

- •В питаннях та відповідях
- •Часть I. Общая патология
- •1.4.Как связана патологическая физиология с другими науками?
- •1.5.Какова взаимосвязь патофизиологии с клиникой?
- •1.6.Какие методы использует медицина в изучении болезней?
- •1.7.Что является основным методом патологической физиологии? в чем его особенность?
- •1.8.Что мы называем экспериментальной моделью болезни?
- •1.9.Какое значение имеет патофизиологический эксперимент как метод изучения болезни у человека?
- •1.10.Какие существуют виды эксперимента?
- •1.11.Назовите этапы проведения патофизиологического эксперимента.
- •1.12.Что такое планирование эксперимента?
- •1.13.Какие существуют методы моделирования болезней?
- •1.14.Какие методы использует патологическая физиология для получения информации об изменениях в организме экспериментальных животных?
- •1.15.Какие методы анализа полученных результатов могут быть использованы в экспериментальных исследованиях?
- •1.16.Где и когда возникла патологическая физиология как учебная дисциплина?
- •1.17.Назовите составные части патологической физиологии как учебной дисциплины.
- •1.18.Какова история развития патологической физиологии в Украине?
- •1.19.Каков вклад а. А. Богомольца в развитие патологической физиологии?
- •1.20.Чем известна научная школа а.А. Богомольца?
- •2.Общая нозология
- •2.1.Какие основные понятия использует общая нозология?
- •2.2.Что такое здоровье?
- •2.3.Что такое норма?
- •2.4.Какое содержание вкладывают в понятие «болезнь»?
- •2.5.Какие существуют определения понятия «болезнь» как философского обобщения?
- •2.6.Чем отличается болезнь от здоровья?
- •2.7.Какие существуют принципы классификации болезней?
- •2.8.Что такое патологический процесс?
- •2.9.Чем различаются понятия «патологический процесс» и «болезнь»?
- •2.10.Дайте определение понятия «типические патологические процессы».
- •2.11.Что такое патологическое состояние?
- •2.12.Как взаимосвязаны патологический процесс и патологическое состояние?
- •2.13.Что такое патологическая реакция?
- •2.14.Какие бывают исходы болезней?
- •2.15.Что такое выздоровление?
- •2.16.Что такое ремиссия, рецидив, осложнение?
- •2.17.Что такое терминальные состояния?
- •2.18.Назовите основные исторически сложившиеся направления в учении о болезни.
- •2.19.Назовите главных представителей гуморального направления в учении о болезни. Как развивается это направление на современном этапе?
- •2.20.Назовите главных представителей солидарного направления в учении о болезни. Кок развивается это направление на современном этапе?
- •2.21.В чем сущность клеточной теории патологии Вирхова?
- •2.22.В чем сущность функционального направления в учении о болезни?
- •3.Общая этиология
- •3.1.Что такое этиология?
- •3.2.Какие философские категории составляют основу этиологии?
- •3.3.Что такое причина болезни? Какими свойствами она обладает?
- •3.4.Сколько причин может иметь болезнь?
- •3.5.Что такое условия возникновения болезни? Как условия могут влиять на развитие болезней?
- •3.6.Как классифицируют этиологические факторы?
- •3.7.Что такое факторы риска? Приведите примеры.
- •3.8.Как влияют социальные факторы на возникновение болезней?
- •3.9. Действием каких факторов объясняют развитие «болезней цивилизации»?
- •3.10.Назовите основные направления общей этиологии. В чем их сущность?
- •4.4.Сочетание каких процессов и явлений составляет основу патогенеза?
- •4.5.Что такое адаптация и компенсация? в чем общность и различие этих понятий?
- •4.6.Как происходит становление адаптации и компенсации? Различают два этапа развития этих состояний.
- •4.7.Что такое причинно-следственные отношения в патогенезе? Какие существуют варианты этих отношений?
- •4.8.Приведите примеры развития патогенеза по типу «порочного круга».
- •4.9.Что такое главное звено патогенеза?
- •4.10.Что понимают под специфическими и неспецифическими механизмами патогенеза?
- •4.11.Какие существуют связи между местными и общими явлениями в патогенезе?
- •4.12.Как взаимосвязаны структурные и функциональные нарушения в патогенезе?
- •5.Патогена дія факторів зовнішнього середовища
- •5.1.Що таке механічна травма? Які загальні порушення можуть обумовлюватись механічною травмою?
- •5.2.Какие патологические процессы могут развиваться при действии на организм термических факторов?
- •5.3.Как изменяется теплопродукция и теплоотдача в разные стадии гипо- и гипертермии?
- •5.4.Какие защитно-компенсаторные реакции развиваются при гипотермии?
- •5.5.Какими собственно патологическими изменениями проявляется стадия декомпенсации при гипотермии?
- •5.6.Какие защитно-компенсаторные реакции развиваются при гипертермии?
- •5.7.Какими собственно патологическими изменениями проявляется стадия декомпенсации при гипертермии?
- •5.8.Какие общие изменения могут возникать при ожогах?
- •5.9.Какие виды ионизирующего излучения могут оказывать патогенное действие на организм?
- •5.10.От чего зависит чувствительность тканей к действию ионизирующей радиации?
- •5.11.В чем сущность прямого повреждающего действия ионизирующей радиации на клетки?
- •5.12.В чем сущность непрямого повреждающего действия ионизирующей радиации на клетки? Что такое радиолиз воды?
- •5.13.Что такое радиотоксины?
- •5.14.Фактори, що сприяють і перешкоджають розвитку променевих ушкоджень.
- •5.15.Захисно-компенсаторні механізми в клітинах направлені на попередження і ліквідацію променевого ураження. Радіопротектори.
- •5.16.Назовите формы и стадии развития острой лучевой болезни.
- •5.17.Какие синдромы наиболее характерны для периода выраженных клинических признаков острой лучевой болезни? Каков их патогенез?
- •5.18.Назовите наиболее важные отдаленные последствия действия на организм ионизирующей радиации.
- •5.19.Вплив зниженого атмосферного тиску.
- •5.20.Когда человек испытывает действие повышенного атмосферного давления? Какие патогенные факторы действуют на организм в этих условиях?
- •5.21.Что такое болезнь декомпрессии и взрывная декомпрессия?
- •5.22.Какие патогенные факторы действуют на организм во время космического полета?
- •5.23.От каких факторов зависит повреждающее действие электрического тока?
- •5.24.Каким местным действием на ткани обладает электрический ток?
- •5.25.Что может быть непосредственной причиной смерти при действии электрического тока на организм?
- •5.26.Чем проявляется патогенное действие на организм инфракрасного и ультрафиолетового излучения?
- •5.27.Что такое фотосенсибилизация?
- •5.28.Что может быть причиной мутации?
- •5.29.Какие нарушения в геноме клеток могут возникать при действии мутагенов?
- •Моногенні спадкові хвороби.
- •5.30.Назовите типы наследования моногенных болезней.
- •5.31.Приведите примеры наследственных болезней, которые передаются по аутосомно-доминантному типу.
- •Приклади спадкових хворіб, які передаються по аутосомно-рецессивному типу.
- •Приклади наследственных болезней, которые передаются по типу, сцепленному с полом.
- •Полігенніхвороби.
- •Хромосомные болезни.
- •Механізми, які лежать в основі геномних мутацій.
- •Синдромы связаны с изменением количества соматических хромосом? Дайте их краткую характеристику.
- •Синдромы связаны с изменением количества половых хромосом?
- •Виды хромосомных мутаций.
- •Клиническими признаками проявлення хромосомних мутацій.
- •Основные методы изучения наследственных болезней.
- •5.32.Что такое конституция? Каково ее значение?
- •5.33.Какие существуют классификации конституциональных типов человека? На чем они основаны?
- •Діатез.
- •Виды диатезів.
- •6.Значение возрастных факторов в патологии. Старение
- •6.1.Назовите виды нарушений внутриутробного развития в зависимости от времени их возникновения.
- •6.2.Назовите основные причины нарушений внутриутробного развития.
- •6.3.Что такое тератогенные факторы?
- •6.4.Что такое критические периоды внутриутробного развития? Какое они имеют значение в патологии?
- •6.5.Что такое старение?
- •6.6.Как классифицируют старение?
- •6.7.Какие факторы влияют на видовую продолжительность жизни?
- •6.8.Какие факторы Влияют на индивидуальную продолжительность жизни человека?
- •6.9.Назовите основные закономерности процесса старения.
- •6.10.Какими структурными изменениями проявляется старение?
- •6.11.Какими функциональными изменениями проявляется старение?
- •6.12.Какие биохимические признаки характерны для старения?
- •6.13.В чем сущность теорий генетически запрограммированного старения?
- •6.14.В чем сущность теорий накопления повреждений, объясняющих причины старения?
- •6.15.Объясните сущность синтетических теорий старения.
- •6.16.Какими механизмами, препятствующими старению, располагает организм?
- •6.17.Какая связь существует между старением и болезнями человека?
- •6.18.Что такое прогерия?
- •6.19.Что такое геропротекция? Какие подходы она использует?
- •7.Роль реактивности в патологии
- •7.1.Что такое реактивность?
- •7.2.Приведите примеры проявлений реактивности на разных уровнях организации живых объектов.
- •7.3.Какие выделяют виды реактивности?
- •7.4.Какие факторы влияют на реактивность организма человека?
- •7.5.Что такое резистентность?
- •7.6.Какие выделяют виды резистентности?
- •7.7.Что такое пассивная и активная резистентность? в чем их различие?
- •7.8.Как взаимосвязаны реактивность и резистентность?
- •7.9.Назовите механизмы неспецифической резистентности, обеспечивающие устойчивость организма к действию инфекционных агентов.
- •7.10.Что понимают под ареактивностью клеток, характеризуя механизмы неспецифической резистентности к инфекциям?
- •7.11.Какие физические и физико-химические факторы являются факторами неспецифической резистентности организма к инфекциям?
- •7.12.Как классифицируются биологические барьеры организма?
- •7.13.Какие функции выполняет физиологическая система соединительной ткани?
- •7.14.Что такое сыворотка Богомольца? Каков механизм ее действия?
- •7.15.Приведите примеры гуморальных факторов неспецифической резистентности организма к действию инфекционных агентов.
- •7.16.Что такое интерфероны?
- •7.17.Что такое система комплемента? Как она активируется?
- •7.18.Назовите функции комплемента и продуктов его активации.
- •7.19.Какие первичные нарушения могут возникать в системе комплемента?
- •7.20.Что такое фагоцитоз? Какие клетки обладают свойствами фагоцитов?
- •7.21.Какие функции присущи клеткам-фагоцитам?
- •7.22.Назовите стадии фагоцитоза.
- •7.23.Что является причиной хемотаксиса фагоцитов?
- •7.24.Какие механизмы обеспечивают прилипание фагоцита к объекту фагоцитоза?
- •7.25.Что такое опсонины? Какие опсонины имеют наибольшее значение для осуществления фагоцитоза?
- •7.30.Как классифицируют нарушения фагоцитоза?
- •7.31.Приведите примеры нарушений фагоцитоза, связанных с особенностями объекта фагоцитоза.
- •7.32.Какие причины могут нарушать процессы опсонизации и вызывать расстройства фагоцитоза?
- •7.33.Какие качественные изменения фагоцитов могут быть причиной нарушений фагоцитоза?
- •8.Нарушения иммунологической реактивности
- •8.1.Что такое иммунологическая реактивность?
- •8.2.Какие механизмы обеспечивает иммунологическую реактивность?
- •8.3.Что такое антигены?
- •8.4.Что такое антитела?
- •8.5.Какие клетки принимают участие в осуществлении иммунных ответов?
- •8.6.Назовите функции в‑лимфоцитов.
- •8.7.Назовите функции т‑лимфоцитов.
- •8.8.Какое участие в осуществлении иммунного ответа принимают макрофаги?
- •8.9.Что такое тимуснезависимые антигены? Каковы механизмы гуморального иммунного ответа на их поступление в организм?
