

1.8. Строение и свойства вмс
 Высокомолекулярные
соединения (ВМС) образуются, как уже
упоминалось, в процессах полимеризации
и поликонденсации. В первом случае
возникают типичные
высокополимерные соединения, во втором
могут образовываться
ВМС, составленные не из одинаковых
группировок атомов и
не являющиеся собственнс полимерами,
например белки.
Поэтому понятие ВМС, строго
говоря, является более
общим, включая, как частное,
высокополимеры. Однако
в современной литературе
это различие в значительной
степени стерлось и в дальнейшем следует
применять
термины: полимер и ВМС
как равнозначные.
Высокомолекулярные
соединения (ВМС) образуются, как уже
упоминалось, в процессах полимеризации
и поликонденсации. В первом случае
возникают типичные
высокополимерные соединения, во втором
могут образовываться
ВМС, составленные не из одинаковых
группировок атомов и
не являющиеся собственнс полимерами,
например белки.
Поэтому понятие ВМС, строго
говоря, является более
общим, включая, как частное,
высокополимеры. Однако
в современной литературе
это различие в значительной
степени стерлось и в дальнейшем следует
применять
термины: полимер и ВМС
как равнозначные.
В
Рис. 4.
Схемы строения высокополимеров.
Полимеры:
а
—линейный;
б —разветвленный; в
—пространственный; г—сшитый.
Количественные изменения М приводят к изменениям качественным — появляются новые свойства, отсутствующие у низкомолекулярных соединений и весьма важные в практическом отношении, например высокая эластичность. Мы видели уже, что структурно-механические свойства являются функцией внутренней структуры дисперсной системы. И в настоящей главе основная задача состоит в установлении связи между строением (структурой) ВМС, с одной стороны, и практически важными свойствами их с другой. Решение этой задачи составляет основу той «молекулярной архитектуры», которая необходима для конструирования полимерных материалов, обладающих требуемыми свойствами.
Существуют несколько типов структуры полимеров, а именно: линейная, разветвленная, пространственная: в отдельный тип можно выделить сшитые структуры (рис. 4).
Линейные полимеры построены из длинных одномерных элементов структуры — отдельных макромолекул или молекулярных блоков (рис. 4, а).
Разветвленные полимеры состоят из цепей с боковыми ответвлениями (рис. 4,6), а в пространственных (сетчатых) полимерах цепи соединены между собой химическими связями в пространстве (рис. 4, в). Пространственные полимеры, цепи которых сшиты короткими мостичными связями, например, атомами О или S, называются сшитыми (рис. 4, г). В качестве примеров названных типов можно назвать, соответственно, натуральный каучук, крахмал, фенолоформальдегидные смолы (клей БФ), эбонит.
Основные особенности строения полимеров, определяющие их свойства: 1) существование двух различных типов связей; 2) гибкость цепей, обусловленная внутренним вращением звеньев.
Рис.
5. Различное пространственное расположение
атомов водорода в молекуле
этана:а—цис-положение;
б—транс-положение.
Внизу — проекции молекулы.
Рис. 5 обой
макромолекулярные цепи. Такая
двойственность определяет
специфику
свойств
ВМС. Поэтому нецелесообразно
относить к полимерам
структуры типа алмаза,
где все связи являются
химическими и, наоборот,
можно считать полимерами
частицы бентонитовой глины (чешуйки
двухмерной, образованной химическими
связями сетки, соединенные между собой
межмолекулярными
силами), графитовые структуры и т. п. В
соответствии
с энергетическим различием различаются
и расстояния между атомами: в цепи они
составляют ~ 1 А, а между соседними цепями
3—5 А.
обой
макромолекулярные цепи. Такая
двойственность определяет
специфику
свойств
ВМС. Поэтому нецелесообразно
относить к полимерам
структуры типа алмаза,
где все связи являются
химическими и, наоборот,
можно считать полимерами
частицы бентонитовой глины (чешуйки
двухмерной, образованной химическими
связями сетки, соединенные между собой
межмолекулярными
силами), графитовые структуры и т. п. В
соответствии
с энергетическим различием различаются
и расстояния между атомами: в цепи они
составляют ~ 1 А, а между соседними цепями
3—5 А.
Вторая особенность строения полимеров определяется гибкостью цепей, связанной со свободой вращения их звеньев. Вращение, полное или ограниченное, происходит при сохранении постоянства величины валентного угла, определяемой σ-связями и равной *
К
Рис. 6.
Зависимость потенциальной
энергии
молекулы
этана
от
величины
угла поворота
метильной группы.
