

7226.1.7. Дайте визначення поняття "анемія".
Анемія — це гематологічний синдром або самостійне захворювання, що характеризується зменшенням кількості еритроцитів і (або) вмісту гемоглобіну в одиниці об'єму крові, а також якісними змінами еритроцитів.
26.1.9. Як класифікують анемії? Наведіть приклади кожного виду анемій.
I. Патогенетична класифікація:
1) постгеморагічні анемії (наприклад, анемія після гострої крововтрати);
2) гемолітичні анемії (наприклад, серпоподібноклітинна);
3) анемії, обумовлені порушеннями еритропоезу (наприклад, залізодефіцитна).
II. За етіологією:
1) спадкові (наприклад, талассмія);
2) набуті (наприклад, хронічна постгеморагічна анемія).
III. За регенеративною здатністю червоного кісткового мозку:
1) регенераторні (наприклад, гостра постгеморагічна анемія);
2) гіперрегенераторні (наприклад, набута гемолітична анемія);
3) гіпорегенераторні (наприклад, залізодефіцитна анемія);
4) арегенераторні (наприклад, апластична анемія).
IV. За колірним показником (КП):
1) нормохромні (КП = 0,85-1; наприклад, гостра постгеморагічна анемія в перші кілька діб після крововтрати);
2) гіпохромні (КП < 0,85; наприклад, залізодефіцитна анемія);
3) гіперхромні (КП > 1; наприклад, ВІ2-фолієводефіцитна анемія).
V. За типом кровотворення:
1) анемії з еритробластичним типом кровотворення (наприклад, залізодефіцитна анемія);
2) анемії з мегалобластичним типом кровотворення (наприклад, В|2-фолієво-дефіцитна анемія).
VI. За клінічним перебігом:
1) гострі (наприклад, анемія після гемотрансфузійного шоку);
2) хронічні (наприклад, гіпопластична анемія).
26.1.11. Що таке постгеморагічні анемії? Як їх класифікують?
Постгеморагічна анемія - це анемія, що розвивається в результаті крововтрати.
Залежно від характеру крововтрати виділяють два види анемій цієї групи: 1) гостру постгеморагічну і 2) хронічну постгеморагічну анемію.
Гостра постгеморагічна анемія виникає після швидкої масивної крововтрати при пораненні судин або їх ушкодженні патологічним процесом.
Хронічна постгеморагічна анемія розвивається внаслідок повторних, часто невеликих крововтрат, викликаних ураженням кровоносних судин при деяких хворобах (дисменорея, виразкова хвороба шлунка, геморой та ін.) і порушенням судин-но-тромбоцитарного і коагуляційного гемостазу (геморагічний діатез). Втрата заліза при частих кровотечах надає цій анемії залізодефіцитний характер.
26.1.14. Опишіть картину крові при хронічній постгеморагічній анемії.
У зв'язку із втратою заліза при частих кровотечах розвиваються гематологічні ознаки залізодефіцитної анемії: зменшується концентрація гемоглобіну й колірний показник, у мазку крові з'являються дегенеративні форми еритроцитів (мікро- і по-йкілоцитоз, гіпохромія). Кількість еритроцитів і гематокрит можуть залишатися без змін.
26.1.19. Назвіть можливі причини й основні механізми внутрішньосудинного гемолізу еритроцитів.
Внутрішньосудиннш гемоліз виникає в кровоносних судинах унаслідок дії факторів, що ушкоджують еритроцити. Ці фактори отримали назву гемолітичних. До них відносять:
а) фактори фізичної природи (механічна травма, іонізуюча радіація, ультразвук, температура);
б) хімічні агенти (гемолітичні отрути);
в) біологічні фактори (збудники інфекційних захворювань, токсини, ферменти);
г) імунні фактори (антитіла).
Механізми внутрішньосудинного гемолізу.
I. Механічний гемоліз. Виникає внаслідок механічного руйнування мембран еритроцитів, наприклад, при роздавлюванні еритроцитів у судинах стопи (маршовий гемоліз).
II. Осмотичний гемоліз. Виникає тоді, коли осмотичний тиск усередині еритроцита більший, ніж осмотичний тиск плазми крові. У цьому випадку вода за законами осмосу надходить в еритроцит, об'єм його зростає, і в кінцевому підсумку відбувається розрив мембрани. Причиною осмотичного гемолізу може бути або зменшення осмотичного тиску середовища, у якому перебувають еритроцити (гіпотонічні розчини), або збільшення осмотичного тиску в самих еритроцитах. Останнє, як правило, пов'язане зі збільшенням концентрації іонів натрію усередині еритроцитів у результаті підвищення проникності їх мембрани або внаслідок порушення роботи Na-K-насосів.
III. Окисний гемоліз. Розвивається внаслідок вільнорадикального окиснення ліпідів і білків плазматичної мембрани еритроцитів. Результатом цього є збільшення проникності еритроцитарної мембрани, що потім веде до реалізації осмотичного механізму гемолізу.
IV. Детергентний гемоліз. Пов'язаний з розчиненням ліпідних компонентів мембрани еритроцитів речовинами-детергентами. Цей вид гемолізу викликають жовчні кислоти (холемічний синдром), жиророзчинні хімічні агенти, деякі токсини бактерій (лецитинази).
V. Комплементзалежний гемоліз. Обумовлений руйнуванням (перфорацією) мембрани еритроцитів активним комплементом. Цей механізм лежить в основі імунного гемолізу.
7326.3. Назвіть види порушень загального об'єму крові. Наведіть приклади.
Порушення об'єму крові виявляють себе гіповолемієїо або гіперволемією — зменшенням або збільшенням об'єму крові, якщо порівнювати з нормою (нормоволемією).
Гіпо- і гіперволемію поділяють на просту (зберігається нормальне співвідношення плазми і клітин крові), поліцитемічну (переважають клітини крові) і олігоци-темічну (переважає плазма).
Крім того, до порушень об'єму крові відносять зміни об'ємного співвідношення між клітинними елементами і плазмою при нормальному загальному об'ємі крові — оліго- і поліцитемічну нормоволемію (гемодилюція і гемоконцентрація). Показником об'ємного співвідношення є гематокрит, що визначає вміст клітинних елементів (переважно еритроцитів) у загальному об'ємі крові (у нормі 0,36-0,48, або 36-48 %).
Гіповолемія проста — зменшення об'єму крові без зміни гематокриту. Виникає відразу після гострої крововтрати і зберігається доти, доки рідина не перейде із тканини в кров.
Гіповолемія олігоцитемічна — зменшення об'єму крові з переважним зменшенням у ній клітин - еритроцитів. Спостерігається при гострій крововтраті в тих випадках, коли надходження крові й тканинної рідини в кровоносне русло не компенсує об'єм і особливо склад крові.
Гіповолемія поліцитемічна—зменшення об'єму крові внаслідок зменшення об'єму плазми при відносному збільшенні вмісту еритроцитів. Розвивається при зневодненні організму (пронос, блювота, посилене потовиділення, гіпервентиляція), шоку (вихід рідини в тканини внаслідок підвищення проникності стінок судин).
Гіперволемія проста— збільшення об'єму крові при збереженні нормального співвідношення між еритроцитами і плазмою. Виникає відразу ж після переливання великої кількості крові. Однак незабаром рідина виходить з кровоносного русла, а еритроцити залишаються, що веде до згущення крові. Проста гіперволемія при посиленій фізичній роботі обумовлена надходженням у загальний кровообіг крові з депо.
Гіперволемія олігоцитемічна — збільшення об'єму крові за рахунок плазми. Розвивається при затримці води в організмі у зв'язку із захворюваннями нирок, при вве-
денні кровозамінників. її можна моделювати в експерименті шляхом внутрішньовенного введення тваринам ізотонічного розчину натрію хлориду.
Гіперволемія поліцитемічна — збільшення об'єму крові за рахунок наростання кількості еритроцитів. Спостерігається при зниженні атмосферного тиску, а також при різних захворюваннях, пов'язаних з кисневим голодуванням (вади серця, емфізема), її розглядають як компенсаторне явище.
Однак при еритремії поліцитемічна гіперволемія є наслідком пухлинного розростання клітин еритроцитарного ряду кісткового мозку.
Олігоцитемічна нормоволемія виникає при анемії внаслідок крововтрати (об'єм крові нормалізувався за рахунок тканинної рідини, а кількість еритроцитів ще не відновилася), при гемолізі еритроцитів, порушеннях гемопоезу.
Поліцитемічна нормоволемія спостерігається при переливанні невеликих кількостей еритроцитарної маси.
26.5. Що таке крововтрата? Які причини викликають крововтрату? Від чого залежать її перебіг і завершення?
Крововтрата - це патологічний процес, що виникає внаслідок кровотечі і характеризується складним комплексом патологічних порушень і компенсаторних реакцій, спрямованих проти зменшення об'єму циркулюючої крові й гіпоксії, обумовленої зниженням дихальної функції крові.
До етіологічних факторів, що викликають кровотечу, відносять:
1) порушення цілісності судин при пораненні або ураженні патологічним процесом (атеросклероз, пухлина, туберкульоз);
2) підвищення проникності судинної стінки (гостра променева хвороба);
3) зниження зсідання крові (геморагічний діатез).
Перебіг і результат крововтрати залежать від особливостей самої кровотечі (швидкості, величини, виду ушкодженої судини, механізму ушкодження); швидкості включення й вираженості компенсаторних реакцій організму; статі, віку, станів, що передують і супроводжують крововтрату (охолодження, травма, захворювання серця, глибокий наркоз). Серйозну небезпеку для життя людини являє втрата 50 % об'єму циркулюючої крові, смертельною є втрата крові понад 60 %.
26.6. Які стадії умовно виділяють у патогенезі гострої крововтрати?
І. Початкова стадія. Характеризується зменшенням об'єму циркулюючої крові -простою гіповолемією, зниженням артеріального тиску, гіпоксією переважно циркуляторного типу.
II. Компенсаторна стадія. Обумовлена здійсненням комплексу захисно-компенсаторних реакцій, спрямованих на ліквідацію наслідків втрати крові.
III. Термінальна стадія. Характеризується наростанням патологічних змін в організмі аж до настання смерті. Розвивається при недостатності компенсаторних реакцій, а також при інтенсивній і швидкій крововтраті, на тлі дії несприятливих факторів (охолодження, велика травма, серцево-судинні захворювання) і при відсутності лікувальних заходів.