- •8.10.Что такое тимусзависимые антигены? Каковы механизмы гуморального иммунного ответа на их поступление в организм?
- •8.11.Опишите механизмы иммунного ответа клеточного типа.
- •8.12.Что такое иммунологическая недостаточность?
- •8.13.Что такое первичная иммунологическая недостаточность? Какие причины ее вызывают?
- •8.14.Как классифицируют первичную иммунологическую недостаточность?
- •8.15.Приведите примеры первичных в-клеточных иммунодефицитов.
- •8.16.Приведите примеры первичных т-клеточных иммунодефицитов.
- •8.17.Приведите примеры комбинированных первичных иммунодефицитов.
- •8.18.Что такое вторичная иммунологическая недостаточность? Какие причины ее вызывают?
- •8.19.Что такое синдром приобретенного иммунодефицита (спид)? Что является его причиной?
- •8.20.Каков патогенез иммунологической недостаточности у больных спиДом?
- •8.21.Какие механизмы могут лежать в основе развития приобретенных иммунных нарушений гуморального типа?
- •8.28.Что такое реакция «трансплантат против хозяина»? При каких условиях она возникает?
- •8.29.Как молено осуществить неспецифическую иммуности-муляцию?
- •8.30.Какие существуют методы иммуносупрессии?
- •9.5.Как классифицируют эндогенные аллергены?
- •9.6.Что такое инфекционные приобретенные эндоаллергены?
- •9.7.Объясните факт существования естественных эндоаллергенов.
- •9.8.Как классифицируют аллергические реакции?
- •9.9.Что такое аллергические реакции немедленного и замедленного типа?
- •9.10.Какие стадии выделяют в патогенезе аллергических реакций?
- •9.11.Какой период времени охватывает иммунологическая стадия аллергических реакций? в чем ее сущность?
- •9.12.Что такое сенсибилизация? Какие существуют ее варианты?
- •9.17.Как в эксперименте можно воспроизвести анафилактический шок?
- •9.18.Что такое феномен Овери? Как его воспроизводят в эксперименте?
- •9.19.Приведите примеры клинических форм аллергических реакций I типа (анафилактических).
- •9.20.Что происходит во время иммунологической стадии аллергических реакций I типа?
- •9.21.Чем характеризуется патохимическая стадия аллергических реакций I типа?
- •9.22.Каким действием обладает гистамин в условиях развития анафилактических реакций?
- •9.23.Какие существуют механизмы, ограничивающие пато-химическую стадию анафилактических реакций?
- •9.24.В чем сущность дополнительного механизма патохимической стадии анафилактических реакций?
- •9.25.Какие местные клинические проявления характерны для анафилактических реакций? Каков механизм их развития?
- •9.26.Назовите основные механизмы развития клинических проявлений анафилактического шока.
- •9.31.Какие механизмы обеспечивают разрушение клеток (цитолиз) во время цитотоксических реакций?
- •9.32.Чем характеризуется патохимическая стадия цитотоксических реакций?
- •9.33.Что происходит во время патофизиологической стадии цитотоксических реакций?
- •9.34.В чем сущность аллергических реакций III типа (им-мунокомплексных) по классификации Кумбса и Джелла?
- •9.35.Как воспроизводят иммунокомплексные реакции в эксперименте?
- •9.36.Приведите примеры клинических форм иммунокомплексных реакций.
- •9.37.Какие антигены и антитела принимают участие в развитии иммунокомплексных реакций?
- •9.38.Какими факторами определяются патогенные свойства циркулирующих иммунных комплексов?
- •9.39.Какие существуют виды циркулирующих иммунных комплексов? Каково их значение в развитии аллергических реакций?
- •9.40.Какие условия способствуют развитию иммунокомплексных повреждений?
- •9.41.Чем характеризуется патохимическая стадия иммунокомплексных реакций?
- •9.42.Что происходит во время патофизиологической стадии аллергических реакций III типа?
- •9.47.Какие биологически активные вещества выделяются во время патохимической стадии аллергических реакций IV типа?
- •9.48.Как классифицируют лимфокины?
- •9.49.Какие механизмы обеспечивают уничтожение анти-геннесущих клеток в реакциях гиперчувствительности замедленного типа?
- •9.50.Что такое аутоаллергические реакции?
- •9.51.Какие механизмы лежат в основе развития аутоал-лергии?
- •9.52.Что такое псевдоаллергические реакции? Приведите примеры.
- •9.53.Назовите основные принципы предупреждения и лечения аллергии.
- •10.Повреждение клетки
- •10.1.Что такое повреждение клетки и какие существуют принципы его классификации?
- •10.2.Какие факторы могут вызывать повреждение клетки?
- •10.3.Какие признаки свидетельствуют о повреждении клетки?
- •10.4.Чем принципиально отличаются два патогенетических варианта повреждения клетки: насильственный и цитопатический?
- •10.5.Какие изменения на молекулярном уровне имеют большое значение в патогенезе повреждения клетки?
- •10.6.В чем сущность пероксидного окисления липидов?
- •10.7.Какие реакции лежат в основе инициации пероксидного окисления липидов?
- •10.8.Какими антиоксидантными системами располагают клетки?
- •I. Ферментные антиоксидантные системы:
- •10.9.В каких случаях происходит активация пол? Активация пол происходит:
- •10.10.Какие механизмы лежат в основе нарушений барьерных функций клеточных мембран при активации пол?
- •10.11.Как нарушается матричная функция мембран в процессе активации пол?
- •10.12.Каким образом повышение активности фосфолипаз способствует повреждению клеточных мембран?
- •10.13.При каких условиях возникает опасность детергентного действия свободных жирных кислот на клеточные мембраны?
- •10.14.В каких случаях ионы кальция вовлекаются в патогенез повреждения клетки? с какими эффектами этих ионов связано их участие в повреждении клеточных структур?
- •10.15.Чем могут быть обусловлены сдвиги в содержании ионов натрия и калия в клетке и какова роль таких сдвигов в патогенезе клеточного повреждения?
- •10.16.Чем может быть обусловлено развитие внутриклеточного ацидоза и какие изменения в клетке могут быть с ним связаны?
- •10.17.Какие изменения белковых молекул имеют значение в патогенезе повреждения клетки?
- •10.18.Какие нарушения функционирования генетического аппарата клетки могут приводить к ее повреждению?
- •10.19.Какие существуют универсальные механизмы повышения проницаемости клеточных мембран при повреждении клетки?
- •10.20.Какие нарушения возникают в клетке в результате повреждения отдельных ее органоидов (плазматической мембраны, митохондрий, эндоплазматического ретикулума, лизосом)?
- •10.21.Какие существуют механизмы гибели клеток? Что такое апоптоз?
- •10.22.Какими защитно-компенсаторными механизмами располагает поврежденная клетка?
- •10.23.Какие существуют подходы к патогенетическому лечению поврежденных клеток?
- •11.Экстремальные состояния
- •11.1.Что такое экстремальные состояния?
- •11.2.Что такое шок?
- •11.3.Назовите основные Виды шока.
- •11.4.Какие механизмы лежат в основе нарушений общей гемодинамики и микроциркуляции при шоке?
- •11.5.Чем определяются тяжелые последствия шока?
- •11.6.Объясните патогенез травматического шока.
- •11.7.Что такое коллапс?
- •11.8.Что такое краш-синдром?
- •11.9.Что такое кома?
- •11.10.Как классифицируют коматозные состояния?
- •11.11.Какие механизмы лежат в основе патогенеза коматозных состояний?
- •12.4.Какие факторы могут быть причиной артериальной гиперемии? Что подразумевают под физиологической и патологической артериальной гиперемией?
- •12.5.Назовите основные механизмы развития патологической артериальной гиперемии.
- •12.6.В чем сущность нейротонического механизма развития артериальной гиперемии?
- •12.7.Объясните нейропаралитический механизм развития артериальной гиперемии.
- •12.8.Какие гуморальные факторы могут вызывать развитие артериальной гиперемии?
- •12.9.Какова роль эндотелия кровеносных сосудов в развитии артериальной гиперемии?
- •12.10.Назовите возможные исходы артериальной гиперемии.
- •12.11.Что такое венозная гиперемия?
- •12.12.Какие факторы могут быть причиной венозной гиперемии?
- •12.13.Какими признаками проявляется венозная гиперемия?
- •12.14.Какие местные и общие нарушения могут быть следствием венозной гиперемии?
- •12.15.Что такое ишемия?
- •12.16.Какие признаки характерны для ишемии?
- •12.17.Назовите основные типы ишемии в зависимости от причины и механизмов ее возникновения.
- •12.18.Чем определяется характер обменных, функциональных и структурных нарушений в ткани при ее ишемии?
- •12.19.Какими последовательными стадиями характеризуется патогенез нарушений в ишемизированной ткани?
- •12.20.Что такое стаз?
- •12.21.Назовите основные варианты стаза и их причины.
- •12.22.Что такое тромбоз?
- •12.23.Назовите три основных фактора, способствующих тромбообразованию (триада Вирхова).
- •12.24.Из каких фаз состоит процесс образования тромба? в чем их сущность?
- •12.25.Какие отрицательные последствия может иметь тромбообразование в условиях патологии?
- •12.26.Что такое эмболия?
- •12.27.Какие выделяют виды эмболии?
- •12.28.Назовите основные причины эмболии экзогенного происхождения.
- •12.29.Назовите основные причины эмболии эндогенного происхождения.
- •12.30.Что понимают под микроциркуляцией? Назовите основные типы нарушений микроциркуляции.
- •12.31.В чем сущность феномена под названием «сладж»?
- •12.32.Как осуществляется обмен воды между плазмой крови и интерстициальной жидкостью?
- •12.33.Как влияют изменения гидростатического и онкотического давления крови и межклеточной жидкости на интенсивность процессов фильтрации – реабсорбции воды в капиллярах?
- •12.34.Что такое недостаточность лимфообращения? Назовите основные ее формы.
- •13.Воспаление
- •13.1.Дайте определение понятия «воспаление».
- •13.2.Почему воспаление называют типическим патологическим процессом?
- •13.3.Назовите внешние признаки воспаления.
- •13.4.Что может быть причиной воспаления?
- •13.5.Какие методы используют при изучении воспаления?
- •13.11.Почему в очаге воспаления развиваются гиперосмия и гиперонкия?
- •13.12.Какие нарушения обмена веществ закономерно возникают в очаге воспаления?
- •13.13.Что такое медиаторы воспаления? Назовите основные их классы,
- •13.14.Какую роль играют лизосомальные факторы в патогенезе воспаления?
- •13.15.Какие факторы могут вызывать дегрануляцию тканевых базофилов в очаге воспаления?
- •13.16.Каким действием в очаге воспаления обладают биогенные амины – продукты дегрануляции тканевых базофилов?
- •13.17.Как происходит активация калликреин-кининовой системы? Назовите основные функциональные эффекты кининов.
- •13.18.Какие медиаторы воспаления являются производными арахидоновой кислоты? Как они образуются и каким действием обладают?
- •13.19.Какова роль лимфокинов и монокинов в патогенезе воспаления?
- •13.20.Какое значение имеет комплемент и продукты его активации в патогенезе воспаления?
- •13.21.Какие продукты активации свертывающей и фибринолитической систем крови могут влиять на патогенез воспаления?
- •13.22.Назовите стадии нарушений местного кровообращения в очаге воспаления. Кто их впервые описал?
- •13.23.Какой механизм лежит в основе кратковременной ишемии в начале воспаления?
- •13.24.Назовите механизмы развития артериальной гиперемии в очаге воспаления.
- •13.25.Какие факторы вызывают переход артериальной гиперемии в венозную в процессе развития воспаления? Можно выделить две группы таких факторов.
- •13.26.Что такое экссудация? Какие механизмы лежат в основе выхода жидкой части крови из сосудов в воспаленную ткань?
- •13.27.Назовите механизмы повышения проницаемости сосудистой стенки при воспалении.
- •13.28.Что является причиной повышения проницаемости сосудистой стенки в очаге воспаления?
