

Векторные системы для клонирования очень крупных фрагментов днк
Векторные системы, способные интегрировать крупные вставки (>100т. п. н.), имеют большую ценность при анализе сложных эукариотических геномов. Без таких векторов не обойтись, например, при картировании генома человека или при идентификации отдельных генов/
Для клонирования фрагментов ДНК размером от 100 до 300 т. п. н. был сконструирован низкокопийный плазмидный вектор на основе бактериофага Р1. Природная форма бактериофага Р1 Е. coli в виде профага не интегрирует в хромосому, а существует в плазмидной форме. Фактически ДНК фага Р1 представляет собой природную фазмиду. Вектор РАС — химерная конструкция, называемая искусственной хромосомой на основе фага Р1
(Р1-artificial chromosomes).
В 1992 году Хируоко Шизуя создал также очень стабильный вектор, способный интегрировать вставки длиной от 150 до 350 т. п. н., на основе F-плазмиды (F-фактора, или фактора фертильности) Е. coli, которая представлена в клетке одной или двумя копиями, с селекционной системой lacZ' векторов pUC. Эта конструкция называется бактериальной искусственной хромосомой (ВАС, англ. bacterial artificial chromosomes) (рис. 27).
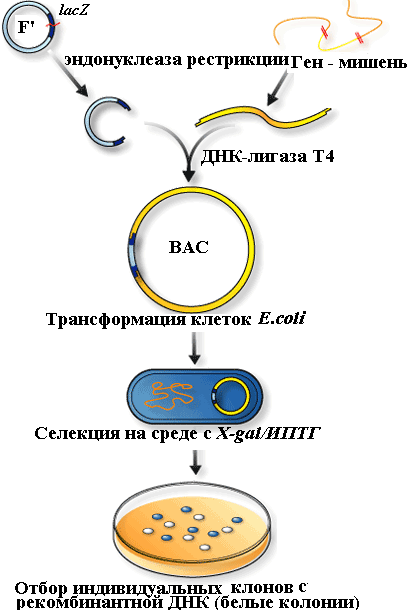
Рис. 27. Клонирование фрагментов ДНК большого размера с помощью ВАС.
Манипуляции процедуры клонирования фрагментов ДНК в клетках E.coli с использованием ВАС-вектора осуществляется в такой же последовательности, как было рассмотрено в случаях с применением плазмидных векторов pBR322 и pUC19 (рис. 20. 22). Особо следует отметить, что трансформация бактериальных клеток с помощью рекомбинантных ДНК на основе ВАС проводится методом электропорации.
Искусственные дрожжевые хромосомы (YAC – yest artificial chromosome). Эта система предназначена для клонирования очень больших фрагментов ДНК (до 2000 т.п.н.), которые потом поддерживаются в дрожжевой клетке как отдельные хромосомы (рис. 28). YAC-система очень стабильна.
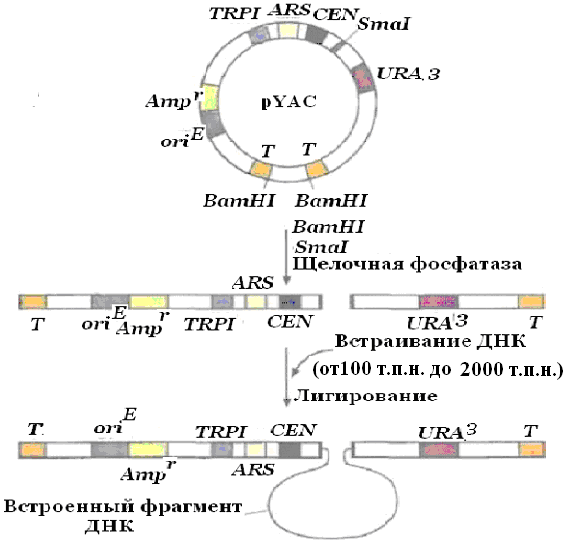
Рис. 28. YAC-система клонирования
YAC-плазмида (pYAC) содержит селективный маркерный ген E. coli (Ampr), сайт инициации репликации, функционирующий в E. coli (oriE); сегмент дрожжевой ДНК, включающий участки URA3, CEN, TRP1 и ARS (CEN - последовательность, выполняюшая центромерную функцию, ARS - дрожжевая автономно реплицирующаяся последовательность, эквивалентная дрожжевому сайту инициации репликации, URA3 - один из генов биосинтеза урацила, TRP1- один из генов биосинтеза триптофана). Т - это теломерные области дрожжевой хромосомы, SmaI — сайт, по которому осуществляется клонирование.
pYAC сначала обрабатывают SmaI, ВатHI и щелочной фосфатазой, а затем сшивают с фрагментом ДНК длиной от 100 до 2000 т.п.н. Конечная генетическая конструкция содержит клонированную ДНК и может стабильно поддерживаться в дрожжевых клетках Ura-Trp-.
Способы трансформации дрожжевых клеток:
Экзогенную ДНК добавляют к клеткам дрожжей, клеточные стенки которых удалены химически или энзиматически (сферопласты).
Клетки перед добавлением чужеродной ДНК обрабатывают ацетатом лития.
Электропорация.
YАС-вектор напоминает хромосому, поскольку он содержит последовательность, функционирующую как сайт инициации репликации ДНК (автономно реплицирующуюся последовательность), сегмент центромерной области дрожжевой хромосомы и последовательности, образующиеся на обоих концах при линеаризации ДНК и действующие как теломеры, обеспечивающие стабильность хромосомы. При встраивании чужеродной ДНК в YAC может происходить нарушение рамки считывания маркерного дрожжевого гена. В результате продукт этого гена не образуется, и при выращивании клеток на специальной среде можно наблюдать цветную реакцию. Кроме того, некоторые YAC-векторы несут селективный маркер, независимый от сайта клонирования.
Вспомогательные методы технологии рекомбинантной ДНК
Химический синтез ДНК
В настоящее время достаточно быстро можно синтезировать искусственные фрагменты нуклеиновых кислот разной длины и любого состава (такие фрагменты называются олигонуклеотидами), а затем соединить их в более длинные цепи с помощью специальных ферментов. Полученные таким образом гены и их фрагменты широко используются в генетической инженерии, биотехнологии, а также для диагностики инфекционных и генетических заболеваний. Так химически синтезированные олигонуклеотиды можно использовать:
для конструирования целых генов или их фрагментов
для амплификации специфических фрагментов ДНК
для направленных мутаций изолированных ДНК
в качестве зондов при гибридизации
в качестве линкеров, облегчающих клонирование.
Напомним, что нуклеиновые кислоты являются нерегулярными биополимерами, состоящими из мономерных структур – нуклеотидов (рис. 29).
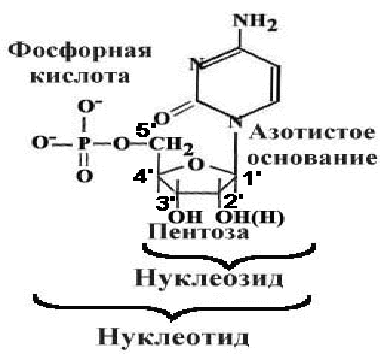
Рис. 29. Состав и строение нуклеотида.
Таким образом:
Нуклеотид = нуклеозид + фосфорная кислота = азотистое основание+ пентоза+фосфорная кислота.
В РНК пентоза - рибоза. В ДНК - дезоксирибоза.
В состав нуклеиновых кислот и олигонуклеотидов входят пять нуклеотидов, отличающихся между собой строением азотистого гетероцикла. Три из них - производные аденина (А), гуанина (G) и цитозина (С) входят в состав и ДНК и РНК, производное тимина (Т) - только в ДНК, урацила (U) - только в РНК. Нуклеотиды в отличие от нуклеозидов содержат остаток фосфорной кислоты. Нуклеотиды соединяются друг с другом в полимерную цепочку с помощью фосфодиэфирных связей (рис. 30). Азотистые основания не принимают участия в соединении нуклеотидов одной цепи.

Рис. 30. Схема соединения нуклеотидов с помощью фосфодиэфирной связи
Задачей химического синтеза нуклеиновых кислот является соединение входящих в их состав мономеров в строго определенной последовательности. Для образования межнуклеотидной связи должно произойти соединение двух нуклеотидов с отщеплением молекулы воды. Образующийся димер реагирует далее со следующим мономером (нуклеотидом) с образованием тримера и т.д. Для проведения этих реакций необходимо выполнение следующих условий.
1. Нужно добиться селективности соединения мономерных звеньев. Например, гидроксильная группа первого нуклеотида должна взаимодействовать с фосфатным остатком второго, но не наоборот, т.к. одним из принципов построения полинуклеотидов в природе является униполярность, т.е. соединение мономеров всегда в направлении 5' →3'. Для этого фрагменты, которые не участвуют в реакции, нужно блокировать с помощью специальных защитных групп.
