем) или некоторыми неорганическими фторидами.
Иод не способен к реакции прямого замещения водорода в алканах ввиду их высокой эндотермичности.
2. Нитрование. Реакцией нитрования называют замещение атома водорода в алкане нитрогруппой. При обычных условиях предельные углеводороды не взаимодействуют с концентрированными кислотами, в том числе и с азотной кислотой. Но при нагревании алканов до 140 0С с разбавленной 10%-ной азотной кислотой с оксидами азота идет реакция нитрования. Впервые такую реакцию осуществил М. И. Коновалов (1888) и она носит его имя. Реакции нитрования сопровождается крекингом углеводородов с образованием ряда нитропроизводных: от нитрометана до нитропроизводного с тем же числом углеродных атомов, что и у исходного углеводорода.
3. Сульфохлорирование алканов проводит смесью диоксида серы (сернистый газ) и хлора. Инициируется этот процесс фотохимически ультрафиолетовым светом. Идет цепная радикальная реакция. Сульфоокисление проводят, действуя на алканы диоксидом серы и кислородом.
4. Окисление. При поджигании на воздухе алканы воспламеняются и горят, превращаясь в диоксид углерода и воду. Окисление парафинов при температуре порядка 150 0С и в присутствии катализаторов приводит к образованию килотсодержащих органических веществ – спиртов, альдегидов, кетонов, кислот. Окисление высших углеводородов парафинового ряда приводит к образованию высших жирных кислот, спиртов, используемых для мыловарения и флотации руд цветных и черных металлов.
5. Крекинг. Процесс расщепления высококипящих предельных углеводородов при температуре 400-600 0С без доступа воздуха на молекулы низших углеводородов носит название крекинга. При крекинге предельных углеводородов происходит гомолитический разрыв углерод-углеродных связей и образование насыщенных и ненасыщенных углеводородов с более короткой цепью.
Под воздействием более высоких температур (1000 0С и выше) в результате пиролиза происходит распад молекул алканов на элементы.
Впервые крекинг нефти осуществлен сотрудником Петербургского политехнического института А. А. Летним (1871-1878), а первый патент на установку для крекинга заявлен инженером В.Г. Шуховым в 1891 г.
В настоящее время различают следующие виды крекинга: жидкофазный, когда мазут подают в печи крекинга в жидком состоянии, парофазный, когда мазут подают в печи в виде пара, и каталитический, при котором сырье разлагается в присутствии катализаторов.
9. Непредельные углеводороды ( название, формула, способы получения и применения)[1.стр. 20-23] Непредельные углеводороды этиленового ряда (олефины, алкены). Строение алкенов. Непредельными углеводородами этиленового ряда (олефинами, алкенами) называют ненасыщенные ациклические углеводороды, содержащие в молекуле одну двойную связь и имеющие общую формулу СпН2п. Название «олефины» происходит от латинского названия этилена gaz olefiant – маслородный газ.
Гомологический ряд этих углеводородов начинается с этилена СН2=СН2. Гомологи этилена образуются путем замещения атомов водорода в этилене на радикалы -СН3, -С2Н5 и т.д. Радикалы, образованные из алканов, называют алкенилами.
Номенклатура. Номенклатура алкенов сходна с номенклатурой парафинов. Названия алкенов нормального строения с двойной связью у крайнего атома углерода образуют от названий соответствующих алканов, заменяя в них суффикс –ан на –илен. Например, соединение СН3-СН=СН2 называют пропилен, а соединение СН3-СН2-СН=СН2 – бутилен и т. д. По заместительной (женевской) номенклатуре двойная связь обозначается суффиксом –ен и цифрой, после нее указывается положение двойной связи в углеродной цепи. Например, углеводород СН3-СН2-СН=СН2 называют бутен-1. В качестве главной цепи выбирают цепь, включающую двойную связь, даже если эта цепь и не будет самой длинной. Нумерацию атомов углерода в цепи проводят так, чтобы атом углерода, от которого начинается двойная связь, получил наименьший номер. Например,
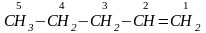
пентен-1
По номенклатуре ИЮПАК положение двойной связи указывают цифрой у атома углерода, но не после, а перед названием углеводорода. Так, углеводород пентен-1 по номенклатуре ИЮПАК надо назвать 1-пентен.
Изомерия.
У этиленовых углеводородов помимо
изомерии цепи наблюдается изомерия
положения двойной связи. В случае
бутилена двойная связь в его молекуле
может располагаться в середине цепи:
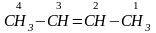 и
тогда углеводород будет называться
бутен-2 по заместительной номенклатуре
или 2-бутен – по Международной
номенклатуре.
и
тогда углеводород будет называться
бутен-2 по заместительной номенклатуре
или 2-бутен – по Международной
номенклатуре.
Кроме того, у этиленовых углеводородов возможна пространственная, или цис-транс-изомерия. Расположение одинаковых заместителей по одну сторону плоскости двойной связи дает цис-изомер, по разные стороны - транс-изомер. При нагревании, освещении или в присутствии катализатора цис- и транс-изомеры могут переходить друг в друга. Как правило, транс-изомеры более устойчивы и имеют более высокую температуру плавления.