- •13.29.Какова динамика повышения проницаемости сосудов при воспалении?
- •13.30.Что такое эмиграция лейкоцитов? Какие лейкоциты и в какой последовательности эмигрируют в очаг воспаления?
- •13.31.Что такое краевое стояние лейкоцитов? Каковы его механизмы?
- •13.32.Что такое адгезивные белки лейкоцитов и эндотелиальных клеток? Что вызывает их появление?
- •13.33.Каким образом лейкоциты преодолевают сосудистую стенку, эмигрируя в воспаленную ткань?
- •13.34.В чем состоит сущность стадии пролиферации в патогенезе воспаления?
- •13.35.Какие факторы вызывают активацию размножения клеток в очаге воспаления?
- •13.36.Какие механизмы лежат в основе развития местных клинических признаков воспаления?
- •13.37.Какие общие проявления характерны для воспаления?
- •13.38.Что такое «белки острой фазы воспаления»? Что является причиной повышения их количества при воспалении?
- •13.39.Как влияют гормоны на течение воспаления? Каков механизм противовоспалительного действия глюкокортикоидов?
- •13.40.Почему воспаление следует считать патологическим процессом, а не защитной компенсаторной реакцией организма?
- •14.4.Что такое естественные и искусственные пирогены?
- •14.5.Что называют экзогенными и эндогенными пирогенами?
- •14.6.Что такое первичные и вторичные пирогены?
- •14.7.Какие существуют доказательства того, что действие первичных пирогенов связано с образованием вторичных?
- •14.8.Какие последовательные процессы составляют сущность патогенеза лихорадки?
- •14.9.Где расположен и что представляет собой с функциональной точки зрения центр терморегуляции?
- •14.10.Каков механизм действия интерлейкина-1 на центр терморегуляции?
- •14.11.Какие существуют доказательства роли простагландинов е в патогенезе лихорадки?
- •14.12.Назовите стадии лихорадки.
- •14.13.Объясните, как происходит повышение температуры тела в I стадию лихорадки.
- •14.14.Объясните механизмы падения температуры тела при завершении лихорадки. Какие существуют варианты такого падения?
- •14.15.Какие типы температурных кривых могут быть характерны для лихорадки? Какие факторы влияют на динамику изменения температуры тела при лихорадке?
- •14.16.Почему лихорадку называют патологическим процессом?
- •14.17.В чем состоит защитно-приспособительное значение лихорадки?
- •14.18.Какие собственно патологические изменения могут возникать при лихорадке?
- •14.19.В чем проявляется влияние лихорадки на обмен веществ?
- •14.20.В чем состоит принципиальное различие между лихорадкой и гипертермией?
- •14.21.Что является основным патогенетическим принципом жаропонижающей терапии?
- •14.22.Приведите примеры использования лихорадки с лечебной целью.
- •15.6.Какие существуют методы экспериментального изучения опухолей?
- •15.7.Назовите основные причины возникновения злокачественных опухолей.
- •15.8.Кто и как впервые доказал роль химических факторов в возникновении злокачественных опухолей?
- •15.9.Как классифицируют химические канцерогены?
- •15.10.Приведите примеры канцерогенов естественного и искусственного происхождения.
- •15.11.Охарактеризуйте канцерогенное действие полициклических ароматических углеводородов (пау).
- •15.12.В чем состоит особенность канцерогенного действия ароматических аминов?
- •15.13.Чем характеризуется канцерогенное влияние нитрозосоединений?
- •15.14.Приведите примеры канцерогенного действия продуктов жизнедеятельности грибов.
- •15.15.Какое содержание вкладывают в понятие «эндогенные канцерогены»?
- •15.16.В каких опытах доказывается возможное участие гормонов в возникновении злокачественного опухолевого роста?
- •15.17.Что такое химические канцерогены прямого и непрямого действия? Дайте их сравнительную характеристику.
- •15.18.Какие свойства химических веществ обусловливают их канцерогенное действие?
- •15.19.Какие стадии проходит химический канцерогенез? в чем их сущность?
- •15.20.Чем объясняется явление опухолевой прогрессии?
- •15.21.Какие физические факторы могут иметь значение в возникновении злокачественных опухолей?
- •15.22.Назовите основные закономерности канцерогенного действия ионизирующего излучения.
- •15.23.Что такое «пластмассовый» канцерогенез? в чем его особенности?
- •15.24.Кем и в каких экспериментах была доказана роль вирусов в возникновении опухолей?
- •15.25.Какие днк-содержащие вирусы являются онкогенными для животных и человека?
- •15.26.Какие рнк-содержащие вирусы являются онкогенными для животных и человека?
- •15.27.Назовите этапы вирусного онкогенеза.
- •15.28.Назовите факторы, от которых зависит трансформирующее действие вирусов на клетку.
- •15.29.Что такое вирусные онкогены?
- •15.30.Что такое протоонкогены?
- •15.31.Как классифицируют вирусные онкогены и протоонкогены?
- •15.32.Что такое клеточные онкогены? Назовите основные механизмы превращения протоонкогенов в клеточные онкогены.
- •15.33.Что такое антионкогены?
- •15.34.Какие молекулярные механизмы могут лежать в основе вирусного онкогенеза?
- •15.35.С какими молекулярными механизмами может быть связан канцерогенез, вызванный действием химических и физических факторов?
- •15.36.Назовите особенности роста злокачественных опухолевых клеток в условиях in vitro.
- •15.37.Назовите основные особенности роста злокачественных опухолей in vitro.
- •15.38.С какой скоростью растут злокачественные опухоли? Какими факторами она определяется?
- •15.39.Какими нарушениями обмена веществ характеризуются злокачественные опухоли?
- •15.40.Что такое инвазивность опухоли? Как злокачественные клетки прорастают в окружающую ткань?
- •15.41.Что такое метастазирование? Как оно осуществляется?
- •15.42.Как влияет опухоль на организм в целом? Почему развивается раковая кахексия?
- •15.43.Какие факторы организма влияют на развитие злокачественных опухолей?
- •15.44.Какие механизмы противоопухолевой защиты существуют в организме?
- •16.3.Какие нарушения химического состава крови свидетельствуют о нарушениях снабжения клеток питательными веществами?
- •16.4.Назовите причины внутриклеточных нарушений энергетического обмена.
- •16.5.Нарушения каких центральных внутриклеточных катаболических путей могут приводить к расстройствам энергообеспечения клеток?
- •16.6.Назовите основные причины нарушений центральных катаболических путей в клетках.
- •16.7.Какие причины могут вызывать развитие витаминной недостаточности в клетках?
- •16.8.Назовите основные причины нарушения биологического окисления в митохондриях клеток.
- •16.9.Что такое разобщение окисления и фосфорилирования? Каковы его механизмы?
- •16.10.При нарушении каких биохимических реакций уменьшается ресинтез атф в клетках?
- •16.11.Какие последствия для клетки вызывает дефицит атф?
- •16.12.Какие нарушения метаболизма в клетках могут быть связаны с первичным дефицитом атф?
- •16.13.Приведите примеры «порочных кругов» в развитии энергодефицитного состояния клеток.
- •17.3.На какие периоды делят патогенез полного голодания с употреблением воды?
- •17.4.Чем характеризуется период неэкономной траты энергии?
- •17.5.Что характерно для второго периода голодания – периода максимального приспособления?
- •17.6.Каковы потери в массе разных органов и тканей во втором периоде голодания?
- •17.7.Дайте характеристику третьего периода голодания.
- •17.8.Какие факторы определяют максимально возможную продолжительность полного голодания с употреблением воды?
- •17.9.Как рассчитать максимально возможную продолжительность полного голодания с употреблением воды?
- •17.10.Что такое абсолютное голодание? в чем его особенность?
- •17.11.Назовите особенности неполного голодания.
- •17.12.Что такое белково-энергетическая недостаточность? Приведите примеры.
- •17.13.Какими клиническими синдромами проявляется белково-энергетическая недостаточность?
- •17.14.Какими нарушениями физиологических функций проявляются строфические изменения в органах и тканях при белково-энергетической недостаточности ?
- •18.3.В каких биохимических процессах, протекающих в организме, используется кислород?
- •18.4.Какие механизмы могут лежать в основе уменьшения напряжения кислорода в тканях?
- •18.5.Назовите причины уменьшения доставки кислорода кровью.
- •18.6.Чем может быть обусловлено уменьшение содержания кислорода в артериальной крови?
- •18.7.Какие изменения могут уменьшать объемную скорость кровотоко в тканях и приводить к гипоксии?
- •18.8.Какие факторы вызывают сдвиг кривой диссоциации оксигемоглобина?
- •18.9.Какие факторы вызывают нарушение диффузии кислорода в тканях?
- •18.10.Что такое гипоксия нагрузки?
- •18.11.Что такое гипоксическая гипоксия? Когда она возникает.
- •18.12.Назовите патогенетические факторы развития горной болезни.
- •18.13.Какие зоны выделяют при подъеме в горы с учетом признаков развивающейся гипоксии?
- •18.23.Какие факторы могут вызывать образование метгемоглобина и, следовательно, развитие кровяной гипоксии? Причинами образования метгемоглобина являются:
- •18.28.На какие группы можно разделить все защитно-компенсаторные реакции, возникающие при гипоксии?
- •18.29.Назовите защитно-компенсаторные реакции организма, направленные на. Увеличение доставки кислорода тканям.
- •18.30.Какие нежелательные последствия может иметь гипервентиляция при гипоксической гипоксии?
- •18.31.Назовите местные (тканевые) реакции, направленные на улучшение обеспечения клеток кислородом в условиях гипоксии.
- •18.32.Назовите защитно-компенсаторные реакции в системах утилизации кислорода при гипоксии.
- •18.33.При каких значениях напряжения кислорода в тканях начинает уменьшаться образование атф в клетках?
- •18.34.Какие механизмы составляют основу гипоксического повреждения клеток?
- •18.35.От каких факторов зависит чувствительность клеток к гипоксии?
- •18.36.Какие периоды характерны для острой гипоксии клеток?
- •18.37.Какова динамика изменений в цнс при острой гипоксии?
- •19.5.Какие изменения углеводного обмена вызывает адреналин?
- •19.12.Дайте определение понятия «гипогликемия».
- •19.13.Какие механизмы могут лежать в основе развития гипогликемии ?
- •19.14.Какими клиническими признаками проявляется гипогликемия? Что такое гипогликемическая кома?
- •19.15.Что такое гипергликемия?
- •19.16.Какие механизмы могут лежать в основе гипергликемии?
- •19.17.Дайте определение понятия «сахарный диабет».
- •19.18.Какие существуют экспериментальные модели сахарного диабета?
- •19.19.Приведите патогенетическую классификацию сахарного диабета.
- •19.20.Дайте сравнительную характеристику сахарного диабета I и II типов.
- •19.21.Каковы причины развития сахарного диабета I типа?
- •19.22.Какие механизмы лежат в основе развития абсолютной инсулиновой недостаточности при сахарном диабете I типа?
- •19.23.Каковы причины развития сахарного диабета II типа?
- •19.24.Опишите патогенез сахарного диабета II типа с ожирением.
- •19.25.Назовите возможные причины внепанкреатической недостаточности инсулина.
- •19.26.Какие виды обмена веществ нарушаются при сахарном диабете?
- •19.27.Объясните механизмы развития гипергликемии при сахарном диабете.
- •19.28.Какие клинические признаки сахарного диабета обусловлены гипергликемией?
- •19.29.Какие нарушения свидетельствуют о расстройствах жирового обмена при сахарном диабете?
- •19.30.Чем проявляются нарушения белкового обмена при сахарном диабете?
- •19.31.Какие нарушения водно-электролитного обмена характерны для сахарного диабета? Каков их патогенез?
- •19.32.Какие нарушения кислотно-основного состояния развиваются при сахарном диабете?
- •19.33.Какие варианты коматозных состояний могут развиваться при сахарном диабете?
- •19.34.Какие осложнения характерны для сахарного диабета?
- •19.35.Какие механизмы могут лежать в основе развития макроангиопатий при сахарном диабете?