2. Сами по себе гидроксильная и фосфатная группы в нуклеотидах не реагируют друг с другом. Для проведения конденсации мономеров нужно активировать фосфатную группу.
3. Поскольку синтез проводится в несколько стадий, каждая реакция должна проходить с очень высоким выходом.
4. Все процессы - введение и удаление защитных групп, активация и конденсация - должны проводиться в мягких условиях, то есть без использования высоких температур, а также концентрированных растворов кислот или щелочей, что могло бы привести к образованию побочных продуктов и разрушению имеющихся и вновь образованных химических связей.
В настоящее время разработано большое число методов синтеза олигонуклеотидов, удовлетворяющих этим условиям, подобраны удобные защитные группы и предложены способы активации фосфатной группы.
Синтез протяженных фрагментов нуклеиновых кислот состоит из большого числа стадий. После проведения каждой химической реакции нужно выделить полученный продукт, очистив его от не вступивших в реакцию исходных веществ и других примесей. Это делает процесс длительным и трудоемким, а также приводит к значительным потерям на каждой стадии выделения.
Американский ученый Роберт Меррифилд в 1962 году предложил оригинальную идею твердофазного метода синтеза, которая позволила резко упростить и ускорить проведение процесса. Идея заключается в том, что первый мономер (нуклеозид) присоединяют к нерастворимому полимерному носителю (твердой фазе), который помещают в колонку-реактор. Раствор, содержащий второй мономер и другие необходимые реагенты, пропускают через колонку. При этом образовавшийся продукт реакции также оказывается присоединенным к твердой фазе. Затем колонку промывают растворителем для удаления непрореагировавших веществ и побочных продуктов, после чего через реактор пропускают следующий мономер, повторяя процедуру многократно до завершения синтеза желаемого продукта. Таким образом, растущая полимерная цепь в процессе синтеза закреплена на твердой фазе, и все реакции с другими компонентами, находящимися в растворе, протекают на поверхности носителя. Этот прием позволяет заменить сложные и трудоемкие процедуры разделения и очистки промежуточных продуктов элементарными операциями промывки полимера.
Синтез включает в себя несколько принципиальных стадий.
Стадия 1. Получение защищенных мономеров, которые являются исходными блоками для построения цепочки олигонуклеотида. Для этого те фрагменты молекул, которые не должны подвергаться химическим превращениям, блокируют специальными защитными группами.
Стадия 2. Присоединение концевого мономера (защищенного нуклеозида) к полимерному носителю. Гранулы носителя с присоединенным концевым мономером вносят в колонку-реактор.
Стадия 3. Удаление защитной группы с концевого мономера. Для этого через колонку пропускают раствор реагента, вызывающего отщепление защитной группы.
Стадия 4. Проведение конденсации, для чего через колонку пропускают раствор второго мономера в смеси с активирующим реагентом.
Стадии 3 и 4 повторяют многократно до получения биополимера необходимой длины. Затем проводят стадии 5 и 6.
Стадия 5. Обработка носителя реагентами, приводящими к отщеплению синтезированного продукта от твердой фазы и удалению всех защитных групп.
Стадия 6. Выделение и очистка синтезированного фрагмента нуклеиновой кислоты с помощью различных хроматографических и электрофоретических методов. (Из-за неполноты протекания реакций к концу синтеза на носителе накапливаются более короткие фрагменты цепи, поэтому необходима тщательная очистка конечного продукта).
Основной вклад в развитие олигонуклеотидного синтеза внес Г. Корана, осуществивший в начале 60-х годов химический синтез фрагментов нуклеиновых кислот заданной последовательности и получивший за эту работу в 1968 году Нобелевскую премию. В 1970 году он впервые синтезировал полный ген аланиновой транспортной РНК.
В 1975 году Р. Летсингер предложил новый метод образования межнуклеотидной связи, на основании которого в начале 80-х годов М. Карузерс разработал так называемый твердофазный амидофосфитный или фосфорамидитный метод синтеза олигонуклеотидов. С этого времени начались бурное развитие этого метода и его автоматизация.
Исходные строительные блоки для синтеза олигонуклеотида – модифицированные дезоксирибонуклеозиды (амидофосфиты или фосфарамидиты). Эти соединения, содержащие трехвалентный фосфор, являются более активными по сравнению с производными пятивалентного фосфора (рис.31).
Амидофосфитные мономеры получают из защищенных нуклеозидов. Модификация состоит в присоединении к аминогруппам дезоксиаденозина и дезоксицидина бензольной группы, а к аминогруппе дезоксигуанозина – изобутиральной. Тимидин, у которого аминогруппа отсутствует, не модифицируют. Такая модификация необходима для защиты нуклеозидов от нежелательных побочных эффектов.
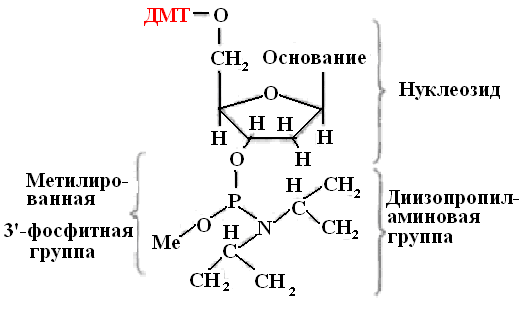
Рис. 31. Структурная формула фосфорамидита
Такие производные всех четырех оснований – A,T,G и C используются для химического синтеза ДНК.
ДМТ – диметокситритил
Ме – метильная группа
Синтез осуществляется в твердой фазе, растущая цепь ДНК фиксируется на твердом носителе, что позволяет проводить все реакции в одной емкости, легко отмывать после каждого этапа ненужные реагенты и добавлять новые в количестве, оптимальном для полного протекания реакции. Первый нуклеозид фиксируют на твердом носителе с помощью химического спейсера (рис 32). Обычно это пористые стеклянные шарики с порами одинакового размера.
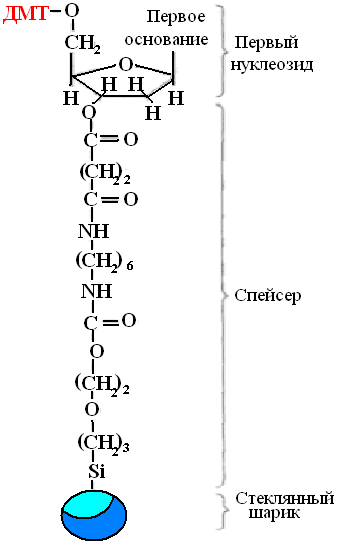
Рис. 32. Первый нуклеозид, фиксированный с помощью спейсера.
После n циклов, проведенных по схеме синтеза (рис. 33), образуется одноцепочечный фрагмент ДНК из n+1 нуклеотида
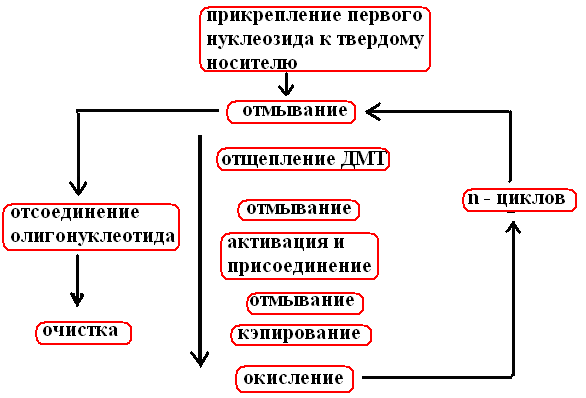
Рис. 33. Схема химического синтеза олигонуклеотида
Цикл начинается после присоединения первого нуклеозида к стеклянному шарику
промывают безводным растворителем (ацетонитрил), остатки которого удаляют продуванием аргона
промывают ТХУ (трихлоруксусная кислота) для отщепления ДМТ (детритилирование)
опять промывают растворителем и ТХУ, продувают аргон и на этом этапе вводят следующий нуклеозид в виде фосфоримидита и тетразол, который активирует фосфорамидит, несвязавшиеся реагенты удаляют продуванием аргона.
! Поскольку по окончании первого этапа не все фиксированные на носителе нуклеозиды оказываются связанными с фосфорамидитом, необходимо предотвратить их взаимодействие с нуклеозидом, добавленным на втором этапе. Для этого непрореагировавшую 5’–ОН группу ацетилируют с помощью уксусного ангидрида и диметиламинопиридина (кэппирование).
!! Нестабильную фосфиттриэфирную связь, образовавшуюся на втором этапе между нуклеотидами с помощью иодной смеси окисляют до пятивалентного фосфаттриэфира.
Все описанные операции проводят до тех пор, пока к растущей цепи в соответствии с программой не присоединится последний нуклеозид.
