

Обработка полученных результатов
1. Определить постоянную калориметра. Для этого на миллиметровой бумаге построить график зависимости температуры от времени: Т = f(t) (рис. 10). Масштаб по оси абсцисс: 1 мин = 1 см. Масштаб по оси ординат: 0,1 0 = 1 см.
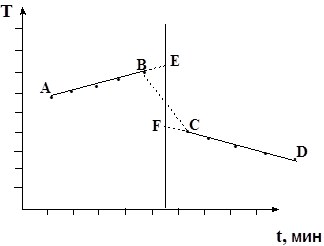
Рис. 10. График изменения температуры при растворении KCl.
Прямая АВ относится к начальному периоду, ВС – к главному, СD – конечному периоду. Точка В – последний отсчет начального периода, точка С – первый отсчет конечного периода. Через середину отрезка ВС провести перпендикуляр в оси абсцисс. Продолжить прямые АВ и CD до пересечением с перпендикуляром. Отрезок EF будет характеризовать действительное изменение температуры в опыте, т.е. ∆T. Крутизна наклона прямых АВ и CD зависит от характера теплообмена с окружающей средой. Определив ∆T, рассчитать постоянную калориметра W по уравнению 5.5:
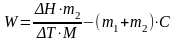 (5.5)
(5.5)
где m1 и m2 – масса воды и растворяемой соли соответственно; ∆H – молярная теплота растворения соли (для KCl ∆H = 18 841 Дж/моль); М – молярная масса растворяемой соли; С – удельная теплоемкость раствора (для раствора KCl С = 4,103 Дж/г·К).
2. Найти изменение температуры для суспензии исследуемого адсорбента ∆T расчетным способом:
 (5.6)
(5.6)
где Т2 – последний отсчет главного периода; Т1 – последний отсчет начального периода; δТ – поправка на потерю тепла в окружающую среду.
Поправку δТ рассчитать по уравнению Щукарева (5.7):
 (5.7)
(5.7)
где n - общее число интервалов главного периода;
Z1 – среднее изменение температуры между двумя отсчетами в начальном периоде (разность первого и последнего отсчета, деленная на число измерений);
Z2 – среднее изменение температуры между двумя отсчетами в конечном периоде (разность первого и последнего отсчета, деленная на число измерений);
Р – число первых интервалов главного периода, когда температура растет быстро ( не менее 0,003 0);
y – число остальных измерений главного периода, когда температура растет медленно.
Если в конечном периоде температура не падает, а повышается, то Z2 становится отрицательной величиной. Если температура в конечном периоде не изменяется, то Z2 = 0.
3. Рассчитать удельную теплоту смачивания Q по уравнению (5.8):
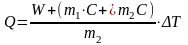 (5.8)
(5.8)
где W – постоянная калориметра; m1 и m2 – масса воды и исследуемого образца соответственно; С – удельная теплоемкость исследуемого вещества.
4. Рассчитать теплоту смачивания 1 м2 поверхности твердого тела q водой по уравнению 5.3 используя справочные данные, приведенные ниже.
5. Найти удельную поверхность исследуемого адсорбента, используя уравнение 5.4.
Таблица 5.1
Поверхностное натяжение воды на границе с воздухом при различных температурах
Температура, оС |
. 103, н/м |
Температура, оС |
. 103, н/м |
10 |
74,22. |
21 |
72,59 |
11 |
74,07 |
22 |
72,44 |
12 |
73,93 |
23 |
72,28 |
13 |
73,78 |
24 |
72,13 |
14 |
73,64 |
25 |
71,97 |
15 |
73,49 |
26 |
71,82 |
16 |
73,34 |
27 |
71,66 |
17 |
73,19 |
28 |
71,50 |
18 |
73,05 |
29 |
71,35 |
19 |
72,90 |
30 |
71,18 |
20 |
72,75 |
35 |
70,38 |
Таблица 5.2
Удельные теплоемкости
№ |
Вещество |
С, Дж/гК |
1 |
Al2O3 |
0,774 |
2 |
SiO2 |
0,741 |
Контрольные вопросы и задания
Чем обусловлен тепловой эффект смачивания?