- •19.36.Как объясняют развитие микроангиопатий при сахарном диабете? Чем они могут проявляться?
- •19.37.Каков патогенез нейропатий при сахарном диабете?
- •19.38.Назовите основные патогенетические принципы лечения сахарного диабета.
- •20.Нарушения жирового обмена
- •20.1.Назовите основные причины нарушений жирового обмена в организме.
- •20.2.Что может быть причиной нарушений переваривания и всасывания липидов в кишках?
- •20.3.Какие изменения состава крови могут быть проявлением нарушений транспорта липидов в организме?
- •20.4.Какими классами представлены липопротеиды плазмы крови?
- •20.5.Дайте сравнительную характеристику разных классов липопротеидов плазмы крови.
- •20.6.Как классифицируют гиперлипопротеинемии?
- •20.7.Какие генетические дефекты могут быть причиной развития первичных (наследственных) гиперлипопротеинемии?
- •20.8.Назовите возможные причины развития вторичных (приобретенных) гиперлипопротеинемий,
- •20.9.Приведите классификацию гиперлипопротеинемий, предложенную экспертами воз.
- •20.10.Приведите примеры развития разных типов гиперлипопротеинемий по классификации воз.
- •20.11.Что такое продукционная и ретенционная гиперлипопротеинемия?
- •20.12.В чем состоит патогенетическое значение гиперлипопротеинемий?
- •20.13.Приведите примеры гиполипопротеинемий. Дайте краткую характеристику.
- •20.14.Что такое «модифицированные» липопротеиды? Приведите примеры. Каково их патогенетическое значение?
- •20.21.Какие механизмы лежат в основе увеличения массы жировой ткани при ожирении?
- •20.22.В чем состоит патогенетическое значение ожирения?
- •20.23.Что такое гиперкетонемия? Какие причины и механизмы лежат в основе ее развития?
- •20.24.Какие гормоны усиливают липолиз в жировой ткани и могут вызывать гиперлипацидемию с последующей гиперкетонемией?
- •20.25.Какие нарушения в организме обусловливает гиперкетонемия?
- •21.Нарушения белкового обмена, обмена аминокислот и азотистых оснований
- •21.1.Что такое положительный и отрицательный азотистый баланс? Приведите примеры.
- •21.6.Что такое продукционная и ретенционная гиперазотемия?
- •21.7.Какие факторы могут вызывать нарушение образования мочевины в печени?
- •21.8.В чем сущность и чем проявляются наследственно обусловленные нарушения обмена фенилаланина?
- •21.9.В чем сущность и чем проявляются наследственно обусловленные нарушения обмена тирозина?
- •21.10.Что такое подагра?
- •21.11.Что является факторами риска подагры?
- •22.4.Назовите основные функциональные эффекты альдостерона.
- •22.5.Что такое ренин-ангиотензинная система? Как она активируется? Назовите основные функциональные эффекты ангиотензина II и ангиотензина III.
- •22.6.Какие факторы стимулируют образование и секрецию предсердного натрийурического гормона (атриопептина) ? Каким действием обладает этот гормон?
- •22.7.Что стимулирует секрецию вазопрессина (антидиуретического гормона)? Каким действием обладает этот гормон?
- •22.8.Какие функциональные эффекты симпатоадренало&ой системы Обусловливают ее участие в защитно-компенсаторных реакциях организма при обезвоживании?
- •22.9.Что такое внеклеточное обезвоживание? Назовите основные его причины.
- •22.10.Что такое изоосмолярное, гипоосмолярное и гиперос-молярное обезвоживание? Приведите примеры.
- •22.11.Какие защитно-компенсаторные реакции развиваются при внеклеточном обезвоживании?
- •22.12.Что такое синдром ангидремии? Какие патогенетические механизмы играют ведущую роль в его развитии?
- •22.13.Что является причиной внутриклеточного обезвоживания? Какие изменения в клетках возникают при этом?
- •22.14.Какими нарушениями на уровне организма проявляется внутриклеточное обезвоживание?
- •22.15.Что такое внеклеточная гипергидрия? Назовите основные ее причины.
- •22.16.Что такое изоосмолярная, гипоосмолярная и гиперосмолярная гипергидрия? Приведите примеры.
- •22.17.Какие защитно-компенсаторные реакции развиваются при внеклеточной гипергидрии?
- •22.18.Что такое отеки? Как они классифицируются?
- •22.19.В развитии каких отеков ведущая роль принадлежит увеличению гидростатического давления крови в капиллярах?
- •22.20.В развитии каких отеков ведущая роль принадлежит уменьшению онкотического давления крови?
- •22.21.Какие отеки относятся к мембраногенным?
- •22.22.Что такое лимфогенные отеки?
- •22.23.Что такое микседематозные («слизистые») отеки?
- •22.24.Как происходит накопление воды в тканях при развитии отеков?
- •22.25.Как влияет ацидоз на разбитие отеков?
- •22.26.В чем сущность опытов Стерлинга, посвященных изучению патогенеза отеков?
- •22.27.Что является причиной развития внутриклеточной гипергидрии? Чем она проявляется на уровне организма?
- •22.28.Какие механизмы могут лежать в основе развития отека клетки при ее повреждении? Каково патогенетическое значение данного явления?
- •22.29.Назовите причины гипернатриемии, защитно-компенсаторные реакции, возникающие при этом, и патогенетическое значение указанного нарушения.
- •22.30.Назовите причины гипонатриемии, защитно-компенсаторные реакции, возникающие при этом, и патогенетическое значение указанного нарушения.
- •22.31.Назовите причины гиперкалиемии, защитно-компенсаторные реакции, возникающие при этом, патогенетическое значение указанного нарушения.
- •22.32.Назовите причины гипокалиемии, защитно-компенсаторные реакции, возникающие при этом, патогенетическое значение указанного нарушения.
- •22.33.Назовите причины и основные проявления нарушений обмена магния в организме.
- •23.Нарушения фосфорно-кальциевого обмена
- •23.1.Какие гормоны осуществляют регуляцию фосфорно-кальциевого обмена в организме?
- •23.2.Как происходит образование гормонально активной формы витамина d?
- •23.3.Назовите основные биологические эффекты гормонально активной формы витамина d.
- •23.4.Какие факторы стимулируют и тормозят секрецию паратирина околощитовидными железами?
- •23.5.Назовите основные биологические эффекты паратирина.
- •23.6.Какие факторы стимулируют и тормозят секрецию кальцитонина?
- •23.7.Назовите основные биологические эффекты кальцитонина.
- •23.8.Что может быть причиной развития гипокальциемических состояний?
- •23.9.Коме защитно-компенсаторные реакции и собственно патологические изменения Возникают при гипокальциемии?
- •23.10.Что такое тетания? Когда она возникает?
- •23.11.Что такое рахит? Какие выделяют патогенетические варианты этого заболевания?
- •23.12.Какие факторы могут быть причиной развития кальципенического рахита?
- •23.13.Объясните механизмы развития основных проявлений кальципенического рахита.
- •23.14.Какие факторы могут быть причиной развития фосфопенического рахита?
- •23.15.Объясните патогенез основных проявлений фосфопе-нического рахита,
- •23.16.Что может быть причиной развития гиперкальциемических состояний?
- •23.17.Какие защитно-компенсаторные реакции и собственно патологические изменения возникают при гиперкальциемии?
- •23.18.Как происходит кальцификация мягких тканей?
- •23.19.Какие причины могут вызывать развитие гипо- и гиперфосфатемии? Какие изменения в организме возникают при этом?
- •24.Нарушения кислотно-основного состояния
- •24.1.Какие буферные системы принимают участие в под-держании кислотно-основного состояния?
- •24.2.Как участвует система внешнего дыхания в регуляции кислотно-основного состояния?
- •24.3.Какие процессы в почках принимают участие в регуляции кислотно-основного состояния?
- •24.4.В чем состоит основное функциональное различие процессов ацидогенеза, происходящих в проксимальных и дистальных извитых канальцах почечных нефронов?
- •24.5.Назовите основные формы нарушений кислотно-основного состояния.
- •24.17.Какие защитно-компенсаторные реакции развиваются при негазовом ацидозе?
- •24.18.Какие изменения показателей кислотно-основного состояния характерны для негазового ацидоза?
- •24.19.Что такое негазовые ацидозы с увеличенной и нормальной анионной разностью?
- •24.25.Как осуществляют коррекцию газового алкалоза?
- •24.26.Какие изменения показателей кислотно-основного состояния характерны для газового алкалоза?
- •24.33.Какие защитно-компенсаторные реакции развиваются при негазовом алкалозе?
- •24.34.Как осуществляют коррекцию негазового алкалоза?
- •24.35.Какие собственно патологические изменения возникают в организме при декомпенсированных алкалозах?
- •24.36.Какая связь существует между нарушениями кислотно-основного состояния и нарушениями электролитного обмена?
- •Часть II. Патологическая физиология органов и систем
- •25.Патологическая физиология системы крови
- •25.1.Какие функциональные, морфологические и регуляторные особенности системы крови влияют на характер, этиологию и патогенез патологических процессов в этой системе?
- •25.2.Какие признаки свидетельствуют о нарушениях в системе крови?
- •25.3.Назовите виды нарушений общего объема крови. Приведите примеры.
- •25.4.Чем обусловлено патогенетическое значение гипо- и гиперволемии?
- •25.5.Что такое кровопотеря? Какие причины вызывают кро-вопотерю? От чего зависят ее течение и исход?
- •25.6.Какие стадии условно выделяют в патогенезе острой кровопотери?
- •25.7.Что является главным звеном патогенеза острой кровопотери?
- •25.8.Какие защитно-компенсаторные реакции закономерно развиваются при кровопотере?
- •25.9.Какие компенсаторные реакции организма направлены на уменьшение объема сосудистого русла в начальной стадии кровопотери?
- •25.10.В чем сущность компенсаторных реакций организма, направленных на увеличение объема циркулирующей крови при кровопотере?
- •25.11.Какие компенсаторные реакции обеспечивают восстановление состава периферической крови при кровопотере?
- •25.12.Какие собственно патологические изменения могут развиваться при кровопотере?
- •25.13.Что такое геморрагический шок? Назовите главные патогенетические звенья его развития.
- •25.14.Нарушения системы эритроцитов
- •25.14.1.Какими количественными изменениями могут проявляться патологические процессы, затрагивающие красный росток крови? Чем они могут быть обусловлены?
- •25.14.2.Какие качественные изменения эритроцитов характерны для нарушений красного ростка крови? Чем они могут быть обусловлены?
- •25.14.3.Какие клетки относятся к регенераторным формам эритроцитов? Дайте их краткую характеристику.
- •25.14.4.Какие дегенеративные изменения могут быть характерны для эритроцитов в условиях патологических процессов, затрагивающих красный росток крови?
- •25.14.5.Какие клетки эритроцитарного ряда относят к клеткам патологической регенерации?
- •25.14.6.Что такое эритроцитоз? Каковы причины и механизмы его развития?
- •25.14.7.Дайте определение понятия «анемия».
- •25.14.8.Какими гематологическими и общими клиническими признаками может проявляться анемия?
- •25.14.9.Как классифицируют анемии? Приведите примеры каждого вида анемий.
- •25.14.10.Какие признаки свидетельствуют о регенераторном характере анемии?
- •25.14.11.Что такое постгеморрагические анемии? Как их классифицируют?
- •25.14.12.Опишите картину крови при острой постгеморрагической анемии.
- •25.14.13.Определите место острой постгеморрагической анемии в разных классификациях анемий.
- •25.14.14.Опишите картину крови при хронической постгеморрагической анемии.
- •25.14.15.Определите место хронической постгеморрагической анемии в разных классификациях анемий.
- •25.14.16.Что такое гемолитические анемии? Как их классифицируют?
- •25.14.17.Какие эндоэритроцитарные факторы могут быть причиной развития гемолитической анемии?
- •25.14.18.Как определить, какие факторы (эндо- или экзоэритроцитарные) являются причиной гемолиза эритроцитов?