!!! Метильные группы удаляют с помощью химической обработки непосредственно в реакционной колонке. Затем отсоединяют олигонуклеотиды от спейсерной молекулы вместе с 3’ –ОН концом и элюируют их из колонки; затем последовательно удаляют бензоильные, изобутиральные и ДМТ-группы. 5’ –конец цепи фосфорилируют ферментативным (полинуклеотидкиназа+АТФ) или химическим методом
Важным преимуществом твердофазного метода синтеза является простота операций. Процесс синтеза состоит из повторяющихся стадий пропускания защищенных мономеров, реагентов и растворителей через колонку с полимерным носителем, к которому присоединена растущая цепь. Это делает возможной автоматизацию процесса.
Автоматические синтезаторы включают в себя реактор, содержащий носитель с присоединенным к нему первым мономером, и сосуды с необходимыми мономерами, реагентами и растворителями. С помощью насоса растворы из этих сосудов поочередно подаются в реактор в соответствии с имеющейся программой и заданной последовательностью синтезируемого фрагмента белка или нуклеиновой кислоты. В настоящее время автоматические синтезаторы пептидов и олигонуклеотидов выпускают различные, в том числе и отечественные, фирмы.
Секвенирование ДНК
Исчерпывающую информацию о молекуле ДНК можно получить, только определив ее нуклеотидную последовательность. Эта процедура называется секвенированием (от англ. sequence). Так, секвенировав ген, т.е. установив последовательность нуклеотидов определенного участка ДНК, часто удается установить его функцию, сравнив эту нуклеотидную последовательность с таковыми для генов, функция которых уже известна. Без данных о нуклеотидной последовательности невозможно проводить исследования по молекулярному клонированию, а также создание экспрессионных рекомбинантных ДНК.
Работы по секвенированию ДНК довольно дорогостоящие. Они требуют специального прецизионного оборудования, особо чистых химических реактивов и биологических препаратов, а также специалистов с высокой профессиональной подготовкой. Потенциал биотехнологии будет реализован в полной мере только тогда, когда ее инструментарий (в том числе и секвенирование геномов) станет столь же доступным и недорогим, как персональные компьютеры. Чтобы снизить стоимость процедуры секвенирования, разработчики новых методов стараются сократить число подготовительных этапов, до предела миниатюризировать оборудование и проводить секвенирование миллионов молекул одновременно
Прежде, чем приступить к описанию методов секвенирования, следует вспомнить основные моменты воспроизведения нуклеотидных последовательностей ДНК, т.е. некоторые принципиальные этапы репликации ДНК.
ДНК клетки, приступающая к делению, претерпевает кардинальные изменения: двойная спираль раскручивается, цепи расходятся. На каждой цепи начинается синтез комплементарных полинуклеотидов, на одной — непрерывный, на второй прерывистый. Его катализирует фермент под названием ДНК-зависимая ДНК-полимераза; другой фермент, ДНК-лигаза, сшивает полинуклеотидные фрагменты в непрерывную цепь. Так из одной молекулы ДНК образуются две.
Многие методы секвенирования ДНК основаны на взаимной комплементарности цепей этой молекулы. Генетический алфавит состоит всего из четырех букв — азотистых оснований аденина (А), цитозина (С), гуанина (G) и тимина (Т). Основания противоположных цепей молекулы ДНК соединяются в соответствии с правилом комплементарности: А образует пару с Т, а С — с G. В результате такого взаимодействия образуется хорошо известная двойная спираль — структура, напоминающая винтовую лестницу (рис. 34).
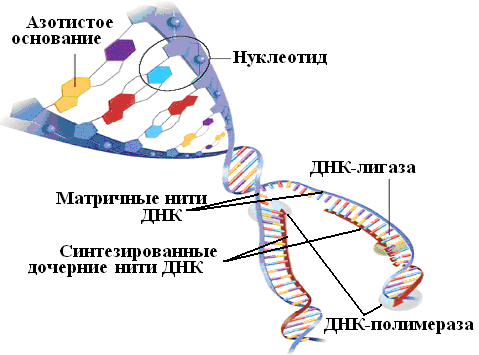
Рис. 34. Схема синтеза дочерних нуклеотидных цепей на обеих цепях ДНК
Живые организмы используют принцип комплементарности при копировании своего генетического материала (репликация) и устранения повреждений в нем (репарация). Он же лежит в основе амплификации (см. ПЦР) целевых фрагментов ДНК и их последующего секвенирования с помощью метода, разработанного в конце 1970-х гг. Ф. Сангером.
Секвенирование ДНК по Сангеру
Перед секвенированием по методу Сангера молекулу ДНК разрезают на фрагменты и клонируют в Escherichia coli. Выделенные из бактериальных клеток фрагменты многократно амплифицируют с помощью полимеразной цепной реакции (рис. 35), как описано в соответствующем разделе данного пособия.
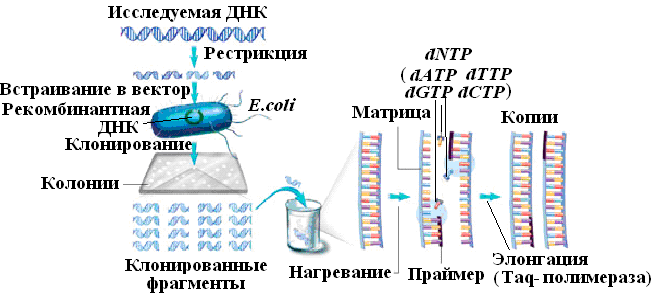
Рис. 35. Схема подготовки фрагментов ДНК с помощью ПЦР для секвенирования
Описание в тексте.
Образец ДНК нагревают до температуры, при которой происходит расхождение цепей. Затем в реакционную смесь добавляют дезоксинуклеозидтрифосфаты (dNTP) и праймер — короткий олигонуклеотид, комплементарный небольшому сегменту ДНК-матрицы. Он гибридизуется с этим сегментом, и ДНК-полимераза последовательно присоединяет к его концу dNTP, комплементарные нуклеотидам копируемой цепи. Процесс многократно повторяют, пока не получат миллионы копий каждого фрагмента.
Раствор с одноцепочечными фрагментами и праймерами распределяют по четырем пробиркам, в каждую из которых добавлены четыре разные dNTP и один из дидезоксинуклеозидтрифосфатов (ddNTP). Дидезоксинуклеотид – это полученный искусственным путем нуклеотид, лишенный 2’ – и 3’ – ОН групп при углеродных атомах сахарного кольца. При присоединении такого нуклеотида во время роста полинуклеотидной цепи синтез останавливается из-за отсутствия 3’ – ОН, к которому Днк-полимераза ковалентно присоединяет фосфатную группу следующего комплементарного матрице нуклеотида. Остановка синтеза ДНК – ключевой этап дидезокси-метода (рис. 36).
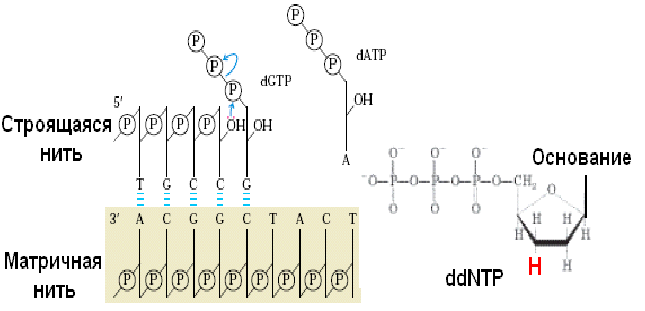
Рис. 36. Остановка синтеза полинуклеотидной цепи с помощью ddNTP
Удлинение гибридизовавшегося с ДНК-фрагментом праймера происходит до тех пор, пока в цепь не включится один из ddNTP (ddATP, ddTTP, ddCTP и ddGTP). В этом месте синтез останавливается, и в результате в каждой из пробирок образуется уникальный набор отрицательно заряженных фрагментов разной длины, оканчивающихся одним из ddNTP.
В случае ручного секвенирования для детектирования синтезированных фрагментов используют радиоактивномеченные dNTP (чаще всего α-Р32 dATP) в составе dNTP во все четырех пробирках, в которых осуществляется ПЦР. В пробирки добавляют формамид, чтобы обеспечить расхождение цепей, и проводят электрофорез в полиакриламидном геле на четырех дорожках. Это позволяет разделить одноцепочечные фрагменты ДНК, даже если они различаются по длине всего на один нуклеотид. На радиоавтографе обнаруживается набор полос, отвечающих меченым фрагментам ДНК, сопоставление которых позволяет прямо “прочитать” нуклеотидную последовательность секвенируемого сегмента ДНК. Нуклеотидная последовательность считывается с радиоавтографа снизу вверх (рис. 37).