26.7. Що є головною ланкою патогенезу гострої крововтрати?
У патогенезі гострої крововтрати центральне місце посідають порушення загальної гемодинаміки — зменшення об 'єму циркулюючої крові (гіповолемія) і обумовлене цим падіння артеріального тиску (гіпотензія) (рис. 94).
Зазначені порушення, з одного боку, є причиною розвитку власне патологічних змін в організмі (анемії, гіпоксії, ацидозу, інтоксикації), з другого- вмикають складний комплекс рефлекторних і гуморальних захисно-компенсаторних реакцій, спрямованих на відновлення й збереження гомеостазу.

Рис. 94. Головна ланка патогенезу гострої крововтрати
26.13. Що таке геморагічний шок? Назвіть головні патогенетичні ланки його розвитку.
Геморагічний шок — це шок, що виникає в результаті інтенсивної гострої крововтрати (рис. 96).
Провідним механізмом його розвитку є зменшення об 'єму циркулюючої крові, що викликає падіння
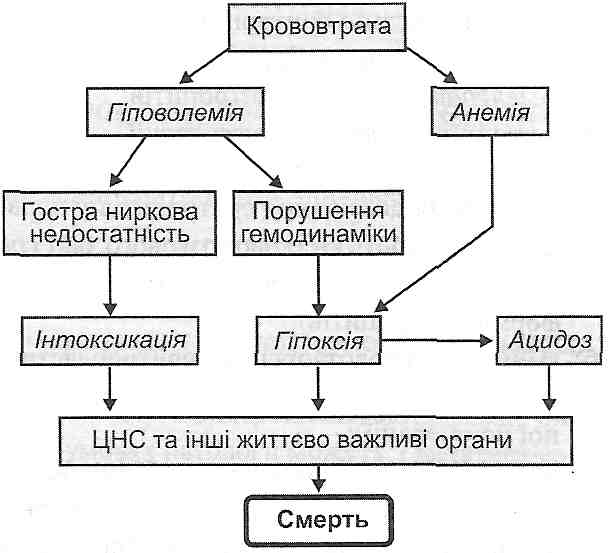
Рис. 96. Схема патогенеза геморагічного шоку
артеріального тиску, порушення мікроциркуляції, розлади кровопостачання життєво важливих органів (головного мозку, серця, нирок). Наслідком цього є розвиток гіпоксії, ацидозу й інтоксикації, що погіршує перебіг шоку, створює "зачаровані кола" у його патогенезі і в кінцевому підсумку веде до смерті.
7426.1.24. Які причини можуть викликати розвиток набутої гемолітичної анемії?
Залежно від причин розвитку виділяють такі види набутої гемолітичної анемії.
I. Анемії, обумовлені механічним ушкодженням еритроцитів.
II. Імунні гемолітичні анемії.
III. Токсичні гемолітичні анемії.
IV. Інфекційні гемолітичні анемії.
V. Набуті мембранопатії.
26.1.25. Наведіть приклади анемій, обумовлених механічним ушкодженням еритроцитів.
1. Механічний гемоліз при протезуванні судин або клапанів серця.
2. "Маршова " гемоглобінурія — травматизація еритроцитів у капілярах стоп під час тривалого маршу.
3. Мікроангіопатична гемолітична анемія (хвороба Мошковича) — травматизація еритроцитів при зіткненні їх з нитками фібрину. Буває при ДВЗ-синдромі.
26.1.26. Що таке імунні гемолітичні анемії? Назвіть можливі причини.
Імунні гемолітичні анемії—це анемії, що виникають за участі специфічних імунних механізмів. Вони обумовлені взаємодією гуморальних антитіл з антигенами, фіксованими на поверхні еритроцитів, і тому є проявом II типу алергічних реакцій за класифікацією Кумбса і Джелла (див. розд. 10).
Залежно від причин розвитку виділяють такі види імунних гемолітичних анемій:
1. Алоімунні (ізоімунні) гемолітичні анемії. їх причиною можуть бути: а) надходження ззовні антитіл проти власних еритроцитів (гемолітична хвороба новонароджених) і б) надходження в організм еритроцитів, проти яких у плазмі є антитіла (переливання крові, не сумісної за групами АВО або Rh).
2. Аутоімунні гемолітичні анемії. Обумовлені утворенням в організмі антитіл проти власних еритроцитів. Це може бути пов'язано або з первинними змінами самих еритроцитів (поява аутоантигенів), або зі змінами в імунній системі (скасування імунологічної толерантності, поява "заборонених" клонів лімфоцитів).
3. Гетероімунні (гаптенові) гемолітичні анемії. Виникають при фіксації на поверхні еритроцитів чужорідних антигенів (гаптенів), зокрема, лікарських препаратів (пеніцилін, сульфаніламіди), вірусів.
26.1.27. Що таке гемолітична хвороба новонароджених?
Гемолітична хвороба новонароджених - це хвороба, що виникає в результаті гемолізу еритроцитів плода й новонародженого, викликаного антитілами матері.
Найчастіше бувають два варіанти гемолітичної хвороби новонароджених: резус-конфлікт і АВО-конфлікт.
Резус-конфлікт. Розвивається у випадку вагітності Шг-матері Ші+-плодом (найчастіше при повторній вагітності). Спочатку відбувається імунізація матері Rh+-epn-троцитами плода, які можуть потрапляти в організм матері під час пологів або при
дефектах плаценти. Найбільш імовірною є імунізація під час пологів, тому резус-конфлікт виникає найчастіше в умовах повторної вагітності Кп+-плодом.
У відповідь на надходження Ші+-еритроцитів в організмі матері синтезуються антитіла проти D-антигену. Ці антитіла (Ig G) здатні проникати через плаценту в організм плода й викликати гемоліз його еритроцитів.
АВО-конфлікт. Найчастіше виникає в ситуаціях, коли мати має групу крові 0(1), а плід - А(ІІ) або В(III). Нормальні ізоаглютиніни в системі АВО належать до класу IgM. Ці антитіла не проникають через плаценту й тому не можуть бути причиною АВ0-конфлікту. Однак у 10 % здорових людей, що мають групу крові 0(1), є антитіла проти аглютиногенів А і В, представлені IgG. Наявність цих антитіл не залежить від попередньої імунізації. Аглютиніни IgG проникають через плаценту і можуть викликати гемоліз еритроцитів плода з групами крові А(П), В(Ш). Серед дітей-первістків гемолітична анемія як результат АВО-конфлікту буває з такою ж частотою, як і у дітей, народжених після других, третіх і наступних пологів, на відміну від резус-конфлікту, при якому частота гемолітичної анемії збільшується зі збільшенням кількості пологів.
26.1.29. Що може бути причиною розвитку токсичної гемолітичної анемії?
Токсичну гемолітичну анемію можуть викликати: а) екзогенні хімічні агенти: миш'яковистий водень, свинець, солі міді, фенілгідра-зин, резорцин та ін.;
б) ендогенні хімічні фактори: жовчні кислоти, продукти, що утворюються при сш вій хворобі, уремії;
в) отрути біологічного походження: зміїна, бджолина, отрута деяких видів паву.
26.1.30. Назвіть можливі причини розвитку інфекційних гемолітичних анемій.
Гемолітичну анемію викликає цілий ряд інфекційних агентів, зокрема, гем тичнйй стрептокок, малярійний плазмодій, токсоплазма, лейшманії.
Причиною гемолізу при інфекційних захворюваннях можуть бути або розм ження збудників в еритроцитах (малярійний плазмодій), або дія токсинів-гемолїзж (гемолітичний стрептокок).
75 II. За причинами виникнення виділяють такі групи анемій:
а) мієлотоксичні. Розвиваються внаслідок ушкодження кровотворних клітин під дією екзогенних (радіація, хімічні агенти, віруси) і ендогенних факторів (імунні фактори, токсичні продукти обміну речовин);
26.1.56. Назвіть можливі причини розвитку залізодефіцитної анемії.
1. Недостатнє надходження заліза в організм:
а) аліментарна анемія в грудних дітей (вигодовування коров'ячим або козячим молоком);
б) порушення всмоктування заліза (резекція шлунка, кишок, гастрити, ентерити).
2. Крововтрати. Це найпоширеніша причина дефіциту заліза в організмі. Найчастіше буває при невеликих, але повторних кровотечах (хронічна постгеморагічна анемія).
3. Посилене використання заліза - вагітність, лактація.
26.1.57. Який патогенез порушень, що розвиваються в організмі у зв 'язку з дефіцитом заліза ?
Недостатність заліза в організмі призводить до порушення синтезу залізовмісних білків, а отже, до розладів функцій, у виконанні яких беруть участь ці білки. Найбільше значення мають такі лінії патогенезу:
1) дефіцит заліза → порушення синтезу гема і гемоглобіну → анемія;
2) дефіцит заліза → порушення синтезу гема → порушення утворення цитохро-мів → розлади клітинного дихання (порушення утилізації кисню) → тканинна гіпоксія;
3) дефіцит заліза → порушення синтезу гема → зменшення активності каталази → порушення функції антиоксидантних систем → активація вільнорадикального окиснення → ушкодження клітин → гемоліз еритроцитів і розвиток дистрофічних змін у клітинах;
4) дефіцит заліза → порушення синтезу гема → зменшення синтезу міоглобіну → зниження пристосувальних можливостей клітин щодо гіпоксії.
26.1.58. Дайте характеристику картини периферичної крові і червоного кісткового мозку при залізодефіцитній анемії.
Для залізодефіцитної анемії характерне зниження концентрації гемоглобіну в периферичній крові і зменшення колірного показника, що свідчить про зменшення насичення кожного окремого еритроцита гемоглобіном. Кількість еритроцитів в одиниці об'єму крові при цьому або трохи зменшується, або залишається без змін.
При біохімічних дослідженнях виявляється зниження вмісту заліза в сироватці крові, а також ступеня насичення залізом трансферину.
У мазку крові зменшується кількість регенераторних форм еритроцитів (ретику-лоцитів, поліхроматофілів) і з'являються дегенеративні форми. Характерні гіпохромія (з'являються анулоцити), анізоцитоз (мікроцитоз), пойкілоцитоз.
У червоному кістковому мозку зменшується вміст сидеробластів і сидероцитів (еритробластів і нормоцитів, що містять гранули заліза), частка яких у нормі становить 20-40 %. У той же час збільшується вміст базофільних і поліхроматофільних форм клітин еритроїдного ряду при одночасному зменшенні оксифільних. Зазначений феномен отримав назву "синього кісткового мозку ".