- •25.14.19.Назовите возможные причины и основные механизмы внутрисосудистого гемолиза эритроцитов.
- •25.14.20.Какие факторы могут обусловливать окислительный гемолиз эритроцитов?
- •25.14.21.Какие нарушения развиваются в организме в результате внутрисосудистого гемолиза эритроцитов?
- •25.14.22.Что такое внутриклеточный гемолиз эритроцитов? Чем он может быть обусловлен?
- •25.14.23.Какие нарушения развиваются в организме в результате внутриклеточного гемолиза эритроцитов?
- •25.14.24.Какие причины могут обусловливать развитие приобретенной гемолитической анемии?
- •25.14.25.Приведите примеры анемий, обусловленных механическим повреждением эритроцитов.
- •25.14.26.Что такое иммунные гемолитические анемии? Назовите возможные причины.
- •25.14.27.Что такое гемолитическая болезнь новорожденных?
- •25.14.28.Какие типы антител могут вызывать гемолиз эритроцитов? Каков его механизм?
- •25.14.29.Что может быть причиной развития токсической гемолитической анемии?
- •25.14.30.Назовите возможные причины развития инфекционных гемолитических анемий.
- •25.14.31.Что такое приобретенные мембранопатии? Приведите примеры.
- •25.14.32.Какие изменения характерны для картины периферической крови и красного костного мозга при приобретенной гемолитической анемии?
- •25.14.33.Что может лежать в основе развития наследственных гемолитических анемий?
- •25.14.34.Чем может быть обусловлено развитие наследственных мембранопатий?
- •25.14.35.Дайте характеристику микросфероцитарной анемии Минковского–Шоффара.
- •25.14.36.Чем может быть обусловлено развитие наследственных ферментопатий?
- •25.14.37.Дайте характеристику глюкозо-6-фосфатдегидро-геназодефицитной анемии.
- •25.14.38.Чем может быть обусловлено развитие наследственных гемоглобинопатий?
- •25.14.39.Дайте характеристику серповидноклеточной анемии.
- •25.14.40.В чем сущность талассемий?
- •25.14.41.Какими клиническими синдромами могут проявляться гвмолитические анемии?
- •25.14.42.Как классифицируют группу анемий, связанных с нарушениями эритропоэза?
- •25.14.43.Что такое гипопластическая анемия? Каковы ее этиология и патогенез?
- •25.14.44.Дайте характеристику картины периферической крови и красного костного мозга при гипопластической анемии.
- •25.14.45.Определите место гипопластической анемии в разных классификациях анемий.
- •25.14.46.Какими синдромами проявляется гипопластическая анемия?
- •25.14.47.Что такое мегалобластические анемии? Приведите примеры.
- •25.14.48.Какова роль витамина в12 и фолиевой кислоты в обеспечении кроветворения?
- •25.14.49.Назовите основные причины недостаточности витамина в и в организме.
- •25.14.50.Каков патогенез нарушений, развивающихся в организме при дефиците витамина в12?
- •25.14.51.Дайте характеристику картины периферической крови и красного костного мозга при в12-фолиеводефицитной анемии.
- •25.14.52.Определите место в12-фолиеводефицитной анемии в разных классификациях анемий.
- •25.14.53.Какими синдромами проявляется в12-фолиеводефицитная анемия?
- •25.14.54.В чем состоит физиологическое значение железа?
- •25.14.55.Как происходит обмен железа в организме?
- •25.14.56.Назовите возможные причины развития железодефицитной анемии.
- •25.14.57.Каков патогенез нарушений, развивающихся в организме в связи с дефицитом железа?
- •25.14.58.Дайте характеристику картины периферической крови и красного костного мозга при железодефицитной анемии.
- •25.14.59.Определите место железодефицитной анемии в разных классификациях анемий.
- •25.14.60.Какими синдромами проявляется железодефицитная анемия?
- •25.14.61.Что такое железо-рефракторная анемия? Каковы ее этиология и патогенез?
- •25.15.Нарушения системы лейкоцитов
- •25.15.1.Дайте общую характеристику нарушений системы лейкоцитов.
- •25.15.2.Как распределяются лейкоциты в организме?
- •II. Периферическая кровь:
- •III. Периферические ткани:
- •25.15.3.Какие показатели используют для характеристики состояния системы лейкоцитов?
- •25.15.4.Какими количественными и качественными изменениями лейкоцитов могут проявляться патологические процессы в организме?
- •25.15.5.Что такое лейкоцитоз? Как классифицируют лейкоцитозы?
- •25.15.6.Приведите примеры физиологического и патологического лейкоцитозов.
- •25.15.7.Что такое реактивный лейкоцитоз? Какие механизмы могут лежать в основе его развития?
- •25.15.8.Какие особенности характерны для перераспределительного лейкоцитоза?
- •25.15.9.Приведите примеры нейтрофильного, эозинофильного, базофильного, лимфоцитарного и моноцитарного лейкоцитозов.
- •25.15.10.Что такое лейкопения? Как классифицируют лейкопении?
- •25.15.11.Какие механизмы могут лежать в основе развития лейкопений, связанных с нарушениями поступления лейкоцитов из красного костного мозга в кровь?
- •25.15.12.Какие механизмы могут лежать в основе развития лейкопений, связанных с сокращением времени пребывания лейкоцитов в периферической крови?
- •25.15.13.Что такое агранулоцитоз?
- •25.15.14.Что такое сдвиг лейкоцитарной формулы?
- •25.15.15.Какие выделяют разновидности сдвига лейкоцитарной формулы влево?
- •25.15.16.Какие дегенеративные изменения могут быть характерны для лейкоцитов в условиях патологии?
- •25.15.17.Что такое гемобластозы и лейкозы?
- •25.15.18.Какие существуют доказательства того, что лейкозы – это заболевания опухолевой природы?
- •25.15.19.Чем лейкозы отличаются от других злокачественных опухолей?
- •25.15.20.На какие классы подразделяют кроветворные клетки?
- •25.15.21.Какие кроветворные клетки могут быть источником лейкоза?
- •25.15.22.Как классифицируют лейкозы?
- •25.15.23.В чем состоит основное патогенетическое различие между острыми и хроническими лейкозами?
- •25.15.24.Какие существуют виды острых лейкозов?
- •25.15.25.Дайте характеристику картины крови при острых лейкозах.
- •25.15.26.Какие существуют виды хронических лейкозов?
- •25.15.27.Дайте характеристику картины крови при хронических лейкозах.
- •25.15.28.Дайте сравнительную характеристику картины крови при остром миелобластном и хроническом миелоцитарном лейкозах.
- •25.15.29.Может ли острый лейкоз с течением времени перейти в хронический, и наоборот, хронический – в острый?
- •25.15.30.Какие факторы могут быть причиной развития лейкозов?
- •25.15.31.Какие существуют доказательства того, что ионизирующая радиация может быть причиной лейкозов?
- •25.15.32.Что свидетельствует о роли химических агентов в возникновении лейкозов?
- •25.15.33.Каково значение вирусов в возникновении лейкозов?
- •25.15.34.Какие факты свидетельствуют о роли наследственного фактора в развитии лейкозов?
- •25.15.35.Как влияют на возникновение лейкозов иммунные факторы?
- •25.15.36.Опишите патогенез лейкозов. Какие стадии развития проходит лейкоз?
- •25.15.37.Какими клиническими синдромами могут проявляться лейкозы?
- •II. Синдромы, связанные с особенностями функционирования лейкозных клеток.
- •III. Синдромы, связанные с метастазированием лейкозных клеток и развитием лейкозных пролифератов в разных органах и тканях.
- •25.16.Нарушения гемостаза
- •25.16.1.Что такое система гемостаза? Какие существуют механизмы остановки кровотечения?
- •25.16.2.Чем обусловлено участие сосудистой стенки в гемостазе?
- •I. Активация механизмов гемостаза:
- •25.16.3.Какова роль тромбоцитов в гемостазе?
- •25.16.4.Как осуществляется сосудисто-тромбоцитарный гемостаз?
- •25.16.5.Как происходит адгезия тромбоцитов?
- •25.16.6.Какие факторы вызывают агрегацию тромбоцитов? Какова динамика этого процесса?
- •25.16.7.Какие механизмы лежат в основе агрегации тромбоцитов?
- •II. Собственно склеивание тромбоцитов.
- •25.16.8.Что представляет собой процесс свертывания крови? Какие факторы принимают в нем участие?
- •25.16.9.Из каких фаз состоит процесс свертывания крови?
- •25.16.10.Как классифицируют нарушения гемостаза?
- •25.16.11.Что такое геморрагические диатезы? Как их классифицируют?
- •25.16.12.Что такое вазопатии? Как они классифицируются?
- •25.16.13.Каковы этиология и патогенез воспалительных вазопатии?
- •25.16.14.Каковы этиология и патогенез диспластических вазопатии?
- •25.16.15.Что такое тромбоцитопения? Какие механизмы могут лежать в основе развития тромбоцитопении?
- •I. Тромбоцитопении, связанные с нарушениями образования тромбоцитов:
- •25.16.16.Каков патогенез нарушений гемостаза в условиях тромбоцитопении ?
- •25.16.17.Что такое тромбоцитопатии? Как их классифицируют?
- •25.16.18.Приведите примеры тромбоцитопатии с разными механизмами нарушений гемостаза.
- •1. Тромбоцитопатии с первичным нарушением адгезии тромбоцитов:
- •3. Тромбоцитопатии с первичным нарушением реакций освобождения содержимого тромбоцитов:
- •25.16.19.Чем могут быть обусловлены нарушения коагуляционного гемостаза – коагулопатии? Коагулопатии могут быть вызваны:
- •25.16.20.Что может быть причиной непосредственного нарушения I фазы свертывания крови?
- •25.16.21.Что может быть причиной непосредственного нарушения II фазы свертывания крови?
- •25.16.22.Что может быть причиной непосредственного нарушения III фазы свертывания крови?
- •1. Дефицит фибриногена:
- •25.16.23.Какие антикоагулянты составляют противосвертывающую систему крови?
- •25.16.24.Чем может быть обусловлено повышение активности противосвертывающей системы крови?
- •25.16.25.Что входит в понятие «фибринолитическая система»?
- •25.16.26.Какие факторы вызывают повышение активности фибринолитической системы крови?
- •25.16.27.Какими клиническими признаками проявляются нарушения коалиционного гемостаза?
- •25.16.28.Что такое двс-синдром?
- •25.16.29.Что может быть причиной двс-синдрома?
- •25.16.30.Что является патогенетической основой развития двс-синдрома?
- •25.16.31.Назовите основные источники поступления в кровь активных протеаз при двс-синдроме.
- •25.16.32.Как разворачивается патогенез двс-синдрома?
- •25.16.33.Что такое тромбофильные диатезы? Что может лежать в основе их развития?
- •26.Патологическая физиология сердца
- •26.1.Что такое недостаточность кровообращения? Чем она может быть обусловлена?
- •26.2.Какие последствия для органов, тканей и организма в целом имеет недостаточность кровообращения?
- •26.3.Что такое недостаточность сердца?
- •26.4.Как классифицируют недостаточность сердца?
- •IV. По патогенезу выделяют:
- •26.5.Дайте краткую характеристику разных патогенетических вариантов недостаточности сердца.
- •26.6.Какие типы перегрузок сердца могут быть причиной развития его недостаточности?
- •26.7.Какие механизмы могут обеспечивать компенсацию сердца при действии на него увеличенных нагрузок?
- •26.8.В чем сущность гетерометрического механизма компенсации сердца?
- •26.9.В чем сущность гомеометрического механизма компенсации сердца?
- •26.10.Чем характеризуется хроноинотропный механизм компенсации сердца?
- •26.11.Какова роль катехоламинов в осуществлении механизмов срочной компенсации сердца?
- •26.12.Назовите варианты долговременной адаптации сердца к действию нагрузок.
- •26.13.Какие механизмы лежат в основе развития гипертрофии сердца?