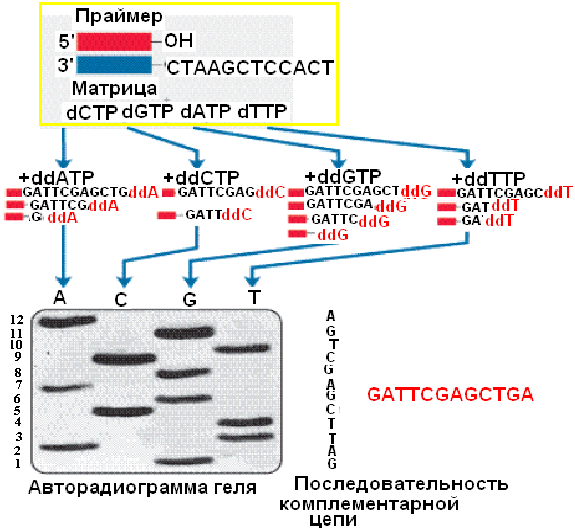
Рис. 37. Визуализация и протоколирование результатов секвенирования с применением изотопной метки.
При проведении автоматического секвенирования, т. е. при использовании специальных приборов – секвенаторов, применяют другие системы детекции. Так, например, каждый из четырех ddNTP метится с помощью специфического флуорофора, в результате чего в каждой из четырех пробирок продукты ПЦР будут иметь в своем составе разный флуорофор (рис. 38).
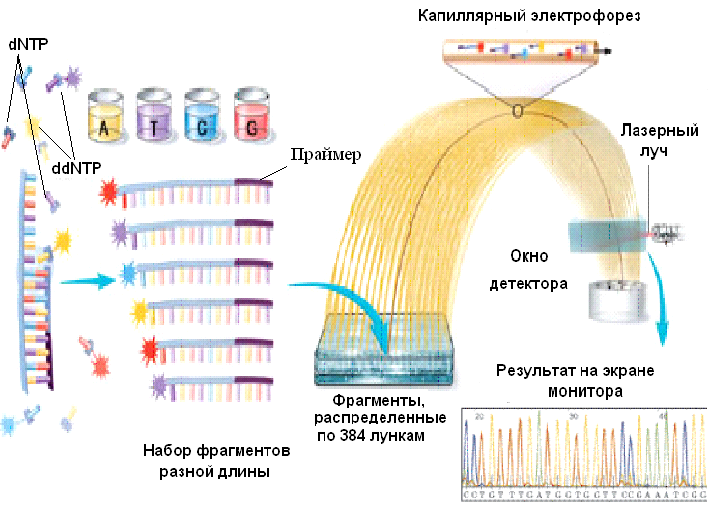
Рис. 38. Принципиальная схема определения первичной последовательности ДНК с помощью автоматического секвенатора.
В результате последнего раунда дупликации, проводимого в особых условиях, получают набор копий фрагментов разной длины, каждый из которых заканчивается флуоресцентно меченым нуклеотидом. Фрагменты разделяют по длине с помощью электрофореза, регистрируют световой сигнал от каждого из них по мере прохождения через детектор и получают нуклеотидную последовательность исходной цепи.
К достоинствам метода Сангера относятся его относительная простота и высокая точность, но, несмотря на последующие усовершенствования, он остается дорогим и трудоемким. Задача создателей альтернативных путей секвенирования состояла в повышении скорости процедуры и ее удешевлении. Для этого нужно было исключить этапы разделения, занимающие много времени, миниатюризировать всю систему, сохранив при этом возможность прочитывать последовательности миллионов фрагментов.
Следует также упомянуть и другие методы секвенирования при помощи биосинтеза, основанные на тех стадиях упомянутого процесса, которые протекают на одиночной цепи секвенируемой ДНК. В одном из таких подходов регистрируется момент присоединения к праймеру, гибридизовавшемуся с ДНК-матрицей, комплементарного нуклеотида (удлинение цепи). В другом - момент сшивания лигазой праймера с олигонуклеотидным зондом, содержащего известный нуклеотид в определенной позиции.
Существуют разные способы регистрации данных процессов, но обычно используется один из двух типов сигналов. Если меткой служит присоединенный к нуклеотиду флуорофор, как было рассмотрено выше, то регистрируется испускаемый им свет определенной длины волны. Флуоресцентное детектирование применяют при секвенировании как методом удлинения цепи, так и методом лигирования.
Другой подход основан на регистрации биолюминесценции, которая инициируется связыванием с белком люциферазой (его синтезируют хорошо знакомые нам светлячки) пирофосфата, высвобождаемого после присоединения к праймеру очередного нуклеотида.
Один из наиболее перспективных методов секвенирования использует для идентификации оснований в молекуле ДНК совсем другой принцип — физическое различие между четырьмя нуклеотидами, А, Т, G и С (рис. 39).
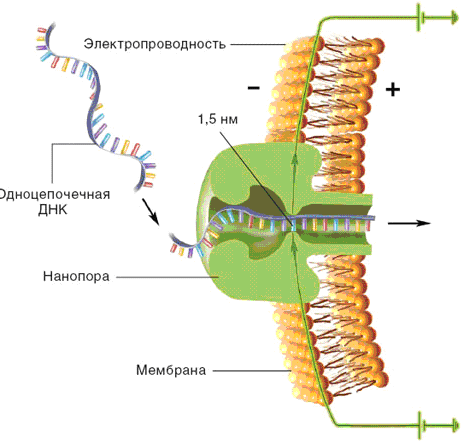
Рис. 39. Схема секвенирования с применением нанопор
Отрицательно заряженная одноцепочечная ДНК проходит через нанометровую пору в мембране, наружная поверхность которой несет отрицательный заряд, а внутренняя — положительный.
Одноцепочечную ДНК протягивают через пору диаметром 1,5 нм, пронизывающую мембрану, и регистрируют изменение электропроводности последней по мере поочередного прохождения нуклеотидов. Каждому типу основания соответствует свое изменение электропроводности, что и позволяет прочитать нуклеотидную последовательность цепи.
Молекулярная гибридизация
Гибридизация нуклеиновых кислот в растворе
Известно, что при нагревании водного раствора ДНК до 100оС, а также при повышении рН до 13 двойная спираль ДНК денатурирует за счет разрушения водородных связей между основаниями антипараллельных комплементарных полинуклеотидных цепей. В 1961 году было обнаружено, что этот процесс обратим: выдерживание ДНК при температуре 65оС вело к восстановлению структуры двойной спирали. Процесс восстановления структуры ДНК называется ренатурацией или гибридизацией. Процесс гибридизации, т.е. образования водородных связей между основаниями нуклеотидов происходит между любыми одноцепочечными полинуклеотидами, если они комплементарны: ДНК-ДНК, РНК-РНК, ДНК-РНК (рис. 40).
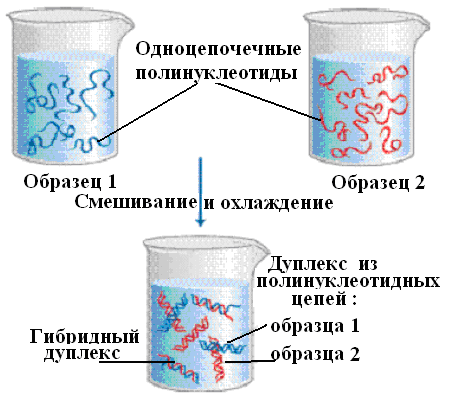
Рис. 40. Схема гибридизации полинуклеотидных цепей
Можно выделить основные этапы проведения экспериментов по гибридизации нуклеиновых кислот:
Двухцепочечную ДНК нагревают в соответствующем буфере. Из-за изменения внешних условий водородные связи между комплементарными азотистыми основаниями становятся термодинамически невыгодными и цепочки полинуклеотидов расходятся.
Препарат денатурированных ДНК затем смешивают с другой денатурированной ДНК, или РНК.
Препараты далее медленно охлаждают, при этом одноцепочечные ДНК отжигаются друг на друга (образуются водородные связи между комплементарными основаниями), при этом образуется “гибридная молекула”.
Анализ скорости отжига одноцепочечных ДНК позволяет оценивать сходства и различия в последовательностях ДНК между видами или особями одного вида. Кроме того, гибридизация нуклеиновых кислот представляет собой высокочувствительный метод выявления специфических последовательностей нуклеотидов и применяется в различных модификациях как в растворе, так и на твердых подложках, например на нитроцеллюлозных фильтрах или нейлоновых мембранах.