Что такое дифференциальная теплота смачивания?
Что такое интегральная теплота смачивания?
Для чего и в каком случае используют показатель теплоты смачивания?
Физический смысл термического коэффициента смачивания.
Лабораторная работа № 6.
Исследование процесса ограниченного набухания полимера
Краткие теоретические сведения
В отличие от процесса растворения низкомолекулярного вещества, при котором происходит в основном диффузия растворяемого вещества в растворитель, начальная стадия процесса растворения высокомолекулярных соединений (ВМС) заключается в диффузии молекул растворителя в объем полимера. Проникновение молекул растворителя в объем полимера сопровождается процессом набухания.
Растворение макромолекул обязательно проходит через стадию набухания, являющуюся характерной качественной особенностью веществ этого типа. При набухании молекулы растворителя проникают в твердый полимер и раздвигают макромолекулы. Последние, из-за своего большого размера, медленно диффундируют в раствор, что внешне проявляется в увеличении объема полимера. Набухание может быть неограниченным, когда конечным его результатом является переход полимера в раствор, и ограниченным, если набухание не доходит до растворения полимера.
Неограниченное набухание – это набухание, заканчивающееся растворением, когда полимер сначала поглощает растворитель, а затем при той же температуре переходит в раствор, образуя однофазную гомогенную систему. Так набухают каучуки в углеводородах, биополимеры в воде. Если растворение полимера останавливается на стадии набухания, то говорят об ограниченном набухании. Так, полимеры пространственного строения могут только ограниченно набухать, образуя студни. Ограниченно набухают полимеры, имеющие химические связи - мостики между молекулами, которые лишают полимер свойства текучести, не позволяют его молекулам оторваться друг от друга и перейти в раствор. Примером ограниченно набухающего полимера с мостиковыми связями между молекулами является вулканизированный каучук, в котором мостиками служат атомы серы или полярные группировки.
Процесс набухания с точки зрения термодинамики характеризуется уменьшением энергии Гиббса ∆G = ∆H ─ T∆ S < 0, и состоит из двух этапов или стадий.
1 стадия – энергетическая, характеризуется сольватацией (гидратацией) полимера: ∆Н < 0; ∆S ≈ 0. При этом |T∆ S| < | ∆H| , поэтому ∆G < 0 за счет уменьшения энтальпии (экзотермическая стадия).
2 стадия – энтропийная, характеризуется активным разрахлением сетки ВМС, увеличением объема полимера, поэтому энтропия возрастает, а энтальпия практически не меняется: ∆Н ≈ 0; ∆S > 0; T∆S > 0.
Уменьшение энергии Гиббса ∆G < 0 происходит за счет возрастания энтропии.
В случае полимеров линейного строения, при изменении условий (температуры, концентрации и др.) ограниченное набухание может перейти в неограниченное, т.е. в растворение полимера. При контакте полимера с растворителем начинается взаимная диффузия молекул растворителя в полимер, а макромолекул полимера − в растворитель. Однако скорость диффузии будет различаться в той же пропорции, что и размеры, а также подвижности диффундирующих частиц. Резкое различие в подвижностях молекул растворителя и макромолекул ВМС является причиной набухания.
Количественной мерой набухания является степень набухания α, которая может иметь объемное или массовое выражение:
 (6.1),
(6.1),
где V0, V, m0, m – соответственно объемы и массы исходного и набухшего полимера.