 илы
отталкивания. Поэтому цис-положение
(рис.5,а), в котором атомы
наиболее сближены, характеризуется
максимальной потенциальной
энергией U,
транс-положение (рис.
5,б)
— минимальной. Зависимость
U
от угла
поворота ф показана на
рис. 6.
илы
отталкивания. Поэтому цис-положение
(рис.5,а), в котором атомы
наиболее сближены, характеризуется
максимальной потенциальной
энергией U,
транс-положение (рис.
5,б)
— минимальной. Зависимость
U
от угла
поворота ф показана на
рис. 6.
Для молекул более сложных картина энергетического профиля соответственно усложняется. Высота энергетического барьера вращения Uo для органических молекул составляет 4— 16 кДж/моль, в зависимости от ближайшего окружения звена (заместители, двойные связи, полярные группы и др.). При небольшом запасе энергии (при низких Г), недостаточном для преодоления барьера, наблюдается неполное ограниченное вращение близ потенциального минимума, которое не выходит за пределы некоторого значения ф, соответствующего U ≈ kT. Благодаря вращению звеньев макромолекула может принимать различные конформации. Конформациями называют энергетически неравноценные формы молекул, переходящие одна в другую без разрыва химической связи путем простого поворота звеньев, в отличие от конфигураций, взаимный переход которых возможен лишь путем разрыва химической σ-связи и образования новой (стереоизомеры). Число конформаций для одной макромолекулы может быть очень большим даже при фиксированных валентных углах, поскольку каждое звено вращается почти независимо от других (рис. 7).
Рис. 8.
Конформации
цепи с фиксированными
валентными
углами.
Рис. 7.
Энергия активации
вращения в зависимости
от угла поворота
звена.
 В
В
![]() (расстояния
между ее концами).
(расстояния
между ее концами).
Гибкость цепи является функцией многих переменных. Она уменьшается с увеличением числа полярных групп, ростом плотности пространственной сетки (матрицы) и с уменьшением температуры.
Рассмотренные особенности строения полимеров приближают нас к поставленной цели — нахождению связи между составом и свойствами. Действительно, высокая гибкость цепи (малые величины ΔU и Uo) позволяет легко растянуть цепь небольшим внешним усилием, после снятия, которого система возвращается на исходный низший энергетический уровень.
Таким образом, причиной свойства — высокой эластичности — является особенность строения — гибкость цепей и блоков, состоящих из цепных молекул.
Для более полного понимания связи между строением и свойствами необходимо рассмотреть фазовые и физические состояния полимеров.
Понятие агрегатного состояния обычно не применяют к полимерам. Как известно, различия между агрегатными состояниями определяются характером движения молекул и плотностью упаковки. Так, для твердых тел вращательное движение отсутствует, а существуют лишь колебательные движения, определяющие упругость и твердость тела. Полимеры не могут находиться в истинно твердом состоянии, как и в состоянии газа; их можно отнести к структурам конденсационно-кристаллизационного типа. Для описания полимеров целесообразно использовать представления о фазовом состоянии вещества. Понятие фазы применяется здесь в структурном смысле и характеризуется порядком взаимного расположения молекул. В соответствии с этим любое вещество— низкомолекулярное и ВМС — находится в одном из трех фазовых состояний — кристаллическом, аморфном или газообразном (последнее для ВМС практически отсутствует).
Кристаллическому состоянию присущ трехмерный дальний порядок в расположении атомов и молекул, аморфному — только ближний порядок и, наконец, газообразному — отсутствие какого-либо порядка.
Для полимеров наиболее характерно аморфное состояние, однако в определенных условиях они могут переходить (частично или полностью) в кристаллическое. Необходимое условие кристаллизации — регулярность строения полимера. Процесс кристаллизации совершается при некоторых оптимальных значениях Т и гибкости цепи, ибо слабое тепловое движение не может обеспечить необходимой ориентации звеньев, а слишком интенсивное — ее нарушает. Температуру, выше которой полимер практически не кристаллизуется, называют температурой кристаллизации. При очень низких Т (ниже —50 °С) кристаллизация полностью прекращается, вследствие затруднений кинетического характера: подвижность звеньев и цепей слишком мала для образования кристаллов за измеримое время.
Процессу кристаллизации способствует механическое растяжение полимера, направляющее ориентацию цепей. Следует отметить, что образование пачек, состоящих из ориентированных цепей, обычно не является фазовым переходом, поскольку при этом не происходит разрыва непрерывности функций и отсутствует скрытая теплота перехода. Пачки не обладают ближним порядком (нет ориентации звеньев) при наличии дальнего (ориентация цепей). В дальнейшем, при регулярном строении полимера, пачки могут сращиваться, образуя плоские ленты. Наслоение лент приводит к образованию трехмерных структур — сферо-литов, превращающихся далее в кристаллы (фазовый переход).