Гибридизация нуклеиновых кислот на твердых подложках
Обобщенная схема гибридизационного анализа на нитроцеллюлозных фильтрах (рис. 41) включает следующие этапы:
Подготовка исследуемой ДНК для фиксации на нитроцеллюлозном фильтре. Обычно исследуемую ДНК фрагментируют с помощью эндонуклеаз рестрикции и фракционируют полученные фрагменты ДНК методом гельэлектрофореза в агарозе (рис. 41 - 1). Гель помещают в щелочной раствор, при этом двойная спираль денатурирует и облегчает связывание отрицательно заряженной ДНК с положительно заряженной мембраной фильтра для дальнейшей гибридизации. При этом разрушаются и остатки РНК.
Нитроцеллюлозный фильтр помещают сверху агарозного геля таким образом, чтобы осуществлялось полное соприкосновение фильтра и геля (рис. 41 – 2). Сверху фильтра кладут несколько слоев фильтровальной бумаги и небольшой груз, чтобы обеспечить возникновение капиллярного тока жидкости через гель на фильтр. Буфер переносится капиллярными силами из участка с высоким содержанием воды (из губки, смоченной щелочным раствором) в зону с низким содержанием воды – на нитроцеллюлозный фильтр. При этом происходит и перенос ДНК из геля на фильтр. Полианионная ДНК связывается с положительно заряженным нитроцеллюлозным фильтром силами ионообменных взаимодействий.
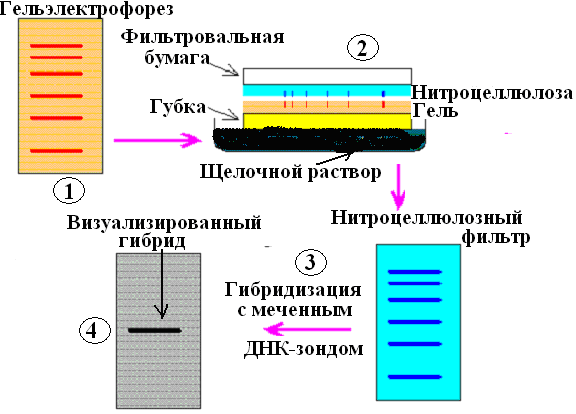
Рис. 41. Схема гибридизации нуклеиновых кислот на нитроцеллюлозных фильтрах (гибридизация по Саузерну)
Для окончательного закрепления ДНК на фильтре его нагревают в вакууме до температуры 80°С в течение двух часов. В случае использования нейлоновых мембран, последнюю освещают ультрафиолетовым излучением.
Нанесение меченой одноцепочечной ДНК-зонда, которая при определенных условиях (температуре и ионной силе) образует водородные связи с ДНК-мишенью (рис. 41 – 3).
Промывание фильтра для удаления избытка несвязавшегося меченого ДНК-зонда.
Детекция гибридных молекул зонд/мишень с помощью специфичной и высокочувствительной системы (радиоактивной, хемилюминесцентной, флуоресцентной) (рис. 41 – 4).
Перенос ДНК из геля на фильтр носит название Саузерн-блоттинга в честь Эдвина Саузерна, который изобрел этот метол. Использующиеся в литературе термины “Нозерн-блоттинг” и “Вестерн-блоттинг” относятся к переносу РНК и белков соответственно. Эти названия — в переводе с английского “северный” и “западный” -не имеют никакого отношения к сторонам света и были придуманы остроумными коллегами Саузерна (Southern — южный). Тем самым они как бы напутствовали Саузерна на разработку новых методов переноса макромолекул, а также четко обозначили, о переносе каких макромолекул идет речь. Кроме того, все увидели, что шутить умеют не только физики, но и биологи.
Гибридизация на чипах
Одним из наиболее точных и современных методов анализа является использование чипов. Они представляют собой пластинки с иммобилизованными мечеными ДНК-зондами. Каждая такая пластинка может содержать несколько десятков тысяч зондов, расположенных в определенной последовательности. Метка проявляется только в спаренных двухцепочечных фрагментах. Если в исследуемом образце есть последовательности, комплементарные последовательностям зонда, то гибридизацию можно определить визуально или с помощью специальных приборов. Как правило, детекторы соединены с компьютером, то есть процедура считывания и обработки информации автоматизирована. Такие ДНК-чипы можно применять для комплексной диагностики инфекционных заболеваний, наследственных дефектов, установления экспрессии тех или иных генов (в этом случае идет гибридизация с мРНК, или кДНК, полученных на мРНК), то есть отслеживания нарушений обмена веществ.
Как исследовать, действительно ли ген существует, то есть транскрибируется ли данный участок ДНК? Для этого ген представляют в чипе частью его последовательности – олигонуклеотидом, который иммобилизован в микроплощадке с определенными координатами на этой матрице (рис. 42 – 1). Этот олигонуклеотид соответствует части экзона, предсказанного компьютером на основе сиквенса геномной ДНК. Чтобы выяснить, действительно геном в данном участке транскрибируется, берется клетка и из нее выделяется суммарная РНК(рис. 42 – 2). Из всех этих молекул РНК получают ДНК-копии, которые флуоресцентно метят и проводят гибридизацию с иммобилизованными на микрочипе олигонуклеотидами (рис. 42 – 3, 4). Если в данных условиях какие-то площадки с олигонуклеотидами «молчат» (они показаны черным), то это значит, что участок геномной последовательности, комплементарной этому олигонуклеотиду, не транскрибируется. Если же площадка матрицы «светится», значит олигонуклеотиды в этой площадке прогибридизовались с флуоресцентно меченым продуктом, то есть соответствующий участок генома транскрибировался и действительно является частью какого-то гена (рис. 42 – 5).
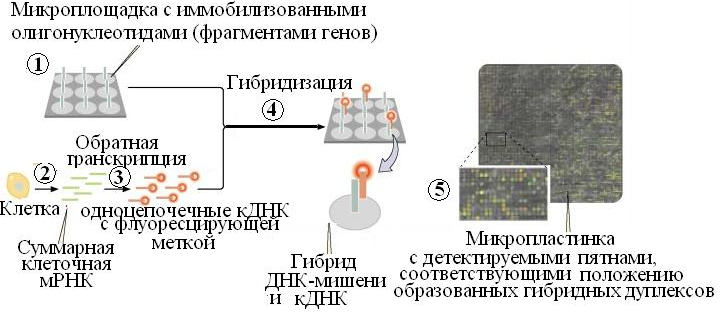
Рис. 42. Схема анализа последовательностей нуклеотидов с помощью ДНК-чипов.
Биологические микрочипы могут быть использованы для установления относительного уровня экспрессии в клетках различных типов для выявления органоспецифических различий (печень – почки), патологических (норма – опухоль), онтогенетических (зародыш – взрослый организм).
Полимеразная цепная реакция
Метод полимеразной цепной реакиции (ПЦР)
Принцип метода полимеразной цепной реакции (ПЦР) (polymerase chain reaction - PCR) был разработан Кэри Мюллисом (фирма “Cetus”, США) в 1983г. Впоследствии он получил за это изобретение Нобелевскую премию. В основе метода ПЦР лежит многократное удвоение определенного участка ДНК. В результате нарабатываются количества ДНК, достаточные для визуальной детекции. В настоящее время ПЦР широко используется как для научных исследований, так и для решения прикладных задач (генотипирование, диагностика инфекционных заболеваний, судебная медэкспертиза и т.д.).
Этот метод представляет собой эффективный способ получения in vitro большого числа копий специфических нуклеотидных последовательностей. Их амплификации (увеличение числа копий) — иногда в миллионы раз — осуществляется в ходе трехэтапного циклического процесса. Для осуществления ПЦР необходимы:
два синтетических олигонуклеотидных праймера (длиной 20-25 нуклеотидов), комплементарные участкам ДНК из противоположных цепей, фланкирующим последовательность-мишень; их 3’–ОН концы должны быть ориентированы навстречу друг другу;
ДНК-матрица длиной от 100 до 35000 п.н. (ДНК или ее часть, содержащая искомый специфический фрагмент);
термостабильная ДНК-полимераза, которая не теряет своей активности при температуре 95° С и выше;
смесь дезоксинуклеозидтрифосфатов (смесь четырех дНТФ является субстратом для синтеза новых комплементарных цепей ДНК);
буферный раствор (реакционная среда, содержащая ионы Mg2+, необходимые для поддержания активности фермента).
Стандартная ПЦР-амплификация представляет собой многократное повторение следующих трех реакций (рис. 43 - 1).
1. Денатурация образца ДНК выдерживанием его при температуре 93 - 95° С в течение по крайней мере 1 минуты. Помимо ДНК в реакционной смеси содержатся в избытке два праймера, термостабильная ДНК-полимераза Taq, выделенная из бактерии Termus aquaticus и четыре дНТФ.
2. Ренатурация или отжиг. Температуру смеси медленно понижают до оптимальной температуры спаривания матрицы ДНК и праймеров. Присоединение праймеров происходит комплементарно к соответствующим последовательностям на границах специфического участка противоположных цепей ДНК. Для каждой пары праймеров существует своя температура отжига, значения которой располагаются в интервале 50-65оС. Время отжига занимает 20-60 сек.