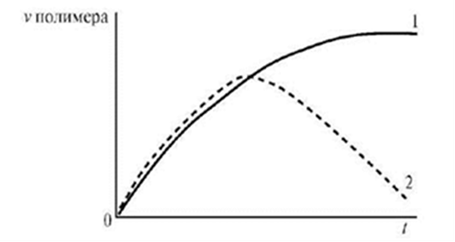
На рисунке 6.1 приведена зависимость степени набухания от времени. Максимальная или равновесная степень набухания определяется природой полимера и растворителя, сродством между ними, густотой пространственной сетки полимера.
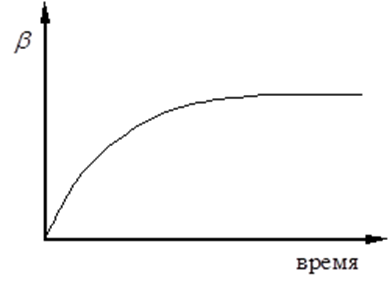
Рис.6.1. Зависимость степени набухания от времени.
Более точным является определение «α» по ее массовому выражению, так как в этом случае результаты измерений не зависят от явления контракции. Контракция заключается в том, что объем раствора (смеси) двух жидкостей оказывается меньше, чем сумма объемов взятых жидкостей.
В зависимости от структуры полимера и температуры набухание может быть ограниченным или неограниченным (рис. 6.2, а). При ограниченном набухании (рис. 3а, кривая 1) степень набухания достигает предельного значения, после чего набухание не зависит от времени (желатин в холодной воде). Для неограниченного набухания характерна зависимость (рис. 6.2 а, кривая 2), проходящая через максимум, после чего α падает до нуля в результате постепенного растворения полимера.
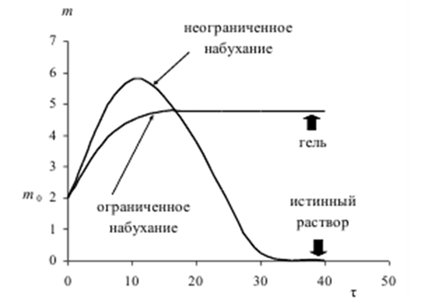
а б
Рис. 6. 2. Кинетика процесса набухания ВМС: а- кривые ограниченного (1) и неограниченного (2) набухания; б- кинетические кривые перехода полимера в гель и раствор.
Ограниченность или неограниченность набухания определяются соотношением энергий связей в полимере с энергией сольватации и энтропийным фактором. В линейных и разветвленных полимерах молекулы связаны ван-дер-ваальсовыми силами, энергия этих связей невелика, поэтому энергия сольватации и энтропийный фактор уже при комнатной температуре превышают их. При таких условиях набухание идет неограниченно. Если между цепями полимера имеются химические связи, то для их разрыва недостаточно бывает энергии сольватации и энтропийного фактора. Набухание протекает ограниченно, и полимер превращается в студень (рис. 6.2,б).
Набухание, как и сольватация, специфично, так как полимер набухает в растворителе, соответствующем его природе. В процессе набухания происходит односторонняя диффузия молекул растворителя в полимер. Это объясняется тем, что крупные макромолекулы, связанные в надмолекулярные структуры, практически не могут переходить в растворитель, а малые и хорошо диффундирующие молекулы растворителя легко проникают в полимер, увеличивая его объем. При набухании отдельные молекулы надмолекулярных структур сольватируются, межмолекулярное взаимодействие значительно ослабевает, в результате чего становится возможной диффузия макромолекул в растворитель (рис. 6.3).

Рис. 6.3. Взаимодействие растворителя с полимером: 1 – межструктурное набухание; 2 – внутриструктурное набухание; 3 – растворение.
Факторы, влияющие на процесс набухания
Процесс набухания имеет химическую природу и, следовательно, специфичен. Полимер набухает только в том растворителе, с которым он взаимодействует, поэтому полярные полимеры набухают в полярных жидкостях (белки в воде), неполярные - в неполярных (каучук в бензине).
Набухание не всегда кончается растворением полимера. Часто после достижения определенной степени набухания процесс прекращается либо из-за ограниченной растворимости ВМС в данном растворителе, либо из-за наличия в полимере поперечных химических связей, препятствующих отрыву макромолекул друг от друга и переходу их в раствор.
