

4. Программа визуализации RasMol
Запуск программы и окна
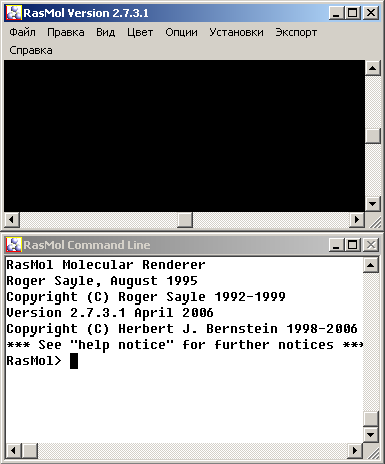
Программа RasMol считывает с определенным образом записанного файла координаты атомов и создает их графическое изображение. Программа функционирует в режиме двух окон - графического и текстового (рис.1). В графическом окне происходит визуализация структур макромолекул, в текстовое окно вводится управляющая информация, которая позволяет изменять масштаб рисунка, цвет, представление молекул, выделять группы атомов, остатков, белковых цепей. Управляющие данные вводятся в виде текстовых команд, описание которых приведено на странице помощи. Графическое окно по умолчанию имеет черный фон. В верхней части этого окна располагается панель меню, имеющая следующие раскрывающиеся меню: 'Файл', 'Правка', 'Вид', 'Цвет', 'Опции', 'Установки', 'Экспорт' и 'Справка'. Кроме этого, основное окошко имеет две полосы прокрутки, которые используются для поворота изображения вокруг вертикальной и горизонтальной осей. Команды могут быть введены с клавиатуры в текстовом окне, даже если активным является графическое окно. Это позволяет вводить команды и менять изображения без переключения окон. Для чтения координатного файла можно пользоваться командой «Открыть» меню «Файл». Можно также открыть PDB файл двойным нажатием на него.
|
Рис.л1.1. Графическое и текстовое окна программы RasMol |
Манипуляции изображением
Вращение молекулы осуществляется передвижением мыши при нажатой левой кнопке или при помощи полос прокрутки справа и внизу. Вращение в плоскости рисунка осуществляется при нажатой правой кнопке мыши с удержанием клавиши Shift. Движение мыши при нажатой левой кнопке и удерживаемой клавише Shift изменяет масштаб изображения.
Для удобства эти функции описаны в приведенной ниже таблице.
Левая кнопка |
Поворот X-Y |
Правая кнопка |
Перемещение X-Y |
Shift+Левая кнопка |
Увеличение |
Shift+Правая кнопка |
Поворот Z |
|
|
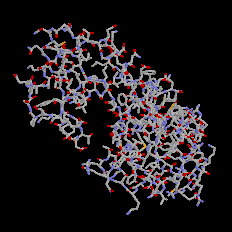
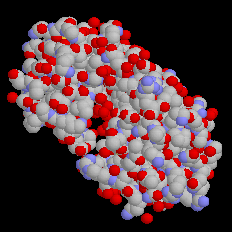
С помощью меню «Вид» можно выбрать форму представления изображения (рис.2). Наиболее используемыми при визуализации кристаллов являются команды «Ван-дер-Ваальсов радиус» и «Атомы и связи». Первая команда заполняет окно атомами в соответствии с их радиусами. При использовании второй команды атомы представляются в виде сфер небольшого диаметра, с пустым пространством между ними. При этом если расстояние между двумя атомами меньше некоторого значения, между ними рисуется связь в виде прямолинейного отрезка.
|
Рис. л1.2. Различные формы представления одной структуры: «Связи», «Ван-дер-Ваальсов радиус», «Атомы и связи» |

5. Построение исходной структуры для моделирования
Для первичного
освоения методов моделирования в данной
работе рассматривается модель идеальной
г.ц.к. решетки металлов. Для построения
исходной структуры используется
программа fcc.exe,
которая позволяет построить расчетную
ячейку г.ц.к. решетки с заданным значением
параметра. Принцип построения исходной
структуры решетки, как и любой другой
системы, достаточно прост,‑ он
заключается в заполнении расчетной
ячейки атомами. При наличии дефектов
это построение может представлять
технически сложную задачу, которая для
каждого случая решается по-своему. В
случае идеальной решетки построение
наиболее просто. Сначала определяются
координаты четырех атомов одной
элементарной ячейки кристалла. Один из
них может лежать в начале координат
(0,0,0), тогда три остальных атома будут
иметь координаты соответственно
![]() ,
,
![]() и
и
![]() .
Координаты всех остальных атомов
расчетной ячейки получаются путем
переноса этих точек на векторы трансляции
решетки.
.
Координаты всех остальных атомов
расчетной ячейки получаются путем
переноса этих точек на векторы трансляции
решетки.
Поскольку программы
молекулярной динамики, в том силе XMD,
требуют, чтобы размеры расчетной ячейки
в каждом направлении превышали удвоенный
радиус обрезания потенциала, невозможно
проводить моделирование системы,
содержащей только одну элементарную
ячейку. В каждом направлении размер
расчетной ячейки Li
должен быть кратным параметру решетки
a0:
![]() (i=1,2,3).
Поскольку
радиус обрезания потенциала обычно
составляет величину около 4-5 Å, а параметр
решетки – примерно 3.5 Å, ki
должны быть не менее 4.
(i=1,2,3).
Поскольку
радиус обрезания потенциала обычно
составляет величину около 4-5 Å, а параметр
решетки – примерно 3.5 Å, ki
должны быть не менее 4.
Программа fcc.exe строит г.ц.к. решетку так, что по осям x,y,z направлены кристаллографические оси [100], [010] и [001], соответственно. Возможен выбор трех размеров расчетной ячейки в единицах периода решетки в соответствующем направлении, то есть чисел ki. Подробная информация о работе этой программы содержится в пояснениях к ней.
Результатом работы программы является координатный файл fcc.in, в котором в формате, требующемся для ввода в XMD, записаны размеры расчетной ячейки и координаты всех атомов:
box 0.35200E+02 0.35200E+02 0.35200E+02
position 4000
1 0.35200E+00 0.35200E+00 0.35200E+00
1 0.21120E+01 0.21120E+01 0.35200E+00
..............................................