3. Синтез ДНК или удлинение праймера. Комплементарное достраивание цепей ДНК происходит от 5’-конца к 3’-концу цепи в противоположных направлениях, начиная с участков присоединения праймеров. Материалом для синтеза новых цепей ДНК служат добавляемые в раствор дезоксирибонуклеозидтрифосфаты (англ. dNTPs). Процесс синтеза катализируется ферментом термостабильной ДНК-полимеразой (Taq-полимеразой) и проходит при температуре 70-72оС. Время протекания синтеза - 20-60 сек.
Образовавшиеся в первом цикле амплификации новые цепи ДНК служат матрицами для второго цикла амплификации, в котором происходит образование искомого специфического фрагмента ДНК (ампликона). (рис. 43 - 3). В последующих циклах амплификации ампликоны служат матрицей для синтеза новых цепей. Таким образом, происходит накопление ампликонов в растворе по формуле 2n, где n-число циклов амлификации. Поэтому, даже если в исходном растворе первоначально находилась только одна двухцепочечная молекула ДНК, то за 30-40 циклов в растворе накапливается около 108 молекул ампликона. Этого количества достаточно для достоверной визуальной детекции этого фрагмента методом электрофореза в агарозном геле.
Процесс амплификации проводится в специальном программируемом термостате (амплификаторе), который по заданной программе автоматически осуществляет смену температур согласно числу циклов амплификации.
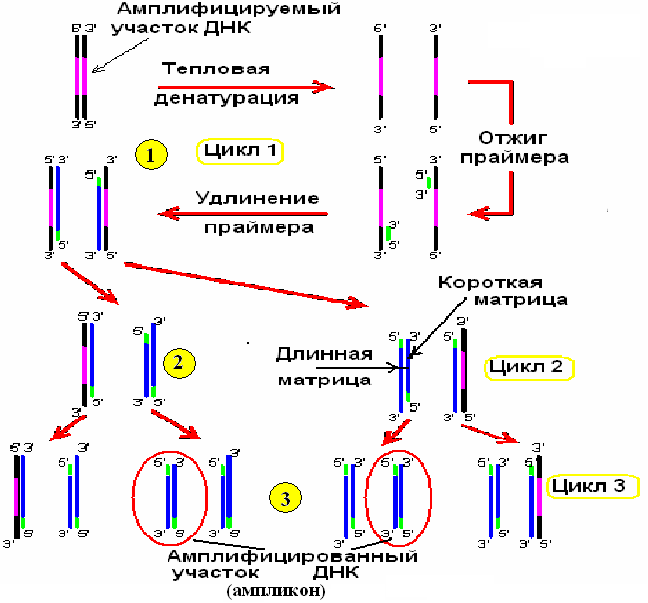
Рис. 43. Схема первых трех циклов полимеразной цепной реакции
Амплифицированный in vitro специфический фрагмент ДНК получают в количествах, достаточных для его прямого секвенирования, т.е. для определения последовательности нуклеотидов в его цепи. Поскольку при этом не требуется промежуточный этап клонирования фрагмента ДНК в генетических векторах, ПЦР иногда называют бесклеточным молекулярным клонированием (cell-free molecular cloning). Автоматизированная процедура Taq-полимеразной цепной реакции, состоящая из 30 и более циклов, занимает 3—4 часа, что существенно быстрее и проще процедуры клонирования определенного фрагмента ДНК в составе рекомбинантных ДНК.
Метод ПЦР произвел настоящую революцию в биотехнологии. Он позволяет в миллионы раз амплифицировать in vitro нужные сегменты ДНК. Некоторые из направлений применения метода ПЦР:
идентификация патогенных микроорганизмов, возбудителей заболеваний человека, животных и растений;
выявление спонтанных мутаций;
внесение специфических мутаций in vitro (направленный мутагенез);
введение адаптеров для создания сайтов рестрикции в амплифицированных фрагментах с дальнейшим клонированием последних, например, в клетках E.coli (рис. 44);
сборка полноразмерных генов из синтетических олигонуклеотидов;
секвенирование ДНК и др.
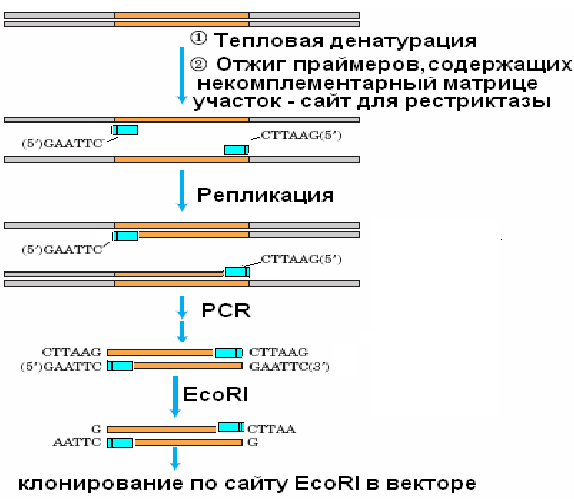
Рис. 44. Введение сайтов рестрикции с помощью ПЦР
ПЦР можно провести всего за один день, ее легко автоматизировать, реакция сравнительно недорога и чрезвычайно специфична.
ПЦР в реальном времени.
В настоящее время широкое распространение в исследовательских и диагностических лабораториях получила новая технология ПЦР — ПЦР в реальном времени (Real-Time PCR). Ее принципиальной особенностью является мониторинг и количественный анализ накопления продуктов полимеразной цепной реакции и автоматическая регистрация и интерпретация полученных результатов (рис. 45)
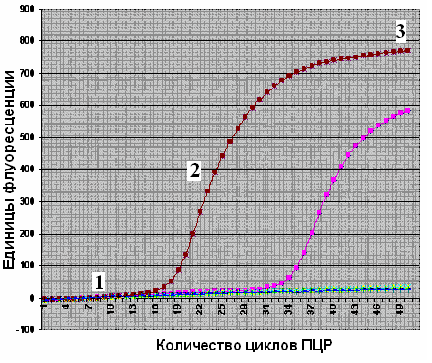
Рис. 45. Кинетическая кривая ПЦР в координатах
1. Стадию инициации (когда ПЦР-продукты еще не
детектируется флуоресцентной меткой).
2. Экспоненциальную стадию (в которой наблюдается
экспоненциальная зависимость количества
флуоресценции от цикла ПЦР).
3. Плато – стадию насыщения.
Real-time PCR - это семейство методик количественного ПЦР со следующими чертами:
определение выхода продукта реакции после каждого цикла амплификации;
построение по этим данным кинетической кривой ПЦР;
определение относительной концентрации субстрата на основании анализа этой кривой.
ПЦР в реальном времени использует флуоресцентно меченые олигонуклеотидные зонды для детекции ДНК в процессе ее амплификации (рис. 46). Детектируемая прибором флуоресценция, прямо пропорциональна количеству ПЦР-продукта, ее обозначают как - репортерная флуоресценця. Механизмы генерации репортерной флуоресценции различаются в зависимости от типа real-time ПЦР.
Детекция сигнала флуоресценции в ходе реакции позволяет наблюдать процесс накопления продукта во время ПЦР, а не после окончания реакции. Сигнал флуоресценции в ходе ПЦР возрастает пропорционально количеству продукта амплификации. Мониторинг сигнала позволяет построить кинетическую кривую реакции, при этом момент заметного увеличения сигнала и отрыва его от фонового – так называемый пороговый цикл – зависит от исходного количества ДНК-мишени. Чем больше количество ДНК в образце, тем раньше наблюдается начало роста сигнала флуоресценции и тем меньше пороговый цикл.
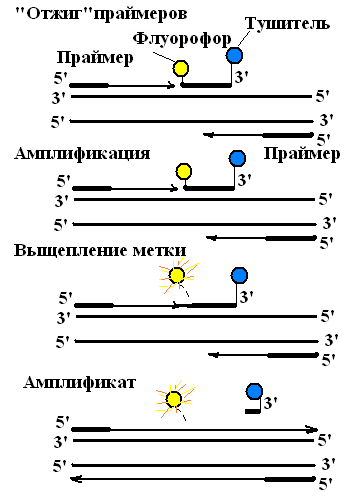
Рис. 46. Схема, представляющая систему использования флуорфоров при проведении ПЦР в реальном времени
Основные преимущества ПЦР в реальном времени по сравнению с методом анализа по конечной точке:
количественный анализ специфической ДНК в широком диапазоне концентраций;
сравнительный количественный анализ нескольких типов ДНК в одной пробирке;
повышение специфичности реакции за счет использования гибридизационных зондов;
обнаружение и определение процентного содержания ДНК с измененной последовательностью;
исключение послеамплификационных манипуляций с продуктом и, как следствие, снижение риска контаминации (загрязнения), экономия времени и сокращение затрат на поддержание ПЦР-лаборатории;
автоматизация и стандартизация ПЦР-анализа.