26.1.59. Визначте місце залізодефіцитної анемії в різних класифікаціях анемій.
|
Класифікація |
Залізодефіцитна анемія |
І. |
За патогенезом |
Анемія, пов'язана з порушеннями еритропоезу |
II. |
За етіологією |
Набута |
III. |
За регенераторною здатністю червоного кісткового мозку |
Гіпорегенераторна |
IV. |
За колірним показником |
Гіпохромна |
V. |
За типом кровотворення |
3 еритробластичним типом кровотворення |
VI. |
За клінічним перебігом |
Хронічна |
26.1.60. Якими синдромами виявляє себе залізодефіцитна анемія?
1. Гематологічний синдром. Охоплює порушення з боку периферичної крові і червоного кісткового мозку.
2. Гіпоксія. Виявляється загальною слабкістю, запамороченнями, серцебиттям, задишкою, непритомностями. Кисневе голодування в умовах дефіциту заліза має два механізми: кров'яний (зменшення кисневої ємності крові) і тканинний (порушення клітинного дихання й утилізації кисню).
3. Синдром трофічних порушень. Виявляється такими ознаками, як сухість і тріщини шкіри, тріщини в кутах рота, ангулярний стоматит, ураження нігтів (зміни форми, стоншення), атрофія сосочків язика (атрофічний глосит), атрофічний гастрит. Вважають, що розвиток зазначених порушень, з одного боку, пов'язаний з гіпоксичним і вільнорадикальним ушкодженням клітин, з другого — з розладами вторинних метаболічних шляхів, у здійсненні яких беруть участь ферменти, що містять залізо.
4. Сидеропенічний синдром. Це специфічний синдром, що виникає при дефіциті заліза. Він виявляється спотвореннями смаку й нюху. Хворі часто їдять крейду, зубний порошок, вугілля, глину, пісок, лід, сирі крупи, тісто, сирий м'ясний фарш. Мають пристрасть до запахів гасу, бензину, ацетону, вихлопних газів автомобілів. Патогенез зазначених порушень досі невідомий.
5. Синдром м'язової слабкості. М'язова слабість, що виникає при дефіциті заліза, завжди більш значна, ніж та, котрої варто було б очікувати, виходячи зі ступеня анемії. Вона виявляється слабкістю і стомлюваністю скелетних м'язів, слабкістю міокарда (міокардіопатія), порушеннями ковтання (дисфагія), порушеннями сечовипускання. Очевидно, що важливе значення в розвитку зазначених симптомів мають гіпоксія (кров'яна і тканинна), а також зменшення вмісту міоглобіну в м'язовій тканині.
76 МЕГАЛОБЛАСТИЧНІ АНЕМІЇ
Мегалобластичні анемії – група анемій, які викликані порушенням синтезу ДНК та РНК в клітинах, внаслідок чого порушується їх розмноження; характеризується мегалобластичним типом кровотворення. Найбільш частими причинами такої патології є дефіцит вітаміну В12 та фолієвої кислоти або ж сумісний дефіцит при синдромі порушення всмоктування в кишках.
В12^ –ДЕФІЦИТНА АНЕМІЯ
Вперше цю патологію описав Адісон в 1849 р., а потім у 1872р. - Бірмер, який назвав її "прогресуючою перніціозною" (злоякісною) анемією.
В12– дефіцитна анемія спостерігається у 1% населення, переважно осіб похилого віку. Захворюваність збільшується після 60 років і складає 1:100, з подвоєнням кожні 10 років. У дітей перніціозна анемія діагностується досить рідко з частотою 1:10000, у віці 30-40 років – з частотою 1:500.
Вітамін В12 (ціанокобаламін) міститься в продуктах тваринного походження – м’ясі, яйцях, сирі, печінці, молоці, нирках. В них ціанокобаламін зв’язаний з білком. При кулінарній обробці, а також у шлунку вітамін В12 звільняється від білка (в останньому випадку - під дією протеолітичних ферментів). Нестача вітаміну В12 в продуктах, голодування чи відмова від вживання продуктів тваринного походження (вегетаріанство) нерідко обумовлює развиток В12 - дефіцитної анемії. Вітамін В12, який надходить до організму з їжею, за пропозицією Кастла (1930), називають "зовнішнім фактором" розвитку анемії. Парієтальні клітини шлунка синтезують термолабільний лужностійкий фактор (його позначають як "внутрішній фактор" Кастла), який являє собою глікопротеїн з молекулярною масою 50 000 - 60 000. Комплекс вітаміну і глікопротеїну зв’язується зі специфічними рецепторами клітин слизової оболонки середньої та нижньої частин клубової кишки и далі надходить в кров. Лише незначна кількість вітаміну В12 (близько 1 %) всмоктується у шлунку без участі внутрішнього фактору. В цілому в добовому раціоні повинно міститися близько 3-7мг ціанокобаламіну. Транспорт вітаміну В12 відбувається за допомогою специфічних транспортних білків- транскобаламінів, які синтезуються в печінці. Запаси вітаміну В12 в організмі досить великі (близько 2 - 5 мг). Основним депо є печінка. Із організму щоденно виводиться з екскрементами близько 2 - 5 мкг цього вітаміну. При значному зменшенні надходження вітаміну В12 зовні або ж зменшенні його всмоктування анемія развивається лише через 3 - 6 років.
Етіологія. Причини, які викликають розвиток названої анемії, можуть бути розділені на три группи:
порушення всмоктування вітаміну В12 в організмі:
-атрофія залоз фундального відділу шлунка (хвороба Аддісона-Бірмера):
-пухлини шлунка (поліпоз, рак);
-захворювання кишечника (термінальний ілеїт, дивертикули, пухлини, нориці);
-оперативні втручання на шлунку, кишечнику (резекція, гастроктомія, езофагоєюноанастомоз);
підвищені витрати вітаміну та порушення утилізації в кістковому мозку:
-кишечні паразити (дифілоботриоз);
-дизбактеріоз кишечника;
-захворювання печінки;
-гемобластози (гострий лейкоз, еритромієлоз, остеомієлофіброз);
недостатнє надходження вітаміну В12 в організм з продуктами харчування (досить рідко).
Патогенез. У клітинах із вітаміну В12 утворюються дві його коферментні форми: метилкобаламін і 5 - дезоксиаденозилкобаламін. Метилкобаламін бере участь у забезпеченні нормального, еритробластичного кровотворення. Тетрагідрофолієва кислота, яка утворюється за участю метилкобаламіну, необхідна для синтезу 5, 10 - метилтетрагідрофолієвої кислоти (коферментна форма фолієвої кислоти), що бере участь в утворенні тимідинфосфату. Останній включається в ДНК еритрокаріоцитів і інших клітин, які інтенсивно діляться (шкіра, слизові оболонки). Недолік тимідинфосфату з порушенням включення в ДНК уридину і оротової кислоти призводить до порушення синтезу і структури ДНК, внаслідок чого порушуються процеси ділення і дозрівання еритроцитів. Вони збільшуються за розмірами (мегалобласти і мегалоцити), у зв’язку з чим подібні на еритрокаріоцити і мегалоцити у ембріона. Але ця подібність лише зовнішня. Еритроцити ембріона повноцінно забезпечують кисневотранспортну функцію. Еритроцити, які утворились в умовах дефіциту вітаміну В12, є результатом патологічного мегалобластичного еритропоезу. Їм властива низька мітотична активність і низька резистентність, коротка тривалість життя. Більша частина їх (до 50 %, в нормі близько 20 %) руйнуються в кістковому мозку. У зв’язку з цим сутт’єво зменшується кількість еритроцитів в периферичній крові. Одночасно із порушенням утворення еритроцитів порушуються гранулоцитопоез і тромбоцитопоез. З’являються клітини гігантських розмірів. До того ж збільшується фагоцитоз нейтр\офілів кістковомозковими нейтрофілами. В окремих випадках до останніх утворюються аутоантитіла, які посилюють нейтропенію у хворих на В12-дефіцитну анемію.
Дефіцит вітаміну В12, а в подальшому метилкобаламіну, призводить до порушення дозрівання епітеліальних клітин травного тракту (вони також швидко діляться), що сприяє розвитку атрофії слизової оболонки шлунка та тонкої кишки з відповідною симптоматикою.
Другий кофермент вітаміну В12 – 5-дезоксиаденозилкобаламін бере участь в обміні жирних кислот шляхом каталізації утворення бурштинової кислоти із метилмалонової. Внаслідок дефіциту вітаміну В12 утворюється надлишок метилмалонової кислоти, яка є токсичною для нервових клітин. Це призводить до порушення утворення мієліну в нейронах головного і спинного мозку (особливо задніх і бічних його стовпів) з подальшим розладом у нервовій системі.
Класифікації. 1 За Міжнародною статистичною класифікацією хвороб та споріднених проблем (десятий перегляд) - МКХ -10 В12– дефіцитна анемія має шифр D51 (D 51.0 - В12- дефіцитна анемія, зумовлена недостатністю внутрішнього фактору: анемії Аддісона-Бірмера та перніциозна (природжена), вроджена недостатність внутрішнього фактору; D 51.1 - В12- дефіцитна анемія внаслідок селективної вітамін В12-мальабсорбції з протеїнурією, синдром Імерслунд-Гресбека, мегалобластична спадкова анемія; D 51.2 - дефіцит транскобаламіну ІІ; D 51.3 - інші вітамін В12- дефіцитні аліментарні анемії, анемія вегетаріанців; D 51.8 - інші вітамін В12- дефіцитні анемії; D 51.8 - вітамін В12- дефіцитна анемія, не уточнена).
Класифікація В12 та фолієво-дефіцитних анемій (Воробйов А.І., 1985).
Екзогенний дефіцит вітаміну В12 (у продуктах харчування);
Дефіцит вітаміну В12 внаслідок ендогенних факторів:
Порушення секреції внутрішнього фактору (гастроглікопротеїну):
після резекції шлунка; гастрит типу А.
Ураження тонкої кишки—анентеральна резекція тонкої кишки.
Конкурентне поглинання вітаміну В12 в кишках: дифілоботріоз; синдром ”сліпої петлі”; целіакія.
Спадкові форми В12 (фолієво)-дефіцитної анемії: Синдром Імерслунд-Гресбека.