Способы получения. Низшие алкены в небольших количествах входят состав нефтяного газа, высшие – в состав некоторых нефтей. Значительные количества алкенов содержатся в жидких продуктах крекинга и пиролиза нефти.
1. Основным источником получения первых четырех членов ряда алкенов – этилена, пропилена, бутиленов и пентиленов – являются газы крекинга и пиролиза нефтепродуктов, а также газы коксования угля (этилен, пропилен). Газы крекинга и пиролиза нефтепродуктов содержат от 15 до 30% олефинов.
2. Значительные количества алкенов могут быть получены дегидрогенизацией алканов при повышенной температуре с катализатором.
3. Алкены часто получают из галогенопроизводных отнятием галогеноводорода при действии спиртового раствора шелочи. Направление реакции дегидрогенирования соответствует правилу Л.М. Зайцева (1875) – галоген взаимодействует с атомом водорода, находящимся у наименее гидрогенизированного атома углерода.
4. В лабораторных условиях наиболее распространенным способом получения алкенов является реакция дегидратации (отнятие воды) спиртов при нагревании с водоотнимающими веществами (концентрированная серная или фосфорная кислота) или пропусканием паров спирта над катализаторами (оксид алюминия, оксид тория). Так, из этилового спирта получают этилен:

5.
Олефины можно получать так называемым
«селективным» гидрированием ацетиленовых
углеводородов над палладиевым
катализатором: СН ≡ СН + Н2
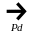 СН2
= СН2
СН2
= СН2
ацетилен этилен
Физические свойства. Первые три представителя – этилен, пропилен, бутилен – газы, от С5 до С17 – жидкости, высшие представители – твердые тела. Температура плавления и кипения, а также, плотность возрастают с увеличением числа углеродных атомов в молекуле алкена. Однако температура кипения их несколько ниже, чем у соответствующих алканов, а плотность несколько выше. Все олефины легче воды и плохо растворимы в ней. Теплота образования этиленовых углеводородов меньше, чем соответствующих алканов.
Химические свойства. В реакциях присоединения двойная связь выступает как донор электронов. Поэтому для олефинов характерна реакция электрофильного присоединения (АЕ). Рассмотрим характерные реакции алкенов.
1. Гидрирование. Непредельные углеводороды легко присоединяют водород по двойной связи в присутствии катализаторов (Pt, Pd, Ni):

пропен пропан
Так как для гидрирования необходима адсорбция молекулы алкена на катализаторе, алкены гидрируются тем легче, чем меньше заместителей у двойной связи (правило С. В. Лебедева).
2. Галогенирование. Алкены при обычных условиях присоединяют галогены, особенно легко хлор и бром. В результате образуются дигалогенопроизводные алканов, содержащие галогены у соседних атомов углерода, так называемые вициальные производные (от лат. vicinus - соседний): СН2 = СН2 + Cl2 → ClСН2 - СН2Cl. Фтор реагирует с
1, 2-дихлорэтан
воспламенением. Иод реагируют медленно и то лишь на солнечном свету. Галогены могут присоединяться к олефинам по радикальному или ионному механизму. Реакция этилена с хлором используется для получения растворителя дихлорэтана и в производстве высокомолекулярного соединения винилхлорида.
3. Гидрогалогенирование. Алкены присоединяют галогеноводороды по реакции
электрофильного присоединения: СН2 = СН2 + НCl → СН3 - СН2Cl
1-хлорпропан
Присоединение галогеноводородов к несимметричным алкенам происходит в соответствии с правилом В.В. Марковникова (1869): при присоединении галогеноводородов к несимметричным алкенам водород присоединяется к более гидрогенизированному атому углерода, а галоген к менее гидрогенизированному, т. е. к тому атому углерода, у которого меньше атомов водорода.
4. Сульфирование. Взаимодействие алкенов с серной кислотой протекает аналогично присоединению галогеноводородов.
5. Гидратация. В присутствии катализаторов (ZnCl2, H2SO4 или H3PO4) при нагревании и давлении алкены присоединяют воду и образуют спирты.
6. Окисление. Алкены легко окисляются. В зависимости от условий окисления образуются разные продукты (диоксид углерода, вода, эпоксиды, гликоли, кетоны, кислоты, озониды).
7. Алкилирование. Сущность этой реакции и состоит в присоединении алкана по двойной связи преимущественно у первичного атома углерода алкена в присутствии серной или фосфорной кислот. Реакции алкилирования используются для получения высокооктанового топлива, применяемого в двигателях внутреннего сгорения.
8. Полимеризация. Реакцией полимеризации называют процесс образования высокомолекулярного веществ (полимера) из молекул низкомолекулярного непредельного вещества путем взаимодействия их между собой за счет разрыва кратных связей. Реакция идет без выделения побочных продуктов. Реакция полимеризации открыта А. М. Бутлеровым (1873).
9. Изомеризация. При высоких температурах или в присутствии катализаторов алкены способны изомеризоваться.