Обратная транскрипция, сопряженная с полимеразной цепной реакцией (ОТ-ПЦР)
Метод ОТ-ПЦР (англ. RT-PCR – reverse transcription polymerase chain reaction) представляет собой метод амплификации специфического фрагмента рибонуклеиновой кислоты. Одноцепочечную молекулу РНК превращают в реакции обратной транскрипции в комплементарную ДНК (кДНК) и далее амплифицируют уже одноцепочечную молекулу ДНК, используя традиционный метод ПЦР. Для превращения последовательности РНК в комплементарную ДНК использут фермент обратную транскриптазу (см. раздел Ферменты).
Экспоненциальная амплификация при помощи ОТ-ПЦР является чувствительной методикой, с помощью которой может быть обнаружено малое количество молекул РНК. ОТ-ПЦР широко используется для диагностики генетических заболеваний и полуколичественного определения специфических молекул РНК в клетке или ткани как индикатор экспрессии соответствующих генов.
Направленный мутагенез
Технология рекомбинантных ДНК позволяет выделять гены любых белков, существующих в природе, экспрессировать их в специфическом хозяйском организме и получать чистые белковые продукты. Однако физические и химические свойства природных белков часто не удовлетворяют условиям, обеспечивающим возможность их промышленного применения. Для создания белков со специфическими свойствами можно использовать подход, основанный на внесении изменений в кодирующие их клонированные гены, т.е. путем осуществления мутагенеза. Это позволяет получать белки с другими, чем у их аналогов свойствами.
Внесение специфических изменений в кодирующие последовательности ДНК, приводящих к определенным изменениям в аминокислотных последовательностях, называется сайт-направленным мутагенезом. В 1993 Смит (Michael Smith) вместе с Мюллисом (Kary Mullis) получили Нобелевскую премию “за фундаментальный вклад в установление направленного мутагенеза, основанного на олигонуклеотидах, и за его развитие для изучения белков”.
В настоящее время в технологии рекомбинантной ДНК наиболее часто используют два методических подхода сайт-направленного мутагенеза. Первый из них (рис. 47) включает встраивание ДНК-мишени в плазмидный вектор, которым трансформируют клетки E. coli, выделяют клонированную и двухцепочечную плазмидную ДНК и денатурируют щелочью, чтобы получить одноцепочечные кольцевые молекулы. Денатурированную ДНК отжигают с синтезированным химическим путем олигонуклеотидом с измененной последовательностью нуклеотидов, например с однонуклеотидной заменой в заранее определенном месте. Гибридизовавшийся олигонуклеотид служит затравкой для синтеза ДНК, а интактная кольцевая молекула ДНК – матрицей. Одноцепочечные разрывы в новосинтезированной цепи зашиваются с помощью ДНК-лигазы Т4. По окончании синтеза и лигирования продуктами реакции трансформируют клетки E. coli. Клетки, несущие мутантный клонированный ген, идентифицируют с помощью гибридизации.
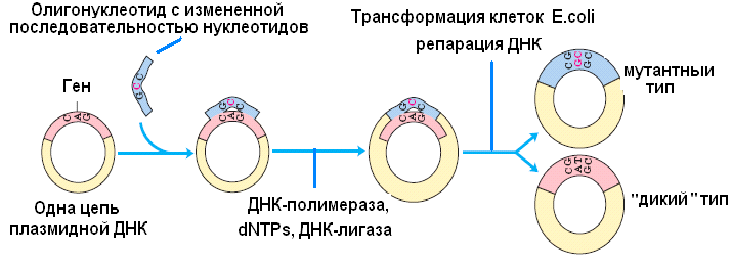
Рис. 47. Сайт-направленный мутагенез с использованием плазмидной ДНК
А второй позволяет проводить сайт-направленный мутагенез с использованием пары праймеров,со встроенным “мутантным” нуклеотидом. Cинтезируют пару праймеров, несущих мутацию, и пару праймеров, комплементарных концам нужного фрагмента ДНК. В ходе первых двух реакций образуются фрагменты ДНК с мутацией, которые объединяют в третьей реакции. Полученный фрагмент вставляют в нужную генно-инженерную конструкцию.
Библиотеки генов
Идентификация нуклеотидных последовательностей, как структурных, кодирующих определенные белки, так и регуляторных, обеспечивающих реализацию генетической информации, является конечной целью многих фундаментальных исследований и базой для прикладных протоколов в биотехнологии. В связи с этим разработаны подходы для создания доступных для работы библиотек ДНК. Создание библиотеки ДНК (банка клонов, банка генов) представляет собой процесс клонирования фрагментов ДНК. Существует два типа библиотек ДНК: геномные библиотеки и кДНК- Идентификация нуклеотидных последовательностей, как структурных, кодирующих определенные белки, так и регуляторных, обеспечивающих реализацию генетической информации, является конечной целью многих фундаментальных исследований и базой для прикладных протоколов в биотехнологии. В связи с этим разработаны подходы для создания доступных для работы библиотек ДНК. Создание библиотеки ДНК (банка клонов, банка генов) представляет собой процесс клонирования фрагментов ДНК. Существует два типа библиотек ДНК: геномные библиотеки и кДНК-библиотеки.
Геномные библиотеки содержат все последовательности ДНК, представляющие полный геном данного организма.
кДНК-библиотеки представляют собой только последовательности комплементарных ДНК, синтезированных на РНК данных клеток.
Создание геномных библиотек. Прокариоты
Разделение геномной ДНК на клонируемые элементы и введение этих элементов в клетки-хозяева дает возможность получить полную библиотеку генов данного организма. Специфика структурной организации генов прокариот и эукариот инициирует разработку и применение различной стратегии для их клонирования.
У прокариот искомая нуклеотидная последовательность (ДНК-мишень или ДНК интереса) часто составляет минимальную часть (0,02%) суммарной хромосомной ДНК. Для экстрагирования последовательности ДНК-мишени разработаны различные экспериментальные подходы, включающие следующие основные этапы:
суммарную ДНК гидролизуют эндонуклеазами рестрикции и каждый из полученных фрагментов встраивают в вектор;
проводят поиск специфической клеточной линии (клона), которая содержит нужную последовательность нуклеотидов;
выделяют ДНК и анализируют.
Один из способов создания библиотеки ДНК состоит в обработке донорной ДНК эндонуклеазой рестрикции, узнающей тетрануклеотиды, например, Sau3AI, которая вносит один разрыв примерно на 256 пар оснований. Гидролиз проводят в таких условиях, чтобы происходило лишь частичное расщепление, так что образуются фрагменты различных размеров.
Глубину гидролиза обычно контролируют изменением времени инкубации или количества фермента. Одни молекулы, представленные на рисунке 48, расщеплены по всем сайтам, имеющимся на данной ДНК для гипотетической рестрицирующей эндонуклеазы (REI-сайтам), другие - только по некоторым.
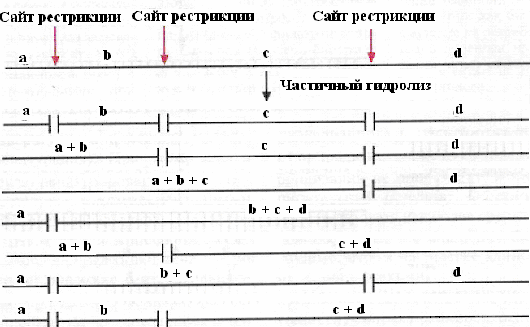
Рис. 48. Схема проведения частичного гидролиза фрагмента ДНК эндонуклеазой рестрикции типа II
Из рисунка 48 видно, что исследователь получает набор фрагментов ДНК всевозможного размера и, что самое ценное, получает возможность экстрагировать последовательности ДНК, содержащие нативные, т.е. не фрагментированные участки. Например, при полном гидролизе с помощью REI исследуемый фрагмент ДНК гидролизуется по всем REI-сайтам, встречающимся на составляющей его нуклеотидной последовательности, образуя фрагменты a, b, c, и d. В случае, когда последовательности нуклеотидов ДНК-мишени содержат сайты для REI, например – между фрагментами a и b, частичный гидролиз дает возможность получить фрагменты a+b и a+b+c, которые содержат непрерывающуюся нуклеотидную последовательность фрагментов a+b. Таким образом, набор фрагментов, получаемых при неполном гидролизе эндонуклеазой REI исследуемой ДНК, дает возможность получить всю последовательность нуклеотидов, представленную такими фрагментами: a+b, a+b+c, b+c+d, c+d b+c и c+d. Полученные таким образом фрагменты ДНК-мишени для создания геномной библиотеки клонируют в векторах на основе фага λ (рис. 49), т.к. последние могут обеспечить клонирование фрагментов ДНК до 20 т.п.н. и эффективность трансформации клеток-реципиентов выше, чем у плазмидных векторов приблизительно в 1000 раз (см. раздел Генетические векторы).