Спадковий дефіцит транскобаламіну II. Спадкове порушення секреції гастроглікопротеїну.
Фолієво-дефіцитні анемії: Екзогенний дефіцит фолієвої кислоти. Дефіцит фолатів у їжі. Підвищена потреба організму: вагітність; алкоголізм і цироз печінки, захворювання, що проходять з підвищеною клітинною проліферацією. Ендогенний дефіцит фолієвої кислоти Стан після резекції тонкої кишки. Спру, целіакія, синдром ”сліпої петлі”. Медикаментозно-індукований дефіцит. Спадкове порушення транспорту фолатів через стінку тонкої кишки.
Клініка. Спостерігаються 3 основні синдроми:
-гастроентерологічний синдром;
-неврологічний синдром;
-синдром макроцитарно-мегалобластичної анемії.
7726.2.5. Що таке лейкоцитоз? Як класифікують лейкоцитози?
Лейкоцитоз - це збільшення кількості лейкоцитів в одиниці об'єму крові понад) 10 • 109/л. Лейкоцитоз не має самостійного значення, він є всього лише симптомом, що супроводжує розвиток багатьох хвороб.
Класифікація лейкоцитозів.
I. Залежно від причин розвитку виділяють фізіологічний і патологічний лейко-1 цитози.
II. Лейкоцитоз може бути абсолютним і відносним. Для абсолютного лейкоцитозу) характерне збільшення абсолютної кількості лейкоцитів в одиниці об'єму крові. Про відносний лейкоцитоз ідеться в тому випадку, коли зростає відносний вміси окремих форм лейкоцитів у периферичній крові.
III. За механізмом розвитку лейкоцитоз буває:
а) реактивним;
б) перерозподільним;
в) пухлинного походження.
IV. Залежно від видів лейкоцитів, вміст яких у крові збільшений, виділяють:
а) нейтрофільний лейкоцитоз (нейтрофільоз);
б) еозинофільний лейкоцитоз (еозинофілія);
в) базофільний лейкоцитоз;
г) лімфоцитарний лейкоцитоз (лімфоцитоз); ґ) моноцитарний лейкоцитоз (моноцитоз).
26.2.14. Що таке зміщення лейкоцитарної формули?
Зміщення лейкоцитарної формули (ядерне зміщення) - це порушення співвідношення між незрілими і зрілими формами нейтрофілів.
При дослідженні лейкограми встановлюють наявність ядерного зміщення ней-трофільних гранулоцитів уліво або вправо. Ця термінологія пов'язана з розташу-
ванням незрілих нейтрофільних гранулоцитів (мієлоцитів, метамієлоцитів, палич-коядерних нейтрофілів) у лівій частині лейкоцитарної формули (формули Арнета й Шилінга), тимчасом як зрілі, сегментоядерні нейтрофіли умовно поставлені в крайнє праве положенні. Збільшення вмісту в крові молодих форм нейтрофільних гранулоцитів свідчить про ядерне зміщення вліво, переважання зрілих нейтрофілів з великою кількістю сегментів (5-6) на тлі зникнення більш молодих клітин - про ядерне зміщення вправо.
7826.2.10. Що таке лейкопенія? Як класифікують лейкопенії?
Лейкопенія — це зменшення кількості лейкоцитів у периферичній крові нижче 4-109/л. Лейкопенія часто виступає симптомом якого-небудь захворювання. Однак є нозологічні одиниці, при яких саме лейкопенія є основним проявом хвороби, вона по суті визначає картину недуги та всі інші симптоми.
Класифікація лейкопепій.
І. За походженням лейкопенії бувають набутими і спадково обумовленими.
Набуті лейкопенії можуть виникати під впливом фізичних (іонізуюча радіація), хімічних (бензол, цитостатики, лікарські препарати), біологічних (віруси гепатиту, інфекційного мононуклеозу) та імунних факторів.
Прикладами спадкових лейкопеній є нейтропенія Костмана, спадкова нейтропе-нія аутосомно-домінантного типу, синдром "лінивих лейкоцитів", циклічна нейтропенія.
її. За видом лейкоцитів, кількість яких зменшена, виділяють:
а) нейтропенію;
б) лімфопенію;
в) еозинопенію.
III. За патогенезом розрізняють:
а) лейкопенії, обумовлені порушенням надходження лейкоцитів із червоного кісткового мозку в кров;
б) лейкопенії, пов'язані зі скороченням часу перебування лейкоцитів у периферичній крові;
в) перерозподільні лейкопенії.
IV. Виділяють цілий ряд клініко-гематологічних синдромів, для яких лейкопенія є провідною ознакою. Серед них агранулоцитоз, гіпопластична анемія, геморагічна алейкія.
26.2.11. Які механізми можуть лежати в основі розвитку лейкопеній, пов'язаних з порушеннями надходження лейкоцитів із червоного кісткового мозку в кров?
1. Ушкодження кровотворних клітин. При цьому розвивається так званамієлотоксична
лейкопенія. Виділяють три основних механізми ушкодження кровотворних клітин:
а) цитолітичний. Пов'язаний з дією на клітини іонізуючої радіації, цитоста-тичних препаратів, імунних факторів (антитіл, Т-лімфоцитів) та ін. Ступінь ураження червоного кісткового мозку при цьому залежить від дози й тривалості дії зазначених факторів;
б) антиметаболічний. У його основі лежить дія агентів, які втручаються в обмін пуринових і піримідинових основ, порушуючи при цьому процеси поділу стовбурових клітин. За таким принципом діють деякі протипухлинні препарати й антибіотики (левоміцетин).
в) ідіосинкразичний. Реалізується при повторному введенні лікарських препаратів, чутливість організму до яких підвищена (ідіосинкразія). Найчастіше це препарати, що містять у своїй структурі бензольні кільця. У випадку ідіосинкразії немає зв'язку між імовірністю розвитку лейкопенії і дозою, а також тривалістю дії лікарських засобів.
2. Порушення мітозу - неефективний лейкопоез. Найчастіше його причиною є:
а) дефіцит необхідних для клітинного поділу речовин, зокрема вітаміну В12 і фолієвої кислоти;
б) порушення регуляції мітозу — дефіцит лейкопоетинів.
3. Порушення дозрівання лейкоцитів. їхню основу можуть складати генетично обумовлені дефекти як самих кровотворних клітин (наприклад, нейтропенія Костмана), так і клітин "мікрооточення" (наприклад, лейкопенія у "сталевих" мишей лінії SL/SI/). При цьому дозрівання клітин крові досягає певної стадії (наприклад, промієлоцитів) і зупиняється.
4= Порушення виходу лейкоцитів із червоного кісткового мозку в кров. Подібні порушення часто пов'язані з генетичними дефектами лейкоцитів, що порушують основні їхні функції й властивості (наприклад, рухливість). Прикладами можуть бути синдром "лінивих лейкоцитів ", нейтропенія "єменських євреїв ".
5. Зменшення плацдарму лейкопоезу. Має місце при заміщенні кровотворної тканини лейкозними клітинами, метастазами пухлин та ін.
26.2.12. Які механізми можуть лежати в основі розвитку лейкопеній, пов 'язаних зі скороченням часу перебування лейкоцитів у
периферичній крові?
1. Деструкція (руйнування) лейкоцитів. Може бути обумовлена:
а) аутоімунними механізмами (ревматоїдний артрит, системний червоний вовчак);
б) гаптеновими механізмами (амідопіринова гостра нейтропенія);
в) гіперспленізмом (підвищенням фагоцитарної активності макрофагів селезінки).
2. Посилене використання лейкоцитів. Цьому передує прискорений вихід лейкоцитів із крові в тканини в умовах хронічного рецидивуючого запалення.
3. Посилене виведення лейкоцитів з організму. Виражена хронічна втрата нейтрофілів спостерігається у курців: під час ранкового кашлю з мокротинням втрачається від 0,5 до 2-Ю8 гранулоцитів і 0,8 - 1,6-108 макрофагів.
26.2.13. Що таке агранулоцитоз?
Агранулоцитоз - це клініко-гематологічний синдром, що характеризується різким зменшенням вмісту нейтрофілів нижче 0,75-109/л при зменшенні загальної кількості лейкоцитів нижче 1'109/л.
В основі розвитку агранулоцитозу можуть лежати два механізми:
а) мієлотоксичний — ураження червоного кісткового мозку;
б) імунний — руйнування клітин гранулоцитарного ряду антилейкоцитарними антитілами.
Агранулоцитоз супроводжується значним ослабленням реактивності організму у зв'язку з порушенням захисної функції лейкоцитів.
79 Лейкози - це злоякісні пухлини, що виникають із кровотворних клітин і первинно уражують червоний кістковий мозок.
26.2.22. Як класифікують лейкози?
I. Залежно від особливостей патогенезу і пов'язаної з ними гематологічної картини лейкози поділяють на гострі й хронічні.
II. Залежно від того, які кровотворні клітини втягуються в пухлинний процес, лейкози поділяють на лімфолейкози (уражується лімфоцитарний паросток), мієло-лейкози (уражується гранулоцитарний паросток), еритромієлози та ін.
III. Залежно від вмісту лейкоцитів у периферичній крові лейкози бувають лейкеміч-ними (виражений лейкоцитоз — від 20— 109/л до 100-109/л), сублейкемічними (помірний лейкоцитоз до 20-109/л), алейкемічними (вміст лейкоцитів не міняється), лейкопенічними (кількість лейкоцитів зменшується).
26.2.21. Які кровотворні клітини можуть бути джерелом лейкозу?
Будь-яка пухлина може розвиватися тільки з тих клітин, що мають первісну здатність до поділу. Не є винятком і лейкози.
Джерелом лейкозів можуть бути клітини І—IV класів, тобто клітини, здатні до проліферації. Клітини V і VI класів (ті, що дозрівають, і зрілі) трансформуватися в лейкозні не можуть, оскільки втратили структури, необхідні для здійснення клітинного поділу.
26.2.18. Які існують докази того, що лейкози - це захворювання пухлинної природи?
1. При лейкозах, як і при злоякісних пухлинах, відбувається безмежний і нерегульо-ваний поділ клітин.
2. Лейкоз, як і будь-яка злоякісна пухлина, виникає з одної-єдиної первинно зміненої, трансформованої клітини. Усі лейкозні клітини, якими б різними вони не були, походять із однієї клітини.