Набухание является экзотермическим процессом, поэтому в соответствии с принципом Ле Шателье, степень набухания увеличивается при уменьшении температуры. Однако скорость набухания при уменьшении температуры уменьшается, так как при более низких температурах диффузия протекает менее интенсивно.
С повышением температуры ограниченное набухание может переходить в неограниченное, т.е. полимер начинает растворяться. Начальная стадия процесса характеризуется положительным тепловым эффектом (выделяется теплота сольватации). В дальнейшем тепловой эффект ∆Н≈0, но зато возрастает энтропия за счет разрыхления сетки, и поэтому свободная энергия Гиббса остается отрицательной:
∆G = ∆H - T∆S < 0. (6.2)
Набухание твердого полимера в растворителе характеризуется степенью набухания (α), выраженной через массу образца ВМС до начала (m0) и после завершения процесса (m):
 (6.3)
(6.3)
Очевидно, что численно равна массе жидкости, поглощенной 1г полимера. Процесс характеризуется также числом набухания n, представляющим собой отношение объема обработанного полимера к объему исходного образца:
 (6.4),
(6.4),
где m0 и m1∞ - массы исходного образца и поглощенного им к концу процесса растворителя, ρ0 и ρ - плотность полимера и растворителя, соответственно.
Определяя степень набухания образца ВМС через различные промежутки времени, получают кривые, характеризующие кинетику процесса. Аналитически эти кривые описываются кинетическим уравнением реакции первого порядка:
 (6.5),
(6.5),
где К - константа скорости набухания, m1τ - количество растворителя, поглощенного образцом к моменту времени τ. Набухание в простейшем случае кинетически протекает как реакция первого порядка, потому что скорость процесса определяется скоростью диффузии растворителя в полимер.
Интегрируя уравнение (6.5), имеем:
 (6.6)
(6.6)
Отсюда следует, что величину К можно определить графически, представив опытную зависимость m1τ, τ в виде прямой линии:
ln(m1∞ - m1τ) = ln(m1∞) - Kτ, (6.7),
тангенс угла наклона которой определяет константу скорости набухания.
О влиянии природы растворителя на степень набухания можно судить по следующим данным, полученным для полистирола в органических жидкостях (α выражено в процентах): хлороформ – 1100, толуол – 1000, циклогексан – 100, гексан – 20, н-гексиловый спирт – 4.
Влияние поперечных связей в макромолекуле на способность к набуханию хорошо иллюстрирует пример с каучуком. Невулканизированный, т.е. не содержащий поперечных связей, каучук неограниченно набухает в углеводородах, образуя растворы. Вулканизированный каучук – резина, содержит поперечные сульфидные связи и набухает ограничено. Эбонит, представляющий собой каучук с очень «густой» пространственной сеткой, набухает значительно меньше. Степень набухания слабо вулканизированного каучука составляет 800-1000, а эбонита не более 150%.
Определяя степень набухания через заданные промежутки времени, можно получить кривые, характеризующие кинетику набухания. Скорость набухания различных полимеров можно сравнивать по наклону касательных, проведенных к кривым из начала координат, а способность полимеров к набуханию следует характеризовать по предельной степени набухания. На процесс набухания полимеров в воде влияет присутствие электролитов и значение рН среды. Влияние электролитов своеобразно прежде всего тем, что влияние оказывают в основном анионы, а катионы – лишь в незначительной степени. Причем одни анионы усиливают набухание, а другие ослабляют:
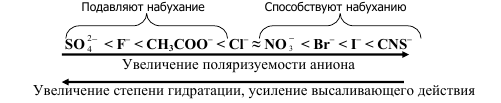
Влияние рН среды на набухание полимера больше всего проявляется в растворах белков, поскольку их молекулы – полиамфолиты.