- •26.14.Какие стадии выделяют в процессе развития компенсаторной гипертрофии сердца? Дайте их характеристику.
- •26.15.Какие особенности гипертрофированного сердца являются предпосылкой развития его декомпенсации?
- •26.16.Чем может быть обусловлено развитие миокардиальной недостаточности сердца?
- •26.17.Что такое аритмии сердца? Как их классифицируют?
- •26.18.Какие аритмии сердца могут возникать в результате нарушения функции автоматизма?
- •26.19.Каковы причины и механизмы развития синусной тахи- и брадикардии?
- •26.20.Какие аритмии возникают в результате нарушения возбудимости миокарда? Каков механизм их развития?
- •26.21.Что такое экстрасистолия? Назовите виды экстрасистол и их основные электрокардиографические характеристики.
- •26.22.Что такое пароксизмальная тахикардия? Чем она характеризуется?
- •26.23.Какие аритмии возникают в результате нарушений функции проводимости миокарда?
- •26.24.Дайте характеристику предсердно-желудочковой блокады сердца.
- •26.25.Чем может быть обусловлено развитие синдрома Вольфа – Паркинсона – Уайта?
- •26.26.Какие аритмии возникают в результате одновременного нарушения функций возбудимости и проводимости?
- •26.27.Какими признаками проявляется мерцательная аритмия? Каковы механизмы ее развития?
- •26.28.Что такое фибрилляция желудочков? Когда она возникает и чем проявляется?
- •26.29.Чем может быть обусловлено развитие нарушений сократительной функции миокарда?
- •26.30.Какие факторы влияют на характер потенциала действия и возбудимость кардиомиоцитов? Какими электрофизиологическими признаками проявляются изменения их возбудимости?
- •26.31.Как влияют изменения Внеклеточной концентрации ионов калия на возбудимость миокарда?
- •26.32.Что такое кардиоплегия? Какие используют подходы к ее осуществлению?
- •26.33.Какие факторы влияют на характер электромеханического сопряжения в миокарде?
- •26.34.Как изменения продолжительности и частоты потенциалов действия влияют на силу сердечных сокращений?
- •26.35.Как влияет внеклеточная концентрация ионов кальция на силу сокращений сердца? Что такое «кальциевый парадокс»?
- •26.36.Какие факторы влияют на состояние систем удаления ионов кальция из кардиомиоцитов и, как следствие, на силу сердечных сокращений? к таким факторам относятся:
- •26.37.От чего зависит сила сокращений отдельных кардиомиоцитов?
- •26.39.Каково значение атф в обеспечении функций клеток миокарда? Чем могут быть обусловлены нарушения энергетического обмена в сердечной мышце?
- •26.40.Дайте сравнительную характеристику гипо- и гиперкальциевого вариантов недостаточности сердца.
- •26.41.Когда развивается внемиокардиальная недостаточность сердца? Какие компенсаторные механизмы включаются при этом?
- •26.42.Какие изменения показателей кардио- и гемодинамики характерны для недостаточности сердца?
- •26.43.Какими клиническими синдромами и признаками проявляется недостаточность сердца?
- •26.44.Какими особенностями характеризуется венечное кровообращение в сердце?
- •26.45.Как осуществляется регуляция венечного кровообращения?
- •26.46.Что такое недостаточность венечного кровообращения? Чем различаются относительная и абсолютная коронарная недостаточность?
- •26.47.Какие патогенетические факторы могут обусловливать развитие абсолютной недостаточности венечного кровообращения ?
- •26.48.Какие механизмы могут лежать в основе развития ишемии миокарда?
- •26.49.Какие патогенетические факторы влияют на миокард в условиях коронарной недостаточности?
- •26.50.Какие последствия для миокарда может иметь недостаточность венечного кровообращения?
- •26.51.Какие нарушения сократительной функции миокарда могут возникать при его ишемии?
- •26.52.Какие механизмы могут обусловливать развитие аритмий при ишемии миокарда?
- •26.53.Что такое реперфузионный синдром? в чем его сущность? Каковы механизмы его развития?
- •26.54.Что такое ишемическая болезнь сердца? Какие существуют ее клинические формы?
- •26.55.Назовите основные причины развития инфаркта миокарда.
- •26.56.Какова роль катехоламинов в развитии инфаркта миокарда?
- •26.57.Какие клинические синдромы характерны для инфаркта миокарда?
- •26.58.Каковы механизмы развития и значение болевого синдрома при инфаркте миокарда?
- •26.59.Что такое кардиогенный шок? в каких формах он может проявляться?
- •26.60.Каков патогенез кардиогенного шока?
- •26.61.В чем сущность резорбционно-некротического синдрома, развивающегося при инфаркте миокарда?
- •26.62.Что такое некоронарогенные некрозы сердца? Как их моделируют в эксперименте?
- •27.Патологическая физиология кровеносных сосудов
- •27.1.Как классифицируют кровеносные сосуды в зависимости от выполняемых функций? Какие патологические процессы характерны для разных типов сосудов?
- •27.2.Как классифицируют склеротические поражения артериальных сосудов?
- •27.3.Какие процессы составляют патогенетическую сущность артериосклероза?
- •27.4.Что такое атеросклероз?
- •27.5.Что такое артериосклероз Менкеберга?
- •27.6.Дайте сравнительную характеристику атеросклероза и артериосклероза Менкеберга.
- •27.7.Чем проявляются склеротические изменения кровеносных сосудов?
- •27.8.Как в эксперименте моделируют атеросклероз?
- •27.9.Что такое факторы риска атеросклероза? Что к ним относится?
- •27.10.Какие существуют концепции патогенеза атеросклероза?
- •27.11.В чем сущность плазменной теории патогенеза атеросклероза?
- •27.12.Какие функции выполняет в организме холестерин? Чем доказывается его роль в развитии атеросклероза?
- •27.13.Какие изменения липопротеидов плазмы крови способствуют развитию атеросклероза?
- •27.14.Каково значение рецепторного аппарата клеток сосудистой стенки в развитии ее атеросклеротических изменений?
- •27.15.Каково значение повреждения эндотелия сосудистой стенки в развитии ее атеросклеротических изменений?
- •27.16.Какими механизмами может быть обусловлено развитие пролиферативных изменений в сосудистой стенке при атеросклерозе?
- •27.17.Какие механизмы могут лежать в основе развития дегенеративных изменений сосудистой стенки при атеросклерозе?
- •27.18.Чем доказывается роль первичных нарушений энергообеспечения сосудистой стенки в развитии ее дегенеративных и склеротических изменений?
- •27.19.Какие механизмы могут лежать в основе собственно склерозирования сосудистой стенки?
- •27.20.Чем могут проявляться нарушения функции резистивных сосудов (сосудов сопротивления)? Какие изменения регуляторных механизмов могут их обусловливать?
- •27.21.Какие нейрогуморальные системы принимают участие в регуляции артериального давления?
- •27.22.Что такое первичная и вторичная артериальная гипертензия?
- •27.23.Какие выделяют гемодинамические варианты артериальной гипертензии?
- •27.24.Какие существуют экспериментальные модели артериальной гипертензии?
- •I. Нарушение функции центральной нервной системы:
- •II. Нарушение мозгового крово- и лимфообращения:
- •III. Нарушение функции депрессорных регуляторных систем:
- •IV. Нарушение функции почек:
- •V. Нарушение гормонального состояния:
- •VI. Нарушение водно-солевого обмена:
- •27.25.Какова этиология первичной артериальной гипертензии?
- •27.26.Какие существуют концепции патогенеза первичной артериальной гипертензии?
- •27.27.Каков патогенез первичной артериальной гипертензии с точки зрения дисрегуляторной концепции ее развития?
- •27.28.В чем сущность мембранной концепции патогенеза первичной артериальной гипертензии?
- •27.29.Как классифицируют артериальную гипотензию? Какие гемодинамические факторы могут лежать в основе ее развития?
- •27.30.Каковы причины и механизмы развития хронической артериальной гипотензии?
- •27.31.Какие общие и местные расстройства гемодинамики могут быть связаны с первичными нарушениями функции емкостных сосудов?
- •28.Патологическая физиология внешнего дыхания
- •28.1.Что такое недостаточность внешнего дыхания?
- •28.2.Как классифицируют дыхательную недостаточность?
- •28.3.Что такое вентиляционная недостаточность дыхания? Чем она проявляется?
- •28.4.Назовите причины вентиляционной недостаточности дыхания.
- •28.5.Какие выделяют патогенетические Варианты вентиляционной недостаточности дыхания?
- •28.6.Чем могут проявляться нарушения центральной регуляции внешнего дыхания?
- •28.7.Что такое периодическое дыхание? Какие известны его формы?
- •28.8.Что такое терминальное дыхание? Чем оно характеризуется?
- •28.9.Что такое одышка? Каковы механизмы ее развития?
- •28.10.Что такое рестриктивная недостаточность дыхания? Каковы ее причины?
- •28.11.Что такое пневмоторакс? Когда он возникает?
- •28.12.Что такое ателектаз?
- •28.13.Что такое обструктивная недостаточность дыхания? Когда она развивается?
- •28.14.Что такое эмфизема легких? Почему при этом заболевании развивается обструктивная недостаточность дыхания?
- •28.15.Что такое бронхиальная астма? Как нарушаются показатели внешнего дыхания при этом заболевании?
- •28.16.Что такое асфиксия? Каков ее патогенез?
- •28.17.Что такое паренхиматозная недостаточность внешнего дыхания? Каковы ее причины и механизмы развития?
- •28.18.Назовите причины нарушения диффузии газов в легких.
- •28.19.Назовите причины нарушений легочной перфузии.
- •28.20.Каковы причины и механизмы развития гипертензии малого круга кровообращения?
- •28.21.Какие механизмы могут лежать в основе развития отека легких?
- •28.22.Какими нарушениями проявляется синдром эмболии малого круга кровообращения?
- •28.23.Какие изменения общих и регионарных вентиляционно-перфузионных отношений могут приводить к нарушениям внешнего дыхания?
- •28.24.Какими клиническими признаками проявляется недостаточность внешнего дыхания?
- •29.Патологическая физиология пищеварения
- •29.1.Что такое недостаточность пищеварения?
- •29.2.Как классифицируют недостаточность пищеварения?
- •29.3.Какие причины могут лежать в основе развития недостаточности пищеварения?
- •29.4.Какие принципы используют в экспериментальном моделировании недостаточности пищеварения?
- •29.5.С помощью каких воздействий на нервную систему моделируют расстройства пищеварения?
- •29.6.Какие нарушения гуморальной регуляции воспроизводят с целью моделирования расстройств пищеварения?
- •29.7.Какими синдромами может проявляться недостаточность пищеварения?
- •29.8.Какими признаками может проявляться диспептический синдром?
- •29.9.Что такое анорексия? Когда она возникает?
- •29.10.Что такое изжога? Каковы ее механизмы?
- •29.11.Что такое отрыжка? Каковы ее механизмы?
- •29.12.Что такое тошнота и рвота? Каковы их механизмы?
- •29.13.Что такое запоры? Когда они возникают?
- •29.14.Что такое метеоризм? Когда он возникает?
- •29.15.Что такое поносы? Чем они могут быть обусловлены?
- •29.16.Что может быть причиной обезвоживания организма при расстройствах функций пищеварения?
- •29.17.Какими нарушениями кислотно-основного состояния могут проявляться расстройства пищеварения?
- •29.18.Чем обусловливается кишечная аутоинтоксикация при нарушениях функций пищеварения?
- •29.19.Какие механизмы могут лежать в основе болевого синдрома при поражениях пищеварительной системы?
- •29.20.Какие нарушения двигательной, секреторной и всасывательной функций пищеварительной системы могут лежать в основе расстройств пищеварения?
- •29.21.Какие причины могут приводить к нарушениям жевания? Какое значение для пищеварения имеют эти нарушения? Причины нарушений жевания:
- •29.22.Что такое кариес зубов? Каковы причины его развития?
- •29.23.Как представляется сегодня патогенез кариеса?
