

Наследственный нефрит
Наследственный нефрит – наследственное поражение гломерулярных базальных мембран, проявляющееся гематурией, прогрессирующей протеинурией, падением почечных функций и часто сочетающееся с нейросенсорной тугоухостью, глазными аномалиями. Сочетание наследственного нефрита с нейросенсорной тугоухостью называют синдромом Альпорта.
В основе заболевания лежат мутации генов 13, 2 и Х–хромосомы, кодирующих цепи коллагена 4 типа, которые находятся во всех базальных мембранах, экспрессируются в ГБМ, мембранах кохлеарного аппарата, передней капсуле хрусталика. Тканевое распределение цепей коллагена 4 типа в человеческих тканях изучается с помощью применения моноспецифических антител.
В настоящее время выделяют три молекулярно–генетические формы данного заболевания:
– Х–сцепленную доминантную;
– аутосомно–рецессивную;
– аутосомно–доминантную.
Х–сцепленный вариант выявляется у 80% больных. У мальчиков заболевание протекает наиболее тяжело, c прогрессированием до хронической почечной недостаточности и глухоты в течение второй и третьей декад жизни. Тяжесть заболевания у гетерозиготных девочек зависит от степени инактивации интактной Х–хромосомы.
Сравнительный анализ аутосомно–доминантного и Х–сцепленного доминантного вариантов показал существенно лучший прогноз у больных с аутосомно–доминантной передачей (средняя продолжительность жизни пациентов 51 год и 25 лет, соответственно).
Аутосомно–рецессивный вариант следует подозревать, когда имеются типичные клинико-патологические особенности заболевания, но отсутствует позитивный семейный анамнез. Хроническая почечная недостаточность и нейросенсорная тугоухость у больных развиваются к 30 годам.
Пример родословной с наследственным нефритом
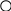

I

I I

I II
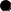
- нефропатия, - тугоухость, - синдром Альпорта.
Клинические особенности наследственного нефрита
Основным признаком заболевания является гематурия. Гематурия может выявляться при рождении и в первые годы жизни у мальчиков. Мужчины имеют персистирующую микрогематурию с эпизодами макрогематурии, проявляющейся часто при респираторных инфекциях, в течение первых 20 лет. Женщины, которые являются гетерозиготами при Х–сцепленной форме, чаще имеют интермиттирующую гематурию. Гематурия персистирует как у мужчин, так и у женщин при аутосомно-рецессивной форме.
Протеинурия обычно отсутствует в течение первых лет жизни, но впоследствии проявляется у мужчин с Х–сцепленным наследственным нефритом и у обоих полов при рецессивной форме. Она увеличивается с возрастом и может достигать нефротического синдрома. Выраженная протеинурия относительно редка у женщин, которые являются гетерозиготами при Х–сцепленной мутации.
Сочетание протеинурии с гипертензией наиболее часто выявляется у мужчин с Х–сцепленной формой заболевания, но, по-видимому, не зависит от половых различий при аутосомно–рецессивной форме. Гипертензия усугубляется с возрастом.
Хроническая почечная недостаточность развивается у всех мальчиков с X–сцепленным наследственным нефритом, но скорость её прогрессии существенно варьирует. Прогноз для больных девочек с Х–сцепленной формой обычно благоприятен. Многие из них очень длительное время находятся в функционально–компенсированной стадии. При аутосомно-рецессивном варианте хроническая почечная недостаточность определяется у всех больных во вторую и третью декады жизни.
Критерии диагностики синдрома Альпорта базируются на определении, по крайней мере, трёх из четырёх основных признаков:
1) семейной гематурии, с прогрессией до почечной недостаточности и без неё;
2) утолщенной и расщеплённой ГБМ;
3) наличия специфического поражения глаз;
4) нейросенсорной тугоухости.
Анамнестические данные
Характерно наличие у родственников подобного заболевания, патологии слуха, зрения. У некоторых нефрит сочетается с невритом слухового нерва; в акушерском анамнезе - спонтанные аборты, мертворождения, нефропатия беременных, патологическое течение беременности. Среди соматических заболеваний преобладают сердечно-сосудистые.
Клинические данные:
Экстраренальные проявления характеризуются
стигмами дизэмбриогенеза: гипертелоризм глаз и сосков молочных желез, высокое (готическое) небо, аномалии прикуса, диспластический рост зубов, аномальная форма ушных раковин и т.д.; часто имеет место сочетание нескольких диспластических признаков у одного больного;
снижением слуха различной степени выраженности;
изменениями со стороны органа зрения: аномалия хрусталика – сферофакия, лентиконус передний и задний, разнообразные катаракты, миопия;
поражениями нервной системы – тремор, миастения, нарушения памяти, снижение интеллекта и др.;
симптомами интоксикации (бледность, слабость, утомляемость, головные боли).
Отечный синдром не характерен, но возможно острое начало с олигурией и отечным синдромом.
Гипертензионный синдром
артериальная гипертония развивается при прогрессировании патологического процесса и снижении функциональной способности почек;
сосудистая гипотония более свойственна начальному периоду заболевания.
Мочевой синдром проявляется преимущественно гломерулярной эритроцитурией, нарастающей при прогрессировании заболевания. Протеинурия и лейкоцитурия наблюдаются реже. Стойкая и значительная протеинурия свидетельствует о прогрессировании патологического процесса. При анализе экскреторных урограмм определяется тенденция к уменьшению размеров почек у детей старшего возраста.
Синдром обменных нарушений проявляется снижением функций проксимального и дистального отделов нефрона, снижением концентрационной способности, при прогрессировании заболевания – водно-электролитным дисбалансом, метаболическим ацидозом, азотемией, анемией и др. При нарастающей динамике развивается полный комплекс ХПН.
Таблица 21
Классификация наследственного нефрита (M. Gubber, R. Habib, 1988)
Вариант |
Тип наследования |
Особенности клинического проявления |
Течение |
Исход |
Первый (с-м Альпорта) |
Доминантный |
Нефрит с гематурией, тугоухостью, поражением глаз |
Прогрессирующее |
ХПН |
Второй |
Доминантный |
Без тугоухости |
Прогрессирующее |
ХПН |
Третий |
Аутосомно-рецессивный |
Семейная доброкачественная гематурия |
Торпидное |
Благоприятный |
Морфологические изменения
При световой микроскопии или прямой иммунофлюоресценции не находят патогномоничных повреждений. Для диагноза может иметь значение непрямая иммунофлюоресценция базальных мембран почек и кожи. Электронная микроскопия наиболее часто выявляет диагностические особенности поражения у пациентов как с нарушением слуха и поражением глаз, так и при их отсутствии.
Патогномоничной структурной особенностью почек при наследственном нефрите является утолщение ГБМ с трансформацией плотного слоя, в котором встречаются чистые электронно–прозрачные поля, содержащие круглые гранулы различной плотности (от 20 до 90 nm в диаметре). Происхождение этих гранул неизвестно, но предполагается, что они представляют собой дегенеративные островки цитоплазмы висцеральных эпителиальных клеток.
Даже в отсутствие выраженной протеинурии часто находят расплавление ножек подоцитов.
Не все больные с наследственным нефритом имеют характерные ультраструктурные особенности. Описываются толстые, тонкие, нормальные и неспецифически поврежденные ГБМ.
Хотя диффузное истончение ГБМ наиболее свойственно доброкачественной семейной гематурии, у некоторых пациентов с этим повреждением возможна прогрессия почечного заболевания и мутация генов коллагена 4 типа.
Экспансия кортикального интерстиция и интерстициальный фиброз редко описываются у мальчиков до 10–летнего возраста.