3. Для лейкозів, як і для інших злоякісних пухлин, характерне явище анаплазії— поява морфологічних, біохімічних та інших властивостей, які наближають лейкозні клітини до ембріональних.
4. Для лейкозів, як і для інших злоякісних пухлин, характерна пухлинна прогресія, тобто набуття із часом лейкозними клітинами все більш і більш злоякісних властивостей.
5. Однаковість причин розвитку лейкозів і злоякісних пухлин. Одні й ті ж етіологічні фактори (фізичні, хімічні, біологічні) здатні викликати виникнення як лейкозів, так і інших злоякісних пухлин.
26.2.19. Чим лейкози відрізняються від інших злоякісних пухлин?
1. У випадку лейкозу неможливо встановити первинну локалізацію пухлини, а отже, радикально видалити її вже на ранніх етапах розвитку.
2. Лейкози - це пухлини, які з самого початку метастазують. Лейкозні клітини легко проникають у кров і розносяться нею по всьому організмі. Вони заселяють все нові й нові ділянки, на яких виникають вторинні лейкозні інфільтрати.
3. Для лейкозів характерна системність ураження. У зв'язку з раннім метастазуванням уражується вся система крові: червоний кістковий мозок, лімфатичні вузли, селезінка, печінка.
4. При лейкозах пригнічується нормальне кровотворення. Це пов'язано з витісненням нормальної кровотворної тканини лейкозними клітинами. З другого боку, має значення токсична дія продуктів лейкозних клітин на клітини гемопоезу.
5. Лейкози — це різновид пухлин, що вражають людину переважно в дитячому і молодому віці.
26.2.33. Яке значення мають віруси у виникненні лейкозів?
У 1908 р. Елерман і Бат уперше отримали лейкоз у курей після введення їм безклітинних фільтратів лейкозних клітин. У 1950 р. Гросс аналогічним чином індукував лейкоз у ссавців (дитинчат мишей). Ці експерименти довели значення вірусів у виникненні лейкозів.
Нині відомо багато вірусів, здатних викликати трансформацію кровотворних !
клітин у пухлинні. їх можна розділити на дві групи.
І. Онкогешіі віруси тварин. Викликають лейкози у різних видів тварин - птахів, мишей, щурів, кішок, мавп. Більшість з них належить до РНК-вмісних вірусів -ретровірусів. Залежно від характеру онкогенної дії їх поділяють на:
а) гостротрансформуючі віруси - викликають розвиток пухлин після короткого латентного періоду (віруси гострих лейкозів тварин). Ці віруси містять j у своїй структурі онкоген;
б) повільнотрансформуючг віруси - викликають розвиток пухлин після тривалого латентного періоду (віруси хронічних лімфолейкозів). Геном цих вірусів не містить онкогена.
її. Онкогенні віруси людиіуі. Сьогодні виявлено два таких віруси.
1. Вірус Епштейна—Барр: ДНК-вмісний вірус сімейства герпес-вірусів. Викликає розвиток лімфами Беркітта у дітей віком від 2 до 14 років у деяких країнах Центральної Африки. Лімфома Беркітта не є в буквальному значенні слова лейкозом, оскільки протікає без первинного ураження червоного кісткового мозку. Джерелом цієї пухлини є підщелепні лімфатичні вузли. Пухлина росте дуже швидко, збільшуючись у масі в 2 рази через кожні 2 дні. Через 6-12 тижнів дитина гине.
2. Вірус Т-клітинної лімфоми—лейкемії людини. Є ретровірусом, належить до так званих Т-лімфотропних вірусів (HTLV-вірусів). Викликає розвиток Т-клітинного лейкозу в Японії, країнах басейну Карибського моря, у Південній Америці, на Алясці. За своїми властивостями подібний до вірусу імунодефіциту людини (ВІЛ), що викликає СНІД.
8026.2.36. Опишіть патогенез лейкозів. Які стадії розвитку проходить лейкоз?
Під впливом онкогенних вірусів, іонізуючої радіації, хімічних речовин відбувається мутація генів або епігеномне порушення регуляції процесу розмноження й дозрівання кровотворних клітин. При цьому в кістковому мозку утворюється клон пухлинних клітин, для яких характерні безмежний ріст і знижена здатність до диференціювання. Швидкий ріст лейкозних клітин призводить до поширення (метастазування) їх по всій системі крові, включаючи кровотворні органи й кров. У лейкозних клітинах, що циркулюють у крові, виявляють однакові хромосомні маркери. Для хронічного мієлолейкозу таким маркером служить "філадельфійська" хромосома.
Нестабільність генотипу лейкозних клітин призводить до виникнення мутацій, як спонтанних, так і обумовлених тривалим впливом канцерогенних факторів, у результаті чого утворюються нові пухлинні клони.
Таким чином, лейкоз проходить дві стадії свого розвитку: 1) моноклонову (відносно більш доброякісну) і 2) поліклонову (злоякіснішу, термінальну). Перехід з першої стадії в другу є показником пухлинної прогресії — лейкозні клітини набувають більшої злоякісності. Вони стають такими, що їх неможна диференціювати ані морфологічними, ані цитохімічними методами, у кровотворних органах і крові збільшується кількість бластних клітин з дегенеративними змінами ядра і цитоплазми. Лейкозні клітини поширюються за межі кровотворних органів, утворюючи лейкозні інфільтрати в різних органах. Унаслідок добору знищуються клітини тих клонів, на які діяли імунна система й гормони організму, цитостатичні засоби (хімічні, гормональні, променеві). Домінують клони пухлинних клітин, найбільш стійких до цих впливів.
26.2.27. Дайте характеристику картини крові при хронічних лейкозах.
Для хронічних лейкозів найчастіше характерні лейкемічний і сублейкемічний варіанти перебігу.
Розглянемо два види хронічних лейкозів, що найчастіше бувають: хронічний мієлоцитарний і хронічний лімфоцитарний.
Хронічний мієлоцитарний лейкоз. Найбільш імовірним джерелом розвитку цього лейкозу є мієлобласти (іноді можуть бути промієлоцити і мієлоцити). Оскільки лейкоз хронічний, то це означає, що лейкозні мієлобласти зберігають здатність до диференціювання у наступні форми. Тому з лейкозної тканини червоного кісткового мозку в кров у великій кількості виходять всі клітини, які походять від мієлобластів, а саме промієлоцити, мієлоцити, метамієлоцити, паличкоядерні І сегментоядернг гранулоцити.
У червоному кістковому мозку переважають клітинні елементи мієлоїдного ряду. Жирова тканина повністю витісняється кровотворною (рис. 109; див. форзац).
Хронічний лімфоцитарний лейкоз. Джерелом його розвитку елімфобласти, які зберегли здатність диференціюватися в наступні форми — пролімфоцити і лімфоцити, і
Тому основна маса лейкозних клітин крові представлена лімфоцитами. їхня 1 кількість у лейкоцитарній формулі становить 80-90 %. Крім лейкозних лімфоцитів, у крові можуть виявлятися пролімфоцити і поодинокі лімфобласти. Характерною є поява так званих тіней Гумпрехта - напівзруйнованих ядер лімфоцитів, що утворюються як артефакт при готуванні мазків крові.
Лімфоцити лейкозного клону (В-лімфоцити) можуть продукувати імуноглобу-ліни однієї специфічності (моноклональні), однак при цьому пригнічується антиті-лоутворення іншими нормальними клонами В-лімфоцитів і поступово розвивається імунологічна недостатність.
У червоному кістковому мозку відбувається майже тотальне заміщення кровотворної тканини лімфоцитами (рис. ПО; див. форзац).
26.2.37. Якими клінічними синдромами можуть виявляти себе лейкози?
Усе різноманіття клінічних ознак лейкозів можна розділити на три групи. І. Гематологічні синдроми, пов'язані із заміщенням нормальної кровотворної тканини лейкозною і пригніченням у зв'язку з цим нормального кровотворення. 1. Панцитопенія ~ зменшення вмісту всіх формених елементів крові. Особливо виражена при гострих лейкозах.
2. Анемія. Основу її патогенезу становить порушення еритропоезу Однак при деяких видах лейкозів певне значення може мати імунний гемоліз еритроцитів (наприклад, при хронічному лімфолейкозі) і кровотечі (геморагічний синдром).
3. Геморагічний синдром. Обумовлений в основному тромбоцитопенією та лейкозними інфільтратами в стінках кровоносних судин.
4. Порушення неспецифічного протимікробного захисту, у зв 'язку з чим зменшується резистентність організму до інфекцій. Основною причиною цього є зменшення вмісту функціонально повноцінних гранулоцитів.
5. Імунологічна недостатність. Розвивається як наслідок лімфопенії (при гострих лейкозах і хронічному мієлолейкозі) або неповноцінності лейкозних лімфоцитів (хронічний лімфолейкоз).
II. Синдроми, пов'язані з особливостями функціонування лейкозних клітин.
1. Гарячка. Показано, що тільки у 7-8 % хворих на лейкози підвищення температури на початку захворювання пов'язане з інфекцією. У більшості ж випадків гарячка має неінфекційне походження.
2. Інтоксикація. Велика кількість лейкозних клітин гине й вивільняє у кров свій вміст. Багато компонентів загиблих клітин мають токсичну дію на центральну нервову систему. Звідси стомлюваність, загальна слабкість, нудота та ін.
3. Аутоімунні процеси. Пов'язані зі змінами, що відбуваються в лімфоци-тарному паростку крові, а саме з появою так званих "заборонених" клонів лімфоцитів, зі зменшенням кількості й функціональної активності Т-супресорів.
III. Синдроми, пов'язані з метастазуванням лейкозних клітин і розвитком лейкозних проліфератів у різних органах і тканинах.
1. Збільшення лімфатичних вузлів, печінки й селезінки.
2. Шкірний синдром. Обумовлений появою в шкірі проліфератів лейкозних клітин - лейкемідів.
3. Виразково-некротичні ураження слизових оболонок (виразково-некротичні стоматит, ангіна, ентеропатії).
4. Кістково-суглобовий синдром, що виявляється болем у кістках і суглобах.
5. Синдром нейролейкозу. Може виявлятися менінгіальним синдромом, синдромом підвищення внутрішньочерепного тиску, різноманітними неврологічними порушеннями: парезами, паралічами, парестезіями. В основі його розвитку — поява лейкозних проліфератів в оболонках головного й спинного мозку, речовині мозку, нервових стовбурах, вегетативних гангліях.