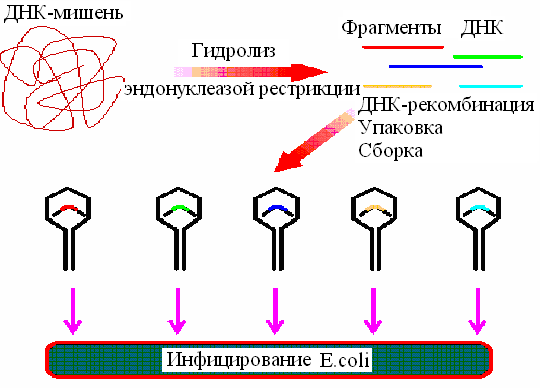
Рис. 49. Схема создания геномной библиотеки с помощью вектора на основе ДНК λ-фага
Следующий после создания библиотеки этап – это поиск клона (клонов), несущего искомую последовательность ДНК. Для этого используют три широко известных метода:
гибридизацию с меченым ДНК-зондом (см. раздел Вспомогательные методы) с последующей визуализацией отобранных образцов (изотопная и неизотопная) (рис. 50);
иммунологический скрининг (рис. 52);
скрининг по активности белка, кодируемого геном-мишенью.
Скрининг с помощью ДНК-ДНК-гибридизации
Клетки E.coli после трансформации высевают на твердую питательную среду, обеспечивающую рост только трансформированных клеток. Клетки из каждой выросшей колонии переносят на твердую подложку (например, нитроцеллюлозный или нейлоновый фильтр), так, чтобы их расположение соответствовало таковому на чашке, затем лизируют и высвободившуюся ДНК подвергают денатурации и фиксируют на фильтре. На фильтр наносят меченый ДНК-зонд и проводят гибридизацию. Смывают с фильтра негибридизовавшийся зонд и проводят детекцию, чтобы определить, какие клетки содержат меченый ДНК-зонд. Идентифицируют на чашке колонии, содержащие искомую ДНК (положительный гибридизационный сигнал), отбирают из соответствующих клонов клетки и культивируют для выделения рекомбинантной ДНК.
Меченые ДНКзонды можно получить разными способами:
методом концевого мечения с помощью полинуклеотидкиназы, осуществляющей присоединение dNTP, в котором в ﻻ-положении по отношению к 5’-углероду дезоксирибозы встроен изотоп фосфора 32Р;
методом ник-трансляции с помощью ДНК-полимеразы I;
методом случайных праймеров, который основан на применении смеси синтетических олигонуклеотидов (олигомеров) в виде затравки для ДНК-полимеразы I (рис. 51). Они представляют собой набор гексануклеотидов, содержащий все возможные комбинации из четырех dNTP.
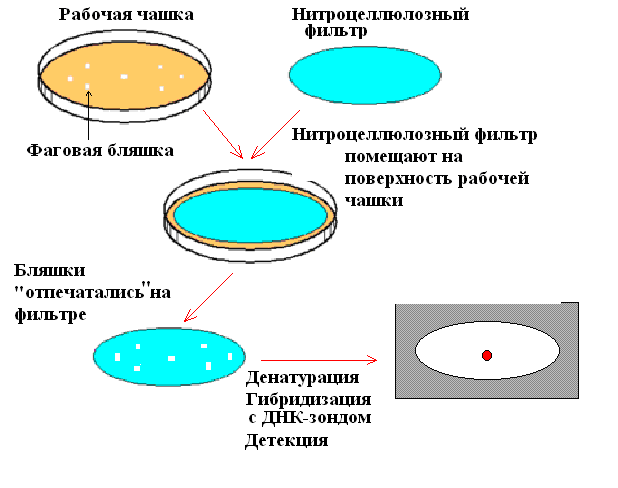
Рис. 50. Скрининг клонов трансформированных клеток E.coli с помощью гибридизации
При использовании методов “ник-трансляции” и “случайных праймеров” в суммарный пул dNTP, так называемых “холодных”, т.е. без метки, вводят один нуклеозидтрифосфот, чаще всего dATP, у которого из трех атомов фосфора - тот, который находится в α-положении по отношении к 5’-углероду дезоксирибозы, представлен изотопом 32Р или 33Р. При встраивании такого меченого субстрата в синтезируемую полинуклеотидную цепь, т.е. при образовании новой фосфодиэфирной связи, отщепляется пиррофосфат – атомы фосфора в γ- и β-положениях, а 32Р остается встроенным в сахарофосфатную цепь.
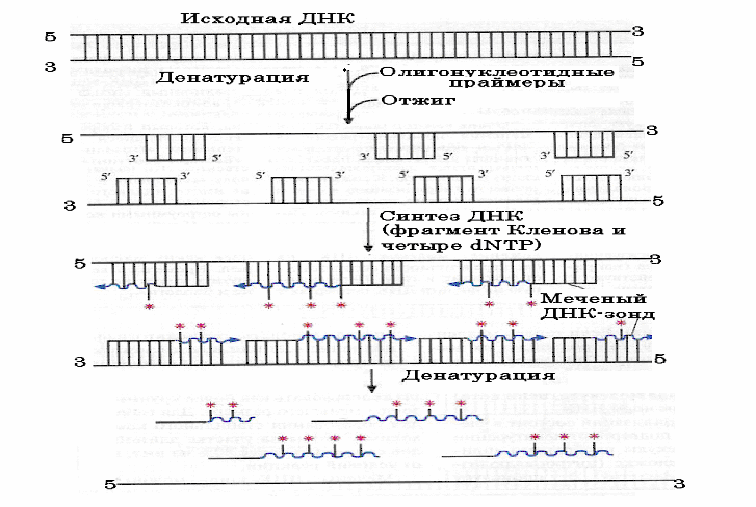
Рис. 51. Схема получения меченых ДНК-зондов с помощью метода “случайных праймеров”
Нерадиоактивные системы детекции используют ферментативное превращение хромогенного или хемолюминисцентного субстрата: первый из них под действием фермента изменяет окраску, а второй – испускает свет.
Последовательности нуклеотидов, которые могут служить ДНК-зондами
для скрининга геномной библиотеки можно получить, по крайней мере, двумя способами: использовать клонированную ДНК близкородственного организма (гетерологичный зонд) либо получить методом химического синтеза, основываясь на известной аминокислотной последовательности белкового продукта искомого гена.
Иммунологический скрининг
Если клонированный ген экспрессируется, то его продукт – весь белок или часть – можно обнаружить иммунологическими методами (рис. 52).
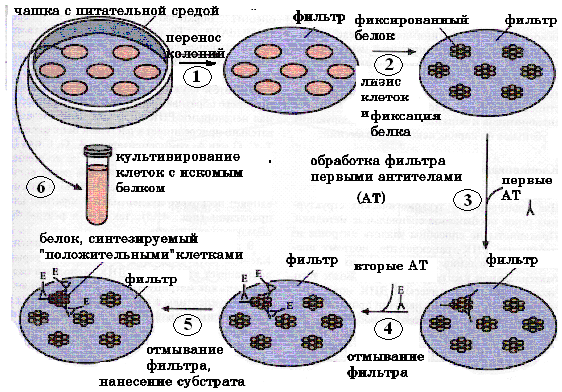
Рис. 52. Скрининг клонов трансформированных клеток E.coli с помощью специфических антител
1-6 – этапы эксперимента (описание см. в тексте)
1. Клетки из каждой выросшей колонии переносят на твердую подложку.
2. Осуществляют лизис клеток и клеточные белки фиксируют на фильтре.
3. Фильтр помещают в раствор, содержащий первые антитела (АТ), которые специфично связываются (преципитируют) только с искомым белком.
4. Несвязавшиеся первые АТ удаляют, наносят на фильтр вторые АТ, специфичные в отношении первых, связанные с ферментом (например, щелочной фосфатазой).
5. Отмывают фильтр от несвязавшихся вторых АТ и помещают в раствор, содержащий субстрат для фермента, при гидролизе которого образуется окрашенный продукт. Гидролиз может произойти только в присутствии вторых антител, к которым присоединен фермент. Таким образом, окрашенные пятна на фильтре будут соответствовать колониям клеток, в которых происходит синтез специфичного белка, т.е. клеткам-трансформантам.
6. Отбирают колонии на чашке, соответствующие окрашенным пятнам на фильтре, и культивируют их. В них может содержаться рекомбинантная ДНК, кодирующая белок, гомологичный первым АТ.