

Вопрос 2:
Структура и функции гемоглобина
Гемоглобины - родственные белки, находящиеся в эритроцитах человека и позвоночных животных. Эти белки выполняют 2 важные функции:
перенос О2 из лёгких к периферическим тканям;
участие в переносе СО2 и протонов из периферических тканей в лёгкие для последующего выведения из организма.
Гемоглобины, так же как миоглобин, относят к гемопротеинам, но они имеют четвертичную структуру (состоят из 4 полипептидных цепей), благодаря которой возникает возможность регуляции их функций.
Гемоглобины взрослого человека
В эритроцитах взрослого человека гемоглобин составляет 90% от всех белков данной клетки.
Гемоглобин А - основной гемоглобин взрослого организма, составляет около 98% от общего количества гемоглобина, тетрамер, состоит из 2 полипептидных цепей
Гемоглобин A2 находится в организме взрослого человека в меньшей концентрации, на его долю приходится около 2% общего гемоглобина. Он состоит из 2
Гемоглобин А1с - гемоглобин А, модифицированный ковалентным присоединением к нему глюкозы (так называемый гликозилированный гемоглобин).
Гемоглобины, синтезирующиеся в период внутриутробного развития плода:
Эмбриональный гемоглобин синтезируется в эмбриональном желточном мешке через несколько недель после оплодотворения. Представляет собой тетрамер . Через 2 нед после формирования печени плода в ней начинает синтезироваться гемоглобин F, который к 6 мес замещает эмбриональный гемоглобин.
Гемоглобин F - фетальный гемоглобин, синтезируется в печени и костном мозге плода до периода его рождения. Имеет тетрамерную структуру, состоящую из 2 -цепей. После рождения ребёнка постепенно замещается на гемоглобин А, который начинает синтезироваться в клетках костного мозга уже на 8-м месяце развития плода.
Гем имеет высокое сродство к оксиду углерода (СО). В водной среде свободный от белковой части гем связывается с СО в 25 000 раз сильнее, чем О2. Высокая степень сродства гема к СО по сравнению с О2 объясняется разным пространственным расположением комплексов Fe2+ гема с СО и О2
В комплексе Fe2+ гема с СО атомы Fe2+, углерода и кислорода расположены на одной прямой, а в комплексе Fe2+ гема с О2 атомы железа и кислорода расположены под углом, что отражает их оптимальное пространственное расположение.
Снижение сродства гемсодержащих белков к СО имеет важное биологическое значение. СО образуется в небольших количествах при катаболизме некоторых веществ, в частности гема. Этот эндогенно образующийся СО блокирует около 1% гемсодержащих белков. Если бы сродство тема к СО не уменьшалось под влиянием белкового окружения, эндогенн
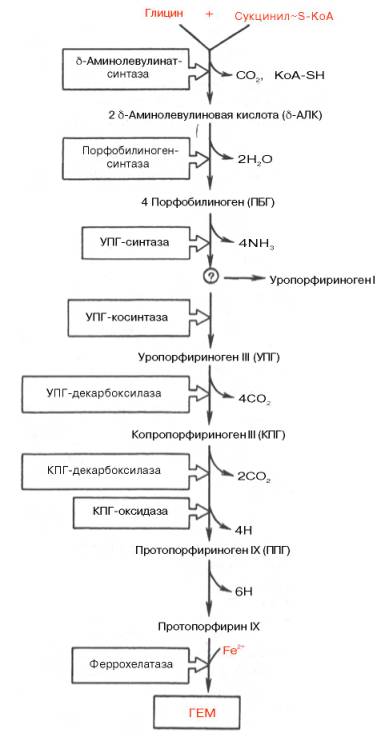
К настоящему времени почти полностью выяснены основные пути образования порфиринов и протопорфиринов, являющихся непосредственными предшественниками гема и хлорофилла. На I стадии, протекающей в 2 этапа, сукцинил-КоА взаимодействует с глицином и образованием δ-аминолевулиновой кислоты (δ-АЛК).
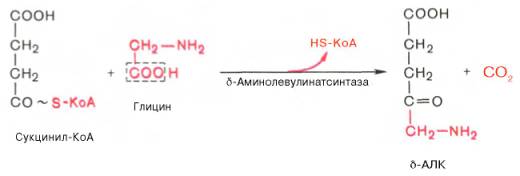
Эту стадию катализирует специфический пиридоксальфосфатзависимый фермент δ-аминолевулинатсинтаза – ключевой, аллостерический фермент синтеза тетрапирролов.
Впервые эта синтаза была обнаружена в эндоплазматической сети клеток печени. Фермент индуцируетсястероидами и другими факторами и ингибируется по типу обратной связи конечным продуктом биосинтеза –гемом.
На II стадии происходит конденсация 2 молекул δ-аминолевулиновой кислоты с образованием первого монопиррольного соединения – порфо-билиногена (ПБГ).