6. Лейкозний пневмоніт. Лейкозні проліферати порушують дихальну функцію легень - розвивається недостатність зовнішнього дихання.
7. Серцева недостатність. Може бути наслідком розмноження лейкозних клітин у м'язі серця.
8181 I. Порушення судинно-тромбоцитарного гемостазу:
а) вазопатії)
б) тромбоцитопенії;
в) тромбоцитопатії.
II. Порушення коагуляційного гемостазу - коагулопатії.
26.3.12. Що таке вазопатії? Як їх класифікують?
Вазопатії—ц& спадково обумовлені або набуті геморагічні діатези, що виникають як наслідок первинних порушень судинної стінки.
Залежно від механізму розвитку вазопатії поділяють на дві групи:
1) запальні вазопатії— васкуліти;
2) диспластичні вазопатії— ураження судин, пов'язані з порушеннями їхньої сполучної тканини (неповноцінність судинної стінки).
26.3.13. Які етіологія і патогенез запальних вазопатій?
Залежно від причин виникнення запальні вазопатії поділяють на:
1) інфекційні васкуліти. Є проявом цілого ряду інфекційних захворювань (вірусних геморагічних гарячок, висипного тифу, сепсису);
2) імунні васкуліти. Розвиваються як наслідок імунокомплексних захворювань (алергічних реакцій III типу за класифікацією Кумбса і Джелла), наприклад, при системному червоному вовчаку, вузликовому періартеріїті, геморагічному васкуліті (хвороба Шенляйн-Геноха);
3) інфекційно-імунні васкуліти. Поєднують обидва попередніх механізми.
У патогенезі запальних вазопатій провідна роль належить ушкодженню ендотелію, що може бути обумовлено:
а) цитопатичною дією ендотеліотропних вірусів;
б) токсинами мікробів, наприклад веротоксином, що його виділяють бактерії кишкової групи; *
в) комплексами антиген-антитіло і комплементом.
Наслідком ушкодження ендотелію судин є:
1) діапедез еритроцитів, що клінічно виявляється точковими крововиливами (пете-хіями);
2) інтенсивне мікротромбоутворення, що викликає порушення мікроциркуляції і живлення тканин;
3) тромбоцитопенія споживання (результат утворення мікротромбів).
26.3.14. Які етіологія і патогенез диспластичних вазопатій?
В основі диспластичних вазопатій лежать набуті або спадково обумовлені порушення сполучної тканини стінки кровоносних судин. Прикладами таких порушень є:
1. Гіповітаміноз С. Аскорбінова кислота— необхідний компонент реакції гідро-ксилювання проліну, унаслідок якої він перетворюється в оксипролін. Ця реакція вважається однією з ключових в утворенні колагену. При гіповітамінозі С з урахуванням сказаного порушується утворення повноцінного колагену (ламкість судин, випадання зубів і т. д.).
2. Телеангіектазії. Це спадково обумовлені локальні дефекти сполучної тканини судин, що обумовлюють стоншення їх стінок і розширення просвіту. Телеангіектазії є джерелом небезпечних для життя кровотеч, особливо при локалізації у внутрішніх органах.
3. Гемангіоми. Це судинні пухлини, які часто кровоточать.
4. Синдром Елерса-Данло. Його основу складають генетично обумовлені дефекти ] колагену.
У патогенезі диспластичних вазопатій мають значення:
1) стоншення стінок мікросудин і розширення їхнього просвіту;
2) неповноцінний локальний гемостаз. Через недостатню кількість або неповноцінність колагену в субендотелії судин порушується адгезія тромбоцитів;
3) легка ранимість судин.
26.3.15. Що таке тромбоцитопенія? Які механізми можуть лежати в основі розвитку тромбоцитопенії?
Тромбоцитопенія — це зменшення вмісту тромбоцитів в одиниці об'єму периферичної крові нижче 150 109/л.
На думку багатьох авторів, геморагічні прояви тромбоцитопенії з'являються при зменшенні кількості тромбоцитів нижче 50 109/л.
За походженням тромбоцитопенії можуть бути спадково обумовленими і набутими.
За механізмом розвитку виділяють такі види тромбоцитопенії. І. Тромбоцитопенії, пов'язані з порушеннями утворення тромбоцитів:
а) мієлотоксичні тромбоцитопенії— виникають унаслідок ушкодження кровотворних клітин. Дуже часто поєднуються з анемією й лейкопенією. Причинами їх розвитку є ті самі фактори, які викликають розвиток гіпопластич-ної анемії;
б) дефіцитні тромбоцитопенії— обумовлені недостатністю вітаміну В12 або фолієвої кислоти;
в) дисрегуляторні тромбоцитопенії— пов'язані з порушенням утворення тромбоцитопоетинів — речовин, що стимулюють утворення тромбоцитів;
г) тромбоцитопенії, пов'язані зі зменшенням плацдарму кровотворення, — розвиваються при лейкозах і метастазах злоякісних пухлин.
її. Тромбоцитопенії, пов'язані з посиленим руйнуванням тромбоцитів. Причиною такого руйнування можуть бути:
а) імунне ушкодження, обумовлене антитромбоцитарними антитілами на власні компоненти кров'яних пластинок або на лікарські препарати, адсорбовані на тромбоцитах. Аутоімунне ушкодження вважають найбільш імовірним механізмом розвитку так званої ідіопатичноїтромбоцитопенічноїпурпури (хвороби Верльгофа);
б) ггперспленізм — гіперфункція селезінки, що супроводжується часто спле-номегалією. У результаті підвищення фагоцитарної активності макрофагів відбувається інтенсивне руйнування всіх формених елементів крові, у тому числі й тромбоцитів;
в) механічне ушкодження тромбоцитів. Часто виникає при гемангіомах і накладенні штучних клапанів серця;
г) набутімембранопатії(гемолітична анемія Маркіафави—Мікеллі). Соматичні мутації кровотворних клітин спричиняються до утворення пулів клітин
(еритроцитів, гранулоцитів, тромбоцитів) з дефектами мембрани. У результаті збільшується чутливість таких клітин до дії комплементу й відбувається їх руйнування. III. Тромбоцитопенії споживання. Виникають у результаті посиленого використання тромбоцитів на утворення тромбів (хвороба Шенляйн—Геноха, хвороба Мошковича, ДВЗ-синдром).
26.3.16. Який патогенез порушень гемостазу в умовах тромбоцитопенії?
У патогенезі геморагічного синдрому при тромбоцитопеніях мають значення:
1) порушення ангіотрофічної функції тромбоцитів, у результаті чого виникають дистрофічні зміни в ендотелії і збільшується ламкість мікросудин. Це веде до збільшення ранимості судин, діапедезу еритроцитів, крововиливів. Останні виявляють себе петехіями на шкірі, кровотечами з ясен і носа, крововиливами в головний мозок і сітківку ока;
2) порушення адгезії й агрегації тромбоцитів. Це викликає порушення формування тромбоцитарного тромбу й призводить до збільшення часу кровотечі (проба Дюка);
3) порушення вторинного спазму ушкоджених артеріол. При тромбоцитопеніях вивільняється мало біогенних амінів (катехоламінів, серотоніну), з дією яких пов'язане скорочення гладких м'язів судин;
4) порушення зсідання крові. Обумовлено недостатнім вивільненням фактора 3 пластинок і тромбостеніну. У результаті порушується І фаза зсідання крові і ретракція згустку.
26.3.17. Що таке тромбоцитопатії? Як їх класифікують?
Тромбоцитопатії—це порушення функціональних властивостей тромбоцитів, йґ якісна неповноцінність. При цьому кількість тромбоцитів може залишатися в нормі J
За походженням тромбоцитопатії бувають спадково обумовленими і набутими.
За характером якісних дефектів кров'яних пластинок тромбоцитопатії поділяють на ендо- і екзотромбоцитарні.
Ендотромбоцитарні тромбоцитопатії обумовлені порушеннями складових частин тромбоцитів. їх, у свою чергу, поділяють на мембранопатії, гранулопатії і фер-ментопатії. Мембранопатії виникають при спадкових аномаліях мембранних пгіко-протеїнів, що виконують функції клітинних рецепторів; при блокаді цих рецепторів аномальними білками плазми крові (парапротеїнами), при ушкодженні мембрани кров'яних пластинок патогенними факторами. Гранулопатії виявляються дефіцитом гранул І і II типів. В основі ферментопатії може лежати зменшення активності ферментів циклу Кребса, гліколізу, порушення функцій АТФ-аз, циклоксигенази і тром-боксансинтетази.
При екзотромбоцитарних тромбоцитопатіях причини порушення функцій тромбоцитів лежать поза кров'яними пластинками. У зв'язку з цим екзотромбоцитарні тромбоцитопатії можуть бути:
а) пов'язаними зі змінами плазми крові (дефіцит плазмових білків, що є плазмовими кофакторами агрегації тромбоцитів);
б) пов'язаними зі змінами в судинній стінці (порушення утворення фактора Вілле-бранда ендотелієм судин, розлади зовнішнього механізму зсідання крові). Залежно від сутності порушень гемостазу виділяють:
1) тромбоцитопатії з первинним порушенням адгезії тромбоцитів^
2) тромбоцитопатії з первинними порушеннями агрегації тромбоцитів;
3) тромбоцитопатії з первинним порушенням реакцій вивільнення вмісту тромбоцитів',
4) тромбоцитопатії, пов'язані з дефіцитом або зменшенням доступності фактора З тромбоцитів.
26.3.18. Наведіть приклади тромбоцитопатій з різними механізмами порушень гемостазу.
1. Тромбоцитопатії з первинним порушенням адгезії тромбоцитів:
а) хвороба Віллебранда — ангіогемофілія. Обумовлена генетичними порушеннями синтезу фактора Віллебранда ендотеліальними клітинами (тип спадкування аутосомно-домінантний);
б) хвороба Бернара—Сульє — макротромбоцитодистрофія. Причиною є спадково обумовлений дефект глікопротеїнів тромбоцитарної мембрани (ГШЬ), що взаємодіють з фактором Віллебранда. При цьому тромбоцити набувають гігантських розмірів. Тип спадкування аутосомно-рецесивний.