- •29.24.Что такое пародонтит? Какие факторы могут лежать в основе его развития?
- •29.25.Каковы причины и значение гиперсаливации?
- •29.26.Каковы причины и значения гипосаливации?
- •29.27.Что такое дисфагия? Назовите ее причины, укажите значение.
- •29.28.Что такое желудочные дискинезии? Какие существуют их варианты?
- •29.29.Назовите причины и значение гипертонических дискинезий желудка.
- •29.30.Что такое пилороспазм?
- •29.31.Назовите причины и значение гипотонических дискинезий желудка.
- •29.32.Назовите типы патологической желудочной секреции.
- •29.33.Чем проявляется желудочная гиперсекреция? Каково ее значение?
- •29.34.Как можно моделировать желудочную гиперсекрецию в эксперименте?
- •29.35.Что такое язвенная болезнь? Какова ее этиология?
- •29.36.Какие существуют теории патогенеза язвенной болезни?
- •29.37.Какие в настоящее время выделяют патогенетические варианты язвы желудка?
- •29.38.Что такое экзогенная язва желудка? Что может быть ее причиной?
- •29.39.Что такое пептическая язва желудка? Как ее моделируют в эксперименте?
- •29.40.Что такое трофические язвы желудка? Как их воспроизводят в эксперименте?
- •29.41.Что такое гипорегенераторные язвы желудка? Чем они могут быть обусловлены?
- •29.42.Чем проявляется желудочная гипосекреция? Каково ее значение?
- •29.43.Как в эксперименте можно моделировать желудочную гипосекрецию?
- •29.44.Каковы причины панкреатической гиперсекреции? Чем она может проявляться?
- •29.45.Что такое острый панкреатит? Какова его этиология?
- •29.46.Что является главным звеном патогенеза острого панкреатита? Какие выделяют патогенетические варианты этого заболевания?
- •29.47.Чем характеризуется первично альтеративный вариант развития острого панкреатита?
- •29.48.Что лежит в основе развития гипертензивного варианта острого панкреатита?
- •29.49.Какие условия вызывают рефлюксный вариант развития острого панкреатита?
- •29.50.Опишите патогенез местных изменений в поджелудочной железе при остром панкреатите.
- •29.51.Какие механизмы лежат в основе развития панкреатического шока?
- •29.52.Какими синдромами проявляется панкреатический шок?
- •29.53.Какие причины вызывают развитие панкреатической гипосекреции? Каково ее значение?
- •29.54.Что такое синдром мальдигестии? Чем он проявляется?
- •29.55.Что такое кишечные дискинезии? Какие существуют их варианты?
- •29.56.Назовите причины и значение гиперкинетических дискинезий кишок.
- •29.57.Чем проявляются гипокинетические дискинезии кишок? Каковы причины их развития и значение?
- •29.58.Что такое кишечная непроходимость? Как она классифицируется?
- •I. Механическая:
- •II. Динамическая:
- •29.59.Какими изменениями в организме проявляется непроходимость кишок? Каков их патогенез?
- •29.60.Какие причины могут вызывать нарушения дефекации? Чем могут проявляться эти нарушения?
- •29.61.Что такое синдром мальабсорбции? Чем могут быть обусловлены нарушения всасывания?
- •29.62.Какие преэнтероцитарные нарушения могут лежать в основе расстройств всасывания в кишках?
- •29.63.Что такое интестинальные ферментопатии? Чем они могут проявляться?
- •29.64.Какие энтероцитарные нарушения могут обусловливать синдром мальабсорбции?
- •29.65.Какие постэнтероцитарные расстройства вызывают нарушение всасывания веществ в кишках?
- •30.Патологическая физиология печени
- •30.1.Какие факторы могут быть причиной поражений печени?
- •30.2.Что такое недостаточность печени? Как она классифицируется?
- •30.3.В каких случаях развивается печеночно-клеточная недостаточность печени? Каковы ее причины? Как ее моделируют в эксперименте?
- •30.4.Что является причиной развития холестатической недостаточности печени? Как ее воспроизводят в эксперименте?
- •30.5.В каких случаях развивается печеночно-сосудистая недостаточность печени? Как ее можно моделировать в эксперименте?
- •30.6.Что такое цирроз печени? Какие патогенетические варианты его выделяют?
- •30.7.Нарушениями каких функций печени может проявляться ее недостаточность?
- •30.8.Какие нарушения углеводного обмена могут развиваться при поражениях печени?
- •30.9.Какие расстройства жирового обмена могут возникать при поражениях печени?
- •30.10.Что такое жировая дистрофия печени? Каков ее патогенез?
- •30.11.Какие расстройства белкового обмена могут возникать при поражениях печени?
- •30.12.Что может быть причиной нарушений белоксинтетической функции печени? Чем проявляются такие нарушения?
- •30.13.Какие нарушения обмена витаминов могут развиваться при поражениях печени?
- •30.14.Как нарушается обмен микроэлементов при поражениях печени?
- •30.15.Какие нарушения в обмене гормонов и биологически активных веществ могут сопровождать развитие недостаточности печени?
- •30.16.Чем могут проявляться нарушения защитной (барьерной) функции печени?
- •30.17.Какие факторы могут вызывать развитие расстройств антитоксической функции печени?
- •30.18.В чем сущность синдрома гепатоцеребральной недостаточности? Чем он может проявляться?
- •30.19.Какие вещества, накапливающиеся в крови при недостаточности печени, являются церебротоксическими?
- •30.20.Какие выделяют патогенетические варианты печеночной комы?
- •30.21.Коков патогенез печеночной комы?
- •30.22.Какими синдромами могут проявляться нарушения экскреторной функции печени?
- •30.23.Как в норме происходит обмен желчных пигментов?
- •30.24.Что такое желтуха? Какие существуют ее виды?
- •30.25.Каков механизм развития гемолитической желтухи? Какие изменения пигментного обмена характерны для этого вида желтухи?
- •30.26.Какие существуют разновидности паренхиматозной (печеночной) желтухи? Как изменяется обмен желчных пигментов при каждой из них?
- •30.27.Каковы причины и механизмы развития механической желтухи? Дайте характеристику нарушений пигментного обмена при этом виде желтухи.
- •30.28.Что такое холемический синдром? в каких случаях он возникает? Чем проявляется?
- •30.29.Что такое ахолический синдром? Чем он проявляется?
- •30.30.Что такое дисхолия? Каковы ее причины? Каков механизм возникновения желчных камней?
- •30.31.Каковы причины, механизмы развития и значение дис-кинезий желчного пузыря и желчных протоков?
- •30.32.Какие функции печени относятся к гемодинамиче-ским? Чем проявляются расстройства этих функций?
- •30.33.Что такое синдром портальной гипертензии? Каковы его причины? Чем он проявляется?
- •30.34.Каковы механизмы развития асцита?
- •30.35.Какие нарушения в системе крови могут развиваться при поражениях печени?
- •31.Патологическая физиология почек
- •31.1.Какими нарушениями гомеостаза могут проявляться поражения почек?
- •31.2.Какие процессы в почках могут нарушаться в условиях поражения этих органов?
- •31.3.Что такое недостаточность почек? Как ее классифицируют?
- •31.4.Каковы причины острой почечной недостаточности? Какие стадии выделяют в ее развитии?
- •31.5.Каковы этиология и патогенез хронической недостаточности почек?
- •31.6.Что такое скорость клубочковой фильтрации? Как ее определяют? Как она меняется при разных видах почечной недостаточности?
- •31.7.Какие механизмы могут лежать в основе нарушений почечных функций?
- •31.8.В чем сущность преренальных нарушений функций почек?
- •31.9.Чем могут быть обусловлены нарушения клубочковой фильтрации?
- •31.10.Какие факторы могут вызывать увеличение клубочковой фильтрации?
- •31.11.Какие механизмы могут лежать в основе нарушений функций почечных канальцев?
- •31.12.В чем сущность постренальных нарушений почечных функций?
- •31.13.Какие количественные и качественные изменения мочи могут наступать при поражениях почек?
- •31.14.Что такое олиго- и анурия? в каких случаях возникает олигурия? Каковы ее последствия?
- •31.15.Что такое полиурия? Каковы механизмы ее развития?
- •31.16.Что такое никтурия? Когда она возникает?
- •31.17.Что такое гипо- и изостенурия?
- •31.18.Каковы механизмы протеинурии?
- •31.19.Что может быть причиной гематурии, лейкоцитурии, цилиндрурии?
- •31.20.Что такое уремический синдром и уремическая кома? Каков их патогенез?
- •31.21.Какие механизмы могут обусловливать развитие отеков при поражениях почек?
- •31.22.Какие нарушения кислотно-основного состояния могут развиваться при поражениях почек?
- •31.23.Какие нарушения фосфорно-кальциевого обмена характерны для недостаточности почек? Каков их патогенез?
- •I. Дистрофические изменения в костях:
- •31.24.Какие виды артериальной гипертензии могут возникать при поражениях почек? Каков их патогенез?
- •31.25.Какие механизмы обусловливают развитие анемии, сопровождающей недостаточность почек?
- •I. Угнетение эритропоэза:
- •31.26.Какими нарушениями гемостаза может проявляться недостаточность почек?
- •31.27.Что такое нефротический синдром? Каков его патогенез?
- •31.28.Какие последствия для организма может иметь потеря белков с мочой при нефротическом синдроме?
- •31.29.Что такое гломерулонефрит? Каковы этиология и патогенез острого гломерулонефрита?
- •31.30.Какие существуют экспериментальные модели острого гломерулонефрита?
- •31.31.Чем может проявляться хронический гломерулонефрит? Каковы его этиология и патогенез?
- •31.32.Что такое пиелонефрит? Что может быть его причиной?
- •31.33.Что такое почечно-каменная болезнь? Каковы ее этиология и патогенез?
- •32.Патологическая физиология эндокринной системы
- •32.1.Что такое истинные гормоны? Какими свойствами они обладают?
- •32.2.Как классифицируют гормоны?
- •32.3.Что такое эндокринная функция? Назовите ее составные части,
- •32.4.Какие существуют типы нарушений эндокринных функций?
- •32.5.Какие выделяют патогенетические варианты нарушений эндокринных функций?
- •32.6.Как осуществляется регуляция эндокринных функций? Чем могут быть обусловлены их дисрегуляторные нарушения?
- •32.7.Какие принципы лежат в основе регуляции эндокринных функций? Как они могут нарушаться?
- •32.8.Чем могут быть обусловлены собственно железистые нарушения эндокринных функций?
- •32.9.Назовите основные причины нарушений биосинтеза гормонов.
- •32.10.Как осуществляется секреция гормонов? Что может обусловливать ее нарушения?
- •32.11.Какие нарушения могут лежать в основе развития периферических расстройств эндокринных функций?
- •32.12.Как осуществляется транспорт гормонов в организме? Какие нарушения эндокринных функций могут быть связаны с его расстройствами?
- •32.13.Как осуществляется метаболическая инактивация гормонов? Какие нарушения эндокринной функции могут быть связаны с расстройствами метаболизма гормонов?
- •32.14.Какие возможны нарушения взаимодействия гормонов с периферическими клетками?
- •32.15.Как происходит реализация биологического действия гормонов на клетки?
- •32.16.Каково значение гипоталамуса в регуляции эндокринных функций?
- •32.17.Какие причины могут вызывать нарушение функции гипоталамо-аденогипофизарной системы?
- •32.18.Какие механизмы могут лежать в основе гипер- и гипофункции гипоталамо-аденогипофизарной системы?
- •32.19.Какие гормоны образуются в аденогипофизе? Какими биологическими эффектами они обладают?
- •32.20.Что такое гипопитуитризм? Какие существуют его формы? Чем он проявляется?
- •32.21.Что такое гиперпитуитризм? Чем он может быть обусловлен?
- •32.22.Какие гормоны выделяются при активации гипоталамо-нейрогипофизарной системы? Какими биологическими эффектами они обладают?
- •32.23.Какие патологические состояния возникают при нарушении функции гипоталамо-нейрогипофизарной системы?