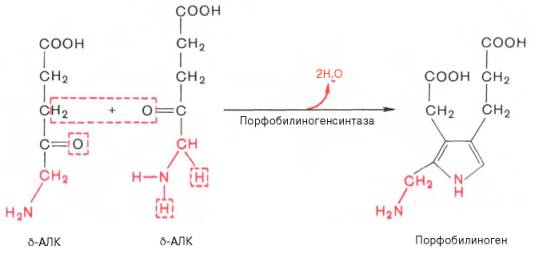
Фермент, катализирующий эту стадию,– порфобилиногенсинтаза также является регуляторным ферментом, подвергаясь ингибированию конечными продуктами синтеза. Предполагают, что механизм этой сложной реакции дегидратации включает образование кетиминной связи (шиффово основание) между кетогруппой одной молекулы δ-аминолевулиновой кислоты и δ-аминогруппой лизина молекулы фермента. В следующей многоступенчатой стадии, катализируемой соответствующими ферментами, из 4 монопиррольных молекул порфобилиногена синтезируется тетра-пиррольный комплекс протопорфирин IX, являющийся непосредственным предшественником гема. Некоторые этапы сложного пути синтеза окончательно не установлены.
В заключительной стадии протопорфирин IX присоединяет молекулу железа при участии феррохелатазы (гемсинтазы), и образуется гем. Последний используется для биосинтеза всех гемсодержащих хромопро-теинов.
Источником железа для этой реакции является ферритин, который считается резервным гемопротеином, откладывающимся в клетках костного мозга, печени и селезенки.
Имеются указания, что, помимо железа, в синтезе гема участвуют некоторые кофакторы, в частности витамин В12, ионы меди, хотя конкретная их роль не раскрыта.
Движущие силы переноса кислорода
- ток крови
- градиент концентраций кислорода между альвеолярным воздухом и межклеточной жидкостью.
В альвеолярном воздухе парциальное давление равно 100 мм. рт. ст. В межклеточной жидкости парциальное давление равно 35 мм. Рт. ст.;
разность концентраций в 65 мм.рт.ст., которая обеспечивает переход кислорода из альвеол в кровь, а из крови - в межклеточную жидкость.
Гемоглобин крови при парциальном давлении, которое имеется в альвеолярном воздухе, насыщается кислородом на 97%.
В венозной крови степень насыщения гемоглобина кислородом равна 64%. При изменении степени насыщения на 33% освобождается 6,6 мл кислорода – такое количество кислорода приносят тканям каждые 100 мл крови.
Транспорт углекислого газа
Примерно 15% углекислого газа, присутствующего в крови, переносится молекулами гемоглобина. Связывание кислорода тесно сопряжено с выдыханием углекислого газа. Это обратимое явление известно как эффект Бора
Гемоглобинопатии (haemoglobinopathia, единственное число; гемоглобин + греческий pathos страдание, болезнь; синонимы гемоглобиноз) — группа наследственных заболеваний, обусловленных наличием в эритроцитах аномальных гемоглобинов либо угнетением синтеза полипептидных цепей нормальных гемоглобинов. К Гемоглобинопатии причисляют как выраженные патологический состояния, протекающие чаще с гемолитической анемией (смотри), так и многочисленные случаи латентного носительства аномальных гемоглобинов или генов талассемии
К наиболее часто встречающимся и известным гемоглобинопатиям относятся серповидно-клеточная анемия, бета-талассемия, персистенция фетального гемоглобина. Гемоглобинопатии классифицируются на качественные и количественные. Качественные обусловлены заменой аминокислот в полипептидных цепях. Замена аминокислоты глутамина 6 на валин в β-цепи приводит к образованию аномального гемоглобина S, что лежит в основе развития серповидно-клеточной анемии. Аномальных гемоглобинов более 300, но не все аномалии проявляются. Первые аномальные гемоглобины назывались буквами латинского алфавита (М, С, Д, S и др.). Но, так как аномальных гемоглобинов много, их названия включают места открытия (Boston, Москва, Волга и др.) или названия госпиталей. Количественные гемоглобинопатии связаны со скоростью синтеза α- или β-полипептидных цепей глобина. Угнетение скорости синтеза α-цепи приводит к развитию α-талассемии, угнетение синтеза β-цепи лежит в основе заболевания β-талассемии. Гемоглобинопатии —наследственные заболевания. Диагностика гемоглобинопатий основывается, кроме клинических данных, на обязательном специальном исследовании электрофорезе гемоглобина. Это исследование проводится не только для больного, но и для ближайших родственников. Данные электрофореза гемоглобина позволяют поставить диагноз талассемии. Для альфа-талассемии характерно обнаружение гемоглобинов-гомотетрамеров Нв-Н и Нв-Bart.Для бета-талассемии характерно повышенное содержание гемоглобина Α2.
Порфирия
наследственное нарушение синтеза гема с повышенным содержанием порфиринов в крови и тканях и усиленным их выделением с мочой и калом. Проявляется фотодерматозом, гемолитическими кризами, желудочно-кишечными и нервно-психическими расстройствами.