2. Тромбоцитопатії з первинними порушеннями агрегації тромбоцитів - диза-грегаційні. Найчастіше буває тромбастенія Гланцмана, що виникає як наслідок дефектів мембранних глікопротеїнів І і II типів, що беруть участь в агрегації. При цьому адгезія тромбоцитів і вивільнення їхніх гранул відбуваються, а агрегація -ні, незважаючи на дію таких потужних агрегантів, як АДФ, адреналін, тромбін. Тип спадкування аутосомно-рецесивний.
3. Тромбоцитопатії з первинним порушенням реакцій вивільнення вмісту тромбоцитів:
а) порушення дегрануляції тромбоцитів — "парез реакції вивільнення ". Виникає, зокрема, при порушенні утворення тромбоксану А2 при дії ацетилсаліцилової кислоти. Показано, одо одноразове приймання аспірину необоротно збільшує час вивільнення гранул від 3,5 до 6 діб, поки не з'являться нові тромбоцити;
б) недостатність накопичення й збереження вмісту гранул тромбоцитів. Виникає, як правило, при генетично обумовлених порушеннях.
Розлади реакцій вивільнення гранул викликають порушення другої хвилі агрегації тромбоцитів. Початкова агрегація кров'яних пластинок закінчується їх дезагрегацією.
4. Тромбоцитопатії, пов'язані з дефіцитом або зменшенням доступності фактора 3 пластинок. У їхній основі можуть лежати або генетично обумовлені дефекти структури цього фактора, або порушення його вивільнення з ушкоджених тромбоцитів. При цьому порушується зсідання крові (коагуляційний гемостаз). Адгезив-но-агрегаційні властивості тромбоцитів не міняються.
8282 В основі розвитку коагулопатій можуть бути:
1) зменшення активності системи зсідання крові;
2) підвищенням активності антикоагулянтної системи;
3) збільшенням активності фібринолітичної системи.
26.3.20. Що може бути причиною безпосереднього порушення І фази зсідання крові?
Залежно від характеру порушень І фази зсідання крові виділяють три групи розладів.
I. Ізольовані порушення зовнішнього механізму активації зсідання. Виникають при дефіциті ф. VII - гіпопроконвертинемії. Цей дефіцит може бути спадково обумовленим (тип спадкування аутосомно-рецесивний) або набутим (гіповітаміноз К, ураження печінки).
II. Ізольовані порушення внутрішнього механізму активації зсідання:
а) дефіцит ф.УШ- гемофілія А. Найчастіше виникає як генетичний дефект коагулянтної частини ф.УШ (тип спадкування зчеплений з Х-хромосомою). Можливе утворення аутоантитіл проти білкових компонентів цього фактора зсідання;
б) дефіцит ф. IX- гемофілія В. Причиною розвитку є спадкова патологія (тип спадкування зчеплений з Х-хромосомою), дефіцит вітаміну К або ураження печінки, антитіла проти ф. IX;
в) дефіцит ф. XI— гемофілія С. Виникає при генетичних порушеннях (тип спадкування аутосомно-рецесивний) або ураженнях печінки;
г) дефіцит ф. ХП. Спадкова патологія, що буває дуже рідко. Завдяки калікре-їн-кініновій системі цей дефект добре компенсується, оскільки запуск внутрішнього механізму зсідання відбувається через зовнішній;
ґ) дефіцит ф. З тромбоцитів. Є наслідком тромбоцитопенії або певних видів тромбоцитопатій.
III. Поєднані порушення зовнішнього і внутрішнього механізмів зсідання. Розвиваються при дефіциті ф. X, що може бути спадково обумовленим (тип спадкування аутосомно-рецесивний) або набутим (гіповітаміноз К, ураження печінки).
26.3.21. Що може бути причиною безпосереднього порушення II фази зсідання крові?
1. Дефіцит ф.ІІ— гіпопротромбінемія. Найчастіше має набутий характер і розвивається внаслідок гіповітамінозу К або уражень печінки.
2. Дефіцит ф. V— парагемофілія. Порушення утворення проакцелерину може бути обумовлено ураженнями печінки або аутоантитілами проти ф.У
26.3.22. Що може бути причиною безпосереднього порушення III фази зсідання крові?
1. Дефіцит фібриногену:
а) афібриногенемія - повна відсутність фібриногену (спадкове захворювання з аутосомно-рецесивним типом спадкування);
б) гіпофібрииогенемія - зменшення синтезу фібриногену в печінці при її ураженнях.
2. Дисфібршшгенемії - якісні порушення фібриногену. Розвиваються як наслідок генетичних дефектів (тип спадкування аутосомно-домінантний). Виявляються утворенням аномального фібрину.
3. Порушення полімеризації фібрину. Розвиваються б результаті утворення комплексів фібриногену з фібрином-мономером і проміжними продуктами, від яких ще повністю не відщепились пептиди А і В. При цьому утворюється так званий "заблокований фібриноген" ("тромбінрезистентний фібриноген"), що не піддається дії тромбіну.
4. Дефіцит ф. ХШ. Виникає як наслідок спадкових порушень (тип спадкування ау-тосомно-рецесивний). виявляється порушеннями перетворення розчинного фібрину (фібрину S) у нерозчинний (фібрин І).
26.3.23. Які речовини складають антикоатулянтну систему крові?
І. Первинні антикоагулянти. Постійно синтезуються в організмі й тому завжди містяться в плазмі крові. До них відносять:
1) антитромбін ПІ- основний універсальний антикоагулянт, що є інгібітором протеаз. Синтезується ендотелієм судин. Пригнічує активність усіх протеолітичних ферментів крові, у тому числі тромбіну, калікреїну, плазміну, ф. ХІІа, ф. ХІа, ф. Ха, ф. ІХа, ф.УІІа;
2) гепарин (антитромбін II) — глікозаміноглікан, що вивільняється тканинними базофілами і базофілами крові при їх дегрануляції. Антикоагулянтні властивості має не сам гепарин, а комплекс гепарину з антитромбіном III. Завдяки гепарину антитромбін III фіксується на поверхні ендотелію судин, де його антикоагулянтні властивості в багато разів зростають;
3) а -антитрипсин, а2-макроглобулін, інгібітор СІ-компонента комплементу - усі вони є неспецифічними інгібіторами протеаз, у тому числі й факторів зсідання крові.
її. Вторинні антикоагулянти. У плазмі крові в нормі не містяться. Утворюються в процесі зсідання крові й фібринолізу. До них відносять:
1) антитромбін І— фібрин, що адсорбує і в такий спосіб інактивує велику кількість тромбіну;
2) продукти фібринолізу. Перешкоджають процесам полімеризації фібрину і утворенню фібринових структур.
26.3.24. Чим може бути обумовлене підвищення активності антикоагулянтної системи крові?
Підвищення активності антикоагулянтної системи крові закономірно виникає при:
1) збільшенні вмісту гепарину в крові — гіпергепаринеміг. Це може бути обумовлено посиленою дегрануляцією тканинних базофілів і базофілів крові, зокрема, при алергічних реакціях І типу за класифікацією Кумбса і Джелла, руйнуванням базо-фільних лейкоцитів при лейкозах, введенням гепарину ззовні;
2) появою "патологічних " антикоагулянтів, до яких відносять антитромбін V, що порушує полімеризацію фібрину-мономеру; "вовчаковий" антикоагулянт, що порушує утворення протромбіназного комплексу; парапротеїни, що унеможливлюють полімеризацію фібрину.
26.3.25. Що входить у поняття "фібринолітична система"?
Фібриноліпшчна система — це система, яка забезпечує лізис (протеоліз) фібрину в кровоносному руслі. У такий спосіб вона бере участь у підтримці рідкого стану крові й у відновленні кровообігу в тромбованих судинах.
До складу системи фібринолізу входять (рис. 119):
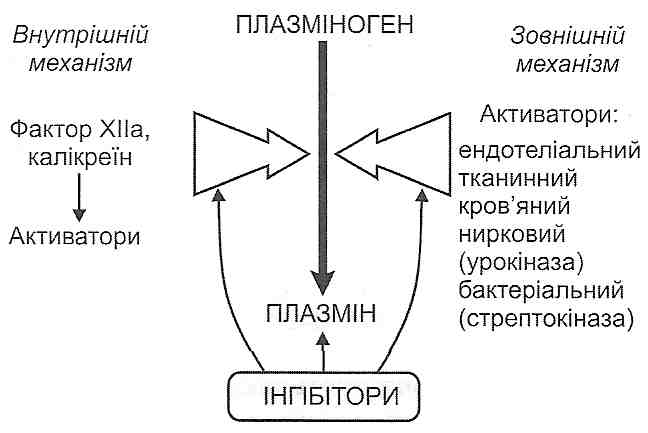
Рис. 119. Схема фібринолізу
1) плазміноген (профібринолізин) — неактивний протеолітичний фермент, що завжди міститься в плазмі крові;
2) плазмін (фібринолізин) — активна форма плазміногену. Утворюється в результаті дії активних протеаз на плазміноген і відщеплення від його молекули пептиду, який "закриває" активний центр;
3) активатори фібринолізу — велика група речовин, які або самі є протеазами і здатні перетворювати плазміноген у плазмін, або викликають появу таких протеаз;
4) інгібітори фібринолізу. До них відносять інгібітори протеаз, серед яких найбільше значення має а2-антиплазмін.
Розрізняють внутрішній і зовнішній механізми активації фібринолізу.
Внутрішній механізм обумовлений активацією фактора XII зсідання крові й утворенням калікреїну, унаслідок чого в крові з'являється велика кількість активаторів фібринолізу.
Зовнішній механізм пов'язаний з надходженням у кров готових активаторів фібринолізу: ендотеліального, клітинного, тканинного (урокіназа), бактеріального (стрептокіназа).
26.3.26. Які фактори викликають підвищення активності фібриполітичної системи крові?
1. Посилене утворення й надходження в кров активаторів фібринолізу. Відбувається при великих ушкодженнях тканин: великі травми, ушкодження клітин токсинами, операційні втручання, лейкози та ін.
2. Зменшення вмісту в крові інгібіторів протеолізу. Має місце при недостатньому їх утворенні або посиленому використанні.
26.3.27. Якими клінічними ознаками виявляють себе порушення коагуляційного гемостазу?
На відміну від порушень судинно-тромбоцитарного гемостазу для коагулопатій характерні не капілярні (точкові) кровотечі, а кровотечі з великих судин — артерій і вен. Такі кровотечі клінічно виявляють себе:
а) гематомами — великими крововиливами в м'язи, під шкіру, у порожнину суглобів (гемартрози);
б) тривалими кровотечами після операційних втручань (видалення зуба та ін.).