- •32.24.Какие гормоны образуются в коре надпочечников?
- •32.25.Как регулируется образование и секреция минералокортикоидов? Назовите основные биологические эффекты альдостерона.
- •3. Непосредственная стимуляция секреции альдостерона высокими концентрациями ионов калия плазмы крови.
- •32.26.Как осуществляется регуляция образования и секреции глюкокортикоидов? Какими биологическими эффектами обладают эти гормоны?
- •32.27.Назовите основные низкодозовые эффекты глюкокортикоидов.
- •32.28.Назовите основные высокодозовые эффекты глюкокортикоидов.
- •32.29.Что такое липокортиновые эффекты глюкокортикоидов? Что к ним относится?
- •32.30.Какие существуют нарушения функции коры надпочечников?
- •32.31.Как классифицируют недостаточность коры надпочечников? Что может быть причиной гипофункции этой эндокринной железы?
- •32.32.Назовите основные проявления недостаточности коры надпочечников.
- •32.33.Что такое гиперальдостеронизм? Какие существуют его виды? Чем он проявляется?
- •32.34.Какие существуют клинические формы гиперфункции пучковой зоны коры надпочечников? Чем они проявляются?
- •32.35.Что такое адреногенитальный синдром? Какие существуют его варианты?
- •32.36.Какие гормоны образуются мозговым веществом надпочечников? Каковы механизмы их действия и биологические эффекты?
- •32.37.Какие существуют нарушения функции мозгового вещества надпочечников?
- •32.38.Что такое стресс и общий адаптационный синдром?
- •32.39.Какие морфологические признаки характерны для общего адаптационного синдрома?
- •32.40.Какие стадии выделяют в развитии стресса?
- •32.41.Какие механизмы участвуют в реализации стресса?
- •32.42.Каково участие разных гормонов в развитии стресса?
- •32.43.Что такое «болезни адаптации"? Какие заболевания к ним относятся?
- •32.44.Назовите гормоны щитовидной железы. Как осуществляется регуляция их образования и секреции?
- •32.45.Каковы механизмы действия и биологические эффекты тиреоидных гормонов?
- •32.46.Назовите основные причины гипотиреоза.
- •32.47.Каков патогенез основных проявлений гипотиреоза?
- •32.48.Назовите основные причины гипертиреоза.
- •32.49.Каков патогенез основных проявлений гипертиреоза?
- •32.50.Что такое зоб? Какие его виды выделяют? Каков патогенез эндемического зоба?
- •32.51.Как осуществляется регуляция образования и секреции паратирина паращитовидными железами? Назовите основные биологические эффекты паратгормона.
- •32.52.Каковы причины и чем проявляется гипопаратиреоз?
- •32.53.Каковы причины и чем проявляется гиперпаратиреоз?
- •32.54.Как осуществляется регуляция образования и секреции мужских половых гормонов? Назовите их основные биологические эффекты.
- •32.55.Каковы причины и основные проявления мужского гипогонадизма?
- •32.56.Каковы причины и основные проявления мужского гипергонадизма?
- •32.57.Какие гормоны относятся к женским половым? Как осуществляется регуляция их образования и секреции?
- •32.58.Назовите основные биологические эффекты женских половых гормонов.
- •32.59.Каковы причины и основные проявления женского гипогонадизма?
- •32.60.Каковы причины и основные проявления женского ги-п ергонадизма ?
- •32.61.В чем сущность эндокринной функции эпифиза? Чем могут проявляться ее нарушения?
- •32.62.Какими изменениями в организме могут проявляться нарушения эндокринной функции поджелудочной железы?
- •Класифікація порушення діяльності нервової системи.
- •I. За анатомічним принципом виділяють:
- •33.Порушення сенсорних ф-цій (чутливості).
- •Виділяють наступні види порушень соматовісцеральної чутливості.
- •Механізми, які лежать в основі порушень соматовісцеральної чутливості.
- •Виникнення синдрому Броун-Секара. Чим він проявляється?
- •Как можно моделировать нарушения двигательных функций нервной системы в эксперименте?
- •Основні синдроми, які характеризують розлади рухової ф-ції нервової системи.
- •Причини і механізми розвитку нарушений нервно-мышечной передачи. Что такое миастения?
- •Что такое периферические параличи и парезы? Чем они характеризуются?
- •Что такое центральные параличи? Чем они характеризуются?
- •Что такое синдром паркинсонизма? Чем он обусловлен и чем проявляется?
- •Гіперкінетичні порушення рухових ф-цій цнс.
- •Основні прояви рухових мозочкових порушень.
- •Судоми. Їх види.
- •Методи експериментального моделювання порушень в вегетативній нервовій системі.
- •Какие конституциональные типы людей выделяют в зависимости от функциональной активности вегетативной нервной системы? Чем они характеризуются?
- •Что такое синдром вегетососудистой дистонии? Какие его варианты существуют?
- •Что такое нервная трофика? Какие существуют механизмы трофических влияний нервов на периферические ткани?
- •Что такое нейродистрофический процесс? Чем он проявляется ?
- •Механизмы лежат в основе развития нейроди-строфического процесса?
- •Какие механизмы могут лежать в основе нарушений интегративных функций центральной нервной системы?
- •Причини і механізми порушення електрофізіологічних процесів в цнс. Порушення електрофізіологічних процесів в нейронах цнс проявляються:
- •Розлади нейрохімічних процесів в цнс, які можуть порушувати її діяльність.
- •Механізми розвитку патологічного збуження та гальмування в нервових центрах.
- •Функціональні зміни в цнс, які зв’язані з патологічним збудженням і гальмуванням.
- •Особливостіенергетичного обміну в головному мозку.
- •Что может быть причиной повреждения нейронов головного мозга?
- •Основные особенности мозгового кровообращения.
- •Какие выделяют формы нарушений мозгового кровообращения? Что такое инсульты?
- •Причини і механізми розвитку набряку мозку.
- •Что может быть причиной внутричерепной гипертензии?
- •Значення пошкодження нейрогліїв для розвитку порушень діяльності цнс.
- •Порушення інтегративної ф-ції цнс.
- •34.Список рекомендованной литературы
- •Часть I. Общая патология
- •Часть II. Патологическая физиология органов и систем
- •Что такое боль? Чем она отличается от других видов чувствительности ?
- •Как классифицируют боль?
- •Что такое соматическая поверхностная боль? На какие виды ее подразделяют?
- •Что такое соматическая глубокая боль? Приведите примеры.
- •Какие виды боли выделяют в зависимости от ее локализации?
- •Назовите возможные причины боли.
- •Что такое хроническая боль? Назовите ее клинические формы.
- •Какие механизмы могут лежать в основе формирования хронической боли?
- •Якими загальними р-ціями организма сопровождается боль?
- •34.1.Что такое антиноцицептивные механизмы? Чем они представлены ?
- •Основні принципи та методи обезболювання.
21.6.Что такое продукционная и ретенционная гиперазотемия?
Гиперазотемия – это увеличение остаточного (небелкового) азота в крови. В норме остаточный азот на 50 % состоит из азота мочевины, около 25 % его приходится на долю аминокислот, остальная часть – на другие азотистые продукты.
Продукционная (печеночная) гиперазотемия возникает вследствие нарушения образования мочевины, детоксикации азотистых продуктов в печени.
Ретенционная (почечная) гиперазотемия является следствием нарушения выделительной функции почек.
21.7.Какие факторы могут вызывать нарушение образования мочевины в печени?
Нарушение синтеза мочевины может наблюдаться на заключительных этапах развития недостаточности печени как один из признаков нарушения ее детоксикационной функции.
Возможны наследственно обусловленные нарушения мочевинообразования. Их причиной может быть недостаточный синтез целого ряда ферментов: аргининсукцинатлиазы (аргининсукцинатурия), карбамоилфосфатсинтетазы и орнитинкарбамоилтрансферазы (аммониемия) и аргининсукцинатсинтетазы (цитруллинурия).
Следствием нарушения синтеза мочевины является накопление аммиака в крови и целый ряд тяжелых клинических проявлений, связанных с этим (подробно см. разд. 31).
21.8.В чем сущность и чем проявляются наследственно обусловленные нарушения обмена фенилаланина?
Наследственно обусловленным нарушением обмена фенилаланина является фенилкетонурия – заболевание с аутосомно-рецессивным типом наследования. Его причиной является генетический дефект фермента фенилаланингидроксилазы, в норме преобразующего фенилаланин в тирозин (рис. 73). При отсутствии указанного фермента окисление фенилаланина идет по пути образования фенилпировино-градной и фенилмолочной кислот. Однако этот путь обладает малой пропускной способностью, и поэтому фенилаланин накапливается в большом количестве в крови, тканях и спинномозговой жидкости, что в первые же месяцы жизни ведет к тяжелому поражению центральной нервной системы и неизлечимому слабоумию.
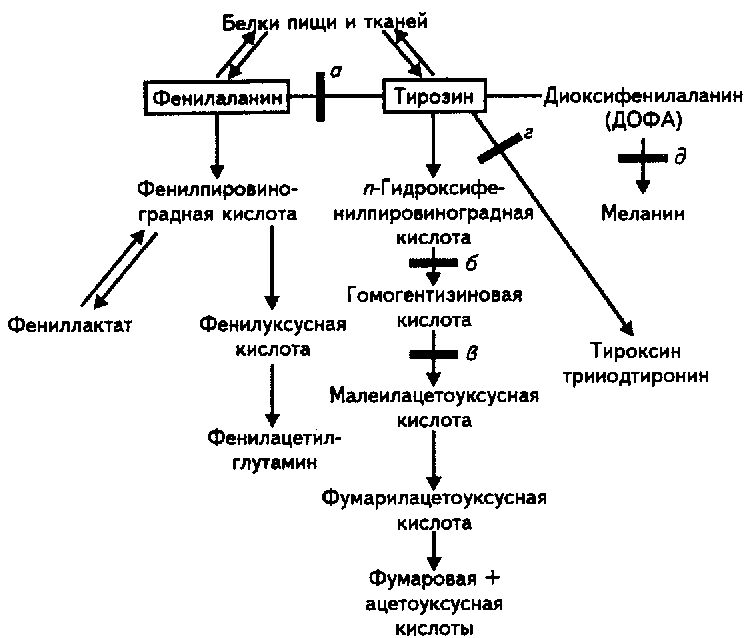
Рис. 73. Блокада путей метаболизма фенилаланина и тирозина: блок а – фенилкетонурия; 6 – тирозиноз; в – алкаптонурия; г – гипо-тиреоз; д – альбинизм
21.9.В чем сущность и чем проявляются наследственно обусловленные нарушения обмена тирозина?
В зависимости от уровня генетических дефектов наследственно обусловленные нарушения обмена тирозина могут проявляться развитием тирозиноза, алкаптонурии, альбинизма. Все эти заболевания наследуются аутосомно-рецессивно.
Тирозиноз возникает вследствие генетического дефекта фермента – оксидазы парагидроксифенилпировиноградной кислоты. В результате она, являясь первым промежуточным продуктом обмена тирозина, не превращается в гомогентизиновую кислоту, накапливается в крови и вместе с тирозином выделяется с мочой.
Алкаптонурия является следствием нарушения синтеза оксидазы гомогентизиновой кислоты, превращающей последнюю в малеилацетоуксусную кислоту. В результате в крови и моче появляется гомогентизиновая кислота. Моча при стоянии на воздухе, а также при добавлении к ней щелочи становится черной, что объясняется окислением гомогентизиновой кислоты кислородом воздуха и образованием алкаптона. Гомогентизиновая кислота из крови проникает в ткани – хрящевую, сухожилия, связки, внутренний слой стенки аорты, вследствие чего появляются темные пятна в области ушей, носа, на склерах. Иногда развиваются тяжелые изменения в суставах.
Альбинизм обусловлен дефицитом фермента тирозиназы. Вследствие этого не образуется красящее вещество кожи и волос – меланин. Организм, лишенный пигмента, становится очень чувствительным к действию ультрафиолетового излучения.