8383. Синдром дисемінованого внутрішньосудинного зсідання крові (ДВЗ-синдром) — це генералізоване зсідання крові всередині судин, що викликає утворення великої кількості мікрозгустків і агрегатів клітин, які порушують мікроциркуляцію в органах і тканинах.
Цей синдром часто характеризують як катастрофу для організму.
26.3.29. Що може бути причиною ДВЗ-синдрому?
Залежно від причин розвитку виділяють такі різновиди ДВЗ-синдрому:
1) інфекційно-септичний (розвивається при сепсисі);
2) посттравматичний (при краш-синдромі, опіковій хворобі, множинних переломах кісток);
3) шокогенний (при всіх видах шоку);
4) хірургічний (після операцій з великою травматизацією тканин);
5) акушерський (при передчасному відшаруванні плаценти, надходженні в кров навколоплідних вод);
6) токсигенний (після укусу змії);
7) пухлинний (при злоякісному пухлинному рості);
8) алергічний (при імунному ушкодженні тканин) та ін.
26.3.30. Що є патогенетичною основою розвитку ДВЗ-синдрому?
В основі патогенезу ДВЗ-синдрому лежить так званий "гуморальний протеазний вибух ", тобто одночасна активація всіх протеолітичних ферментів плазми крові, що входять до складу чотирьох позаклітинних біохімічних систем (рис. 120):
а) системи зсідання крові;
б) фібринолітичної системи;
в) калікреїн-кінінової системи;
г) системи комплементу.
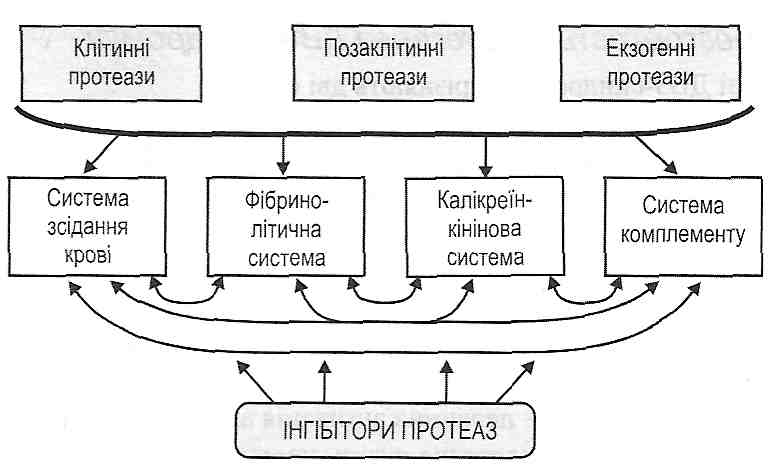
Рис. 120. "Гуморальний протеазний вибух"
Основний принцип активації позаклітинних протеаз — відщеплення пептидів, що закривають їхні активні центри. Утворення активних протеолітичних ферментів крові має свої особливості:
а) можлива самоактивація ферментів — активний фермент, впливаючи на неактивну форму, переводить її в активну;
б) одні активні протеази здатні активувати інші (перехресна активація)',
в) ланцюговий характер активації. Теоретично поява навіть однієї молекули активної протеази може викликати активацію всіх наявних протеаз крові.
Однак у нормі реакції активації протеолітичних ферментів мають обмежений характер, що пояснюється існуванням великої групи інгібіторів протеаз.
При патології, коли у кров надходять великі кількості активних протеаз, потужність існуючих інгібіторів може виявитися недостатньою. Отоді й виявить себе ланцюговий характер активації протеолітичних систем плазми крові. Така активація набуває генералізованого характеру, втягує всі протеази крові - відбувається "гуморальний протеазний вибух".
26.3.31. Назвіть основні джерела надходження в кров активних протеаз при ДВЗ- синдромі.
Існує три основних джерела надходження протеаз у кров.
I. Ушкоджені клітини. Має значення гостре ушкодження великої кількості клітин, з яких у позаклітинний простір і кров надходять лізосомні протеази, тканинний тромбопластин.
Запалення як місцевий процес, що виникає при ушкодженні клітин, обмежує надходження продуктів розпаду в кров, локалізуючи ушкодження і в такий спосіб попереджаючи розвиток ДВЗ-синдрому.
II. Надходження в кров великої кількості позаклітинних протеаз; наприклад трипсину при гострому панкреатиті, ферментів, що містяться в навколоплідних водах.
III. Екзогенні протеази. їхніми джерелами можуть бути бактеріальні клітини при сепсисі, зміїна отрута та ін.
26.3.32. Як розгортається патогенез ДВЗ-синдрому?
У патогенезі ДВЗ-синдрому розрізняють дві фази.
I фаза - фаза гіперкоагуляції і агрегації тромбоцитів. Основу цієї фази становить генералізована активація системи зсідання крові, тобто утворення тромбіну (тромбінемія), що призводить до утворення фібрину і агрегатів тромбоцитів.
Існує три механізми запуску цієї фази:
1) ферментативний механізм - надходження в кров великої кількості активних протеаз і тканинного тромбопластину;
2) контактний механізм - активація ф. XII при контакті його з чужорідними поверхнями (екстракорпоральний кровообіг, гемодіаліз, штучні клапани серця);
3) тромбоцитарний механізм - первинна активація агрегації тромбоцитів при гене-ралізованому ушкодженні ендотелію судин, порушеннях реологічних властивостей крові, гострому внутрішньосудинному гемолізі еритроцитів.
У результаті реалізації зазначених механізмів утворюється велика кількість мі-крозгустків і агрегатів клітин, що призводить до розладів мікроциркуляції, розвитку сладж-синдрому (див. розд. 13).
Усе це веде до появи таких клінічних проявів, як гіпоксія, ацидоз, інтоксикація продуктами розпаду, гостра недостатність зовнішнього дихання (мікрозгустками закупорюються капіляри легень), гостра ниркова недостатність (забиваються капіляри клубочків), порушення мозкового кровообігу.
II фаза - фаза гіпокоагуляції (геморагічний синдром). Ця фаза розвивається як наслідок виснаження механізмів судинно-тромбоцитарного і коагуляційного гемостазу.
У її виникненні мають значення:
а) зменшення активності системи зсідання крові (споживання факторів І, V, VIII);
б) активація фібринолітичної системи (надходження в кров великої кількості активаторів фібринолізу);
в) підвищення антикоагулянтної активності крові за рахунок утворення продуктів фібринолізу;
г) розвиток тромбоцитопенії споживання;
ґ) підвищення проникності стінки судин (має значення утворення великих кількостей кінінів). Фаза гіпокоагуляції клінічно виявляє себе масивними кровотечами, що їх важко зупинити.
8484
8427.1. Що таке недостатність кровообігу? Чим вона може бути обумовлена?
Недостатність кровообігу - це стан, при якому серцево-судинна система не може забезпечити органи й тканини організму необхідною кількістю крові. Є найчастішим проявом різних порушень функцій системи кровообігу.
Недостатність кровообігу може бути обумовлена:
1) недостатністю серця;
2) недостатністю кровоносних судин;
3) серцево-судинною недостатністю, тобто одночасною недостатністю серця і судин.
27.3. Що таке недостатність серця?
Недостатність серця - це патологічний стан, обумовлений нездатністю серця забезпечити кровопостачання органів і тканин відповідно до їхніх потреб.
Це стан, при якому навантаження на серце перевищує його здатність виконувати роботу. Він виявляє себе тим, що серце не здатне переміщати в артеріальне русло всю кров, що надходить до нього по венах.
27.4. Як класифікують недостатність серця?
I. Залежно від клінічного перебігу розрізняють гостру і хронічну недостатність серця.
II. За вираженістю клінічних проявів недостатність серця може бути прихованою (компенсованою) і явною (декомпенсованою).
III. Залежно від порушення функції переважно того чи того відділу серця розрізняють лівоишуночкову, правошлуночкову і тотальну недостатність серця.
IV. За патогенезом виділяють:
а) недостатність серця від перевантаження;
б) міокардіальну недостатність серця;
в) позаміокардіальну недостатність.
27.6. Які типи перевантажень серця можуть бути причиною розвитку його недостатності?
Виділяють два типи перевантажень серця.
1. Перевантаження об'ємом виникає тоді, коли до серця або до окремих його порожнин притікає збільшений об'єм крові. У цих умовах серце або його відділ, що зазнає перевантаження, мають переміщати збільшений об'єм крові в артеріальну систему. Це досягається збільшенням хвилинного об'єму серця відповідно до збільшеного венозного повернення.
Перевантаження об'ємом виникає при:
а) збільшенні венозного повернення крові до серця, зокрема при збільшенні об'єму циркулюючої крові (гіперволемія) або збільшенні тонусу венозних судин (зменшення ємності венозної системи);
б) вадах серця — недостатності його клапанів. Так, при недостатності аортального і двостулкового клапанів розвивається перевантаження лівого шлуночка, при недостатності клапана легеневої артерії і тристулкового клапана - перевантаження правого шлуночка.
2. Перевантаження опором виникає тоді, коли серце або окремі його відділи змушені виконувати роботу проти збільшеного опору, що перешкоджає переміщенню всієї крові в артеріальну систему. При перевантаженні опором серце має зберегти свій хвилинний об'єм, незважаючи на збільшений опір вигнанню крові.
Перевантаження опором розвивається при:
а) збільшенні артеріального тиску (збільшенні периферичного судинного опору). При гіпертензії великого кола кровообігу перевантаження опором діє на лівий шлуночок, а при гіпертензії малого кола - на правий шлуночок;
б) вадах серця — стенозах клапанних отворів. 'Так, при стенозі отвору аорти розвивається перевантаження лівого шлуночка, при стенозі отвору двостулкового клапана - лівого передсердя, при стенозі отвору легеневої артерії - правого шлуночка, при стенозі отвору тристулкового клапана - правого передсердя.
27.7. Які механізми можуть забезпечувати компенсацію серця при дії на нього збільшених навантажень?
При дії на серце навантажень об'ємом і опором збільшення роботи серця забезпечується двома типами компенсаторних механізмів.
I. Негайні механізми компенсації серця. До них відносять:
а) гетерюметричтш механізм;
б) гомеометричний механізм;
в) хроноінотропний механізм;.
г) інотропна дія катехоламінів.
II. Механізми довгострокової адаптації серця - гіпертрофія міокарда.