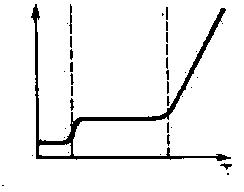
Типичным для полимеров, как было уже отмечено, является аморфное фазовое состояние, которому соответствуют три различных физических состояния линейных полимеров: стеклообразное, высокоэластическое и вязкотекучее *.
Стеклообразное состояние достигается посредством охлаждения полимера ниже температуры стеклования Tg, при которой полимер полностью отвердевает без образования кристаллической решетки. В этом состоянии возможны только колебания атомов около положения равновесия; вращение звеньев (даже ограниченное) и перемещение цепей отсутствуют.
При температурах выше Tg полимер переходит в высокоэластическое состояние, для которого характерны колебательные движения звеньев (ограниченное вращение), позволяющее макромолекуле изгибаться и обратимо изменять эффективную длину цепи r под действием внешней нагрузки, т. е. проявлять свойство эластичности.
Дальнейшее повышение Т приводит к освобождению цепей, которые приобретают способность к тангенциальному перемещению (пластическому течению) при нагрузке, превышающей некоторый силовой барьер. Полимер переходит при этом в вязкотекучее состояние при температуре, называемой температурой текучести Tf. При охлаждении полимер проходит рассматриваемые состояния в обратном порядке.
Tg
Tf
Рис. 9.
Термомеханическая
кривая.
 В
В
Рассмотренные представления о строении полимеров и фазовых состояниях позволяют понять природу упругопластических свойств.
Одно из важнейших свойств полимеров — высокая эластичность— отсутствует у низкомолекулярных веществ, металлов, кристаллических тел (обладающих упругостью). Она отличается от упругости прежде всего порядком величины, а также релаксационным характером явления.
Ниже сопоставлены значения модулей упругости твердых тел с модулями высокоэластичных полимеров:
Вещество Е, кгс/мм2
Сталь 2 ∙104
Медь 1 ∙ 104
Кварц 1 ∙ 104
Полимеры 2∙10-2
Приведенные данные свидетельствуют, что модуль высокоэластичности полимеров меньше модуля упругости твердых тел на 6 порядков. Это различие связано с тем, что природа упругости имеет энергетический характер, а эластичности — энтропийный.
Изменение
свободной энергии при растяжении равно
произведению силы f
на
деформацию dl.
dA
= f∙dl.
Поскольку
dA
= dU
— TdS,
мы
можем записать, при
Т
= const:
![]() .
.
При
деформации кристаллов происходит
смещение атомов, изменение межатомных
расстояний, а следовательно, увеличение
объема и потенциальной энергии.
Энтропия в этом процессе существенно
не меняется, поэтому
![]() .
При
эластической деформации, наоборот,
расстояние между атомами, объем и
потенциальная энергия межатомного
взаимодействия остаются постоянными.
Изменяется
пространственная ориентация звеньев
цепи; при этом увеличивается расстояние
между ее концами:
.
При
эластической деформации, наоборот,
расстояние между атомами, объем и
потенциальная энергия межатомного
взаимодействия остаются постоянными.
Изменяется
пространственная ориентация звеньев
цепи; при этом увеличивается расстояние
между ее концами:
![]() и
и![]()
С ростом длины цепи, при f > 0, энтропия уменьшается. Физический смысл этого уменьшения заключается в том, что чем более вытянута цепь, тем меньше число возможных конформаций, определяющее термодинамическую вероятность w, с которой энтропия связана соотношением Больцмана:
S = k∙lnw
Замедленность конформационных переходов в наиболее вероятное состояние (исходное) после снятия нагрузки определяет релаксационный характер процесса. Время релаксации τ полимеров в высокоэластическом состоянии равно 10-4— 10-6 с; оно значительно превышает т низкомолекулярных жидкостей (~ 10- 10 с).
Построение кривых γ— t по экспериментальным данным позволяет определить упругопластические параметры полимеров методами, общими для структурированных систем.
Высокая эластичность характерна для макромолекул неполярных, обладающих большой термодинамической гибкостью цепи (полиизопрен, полибутадиен и др.). Введение небольшого числа полярных групп не нарушает эластичности (полихлоропрен), изменяя лишь величину модуля. Однако для полимеров сильно полярных или содержащих крупные заместители (полистирол) эластичность проявляется лишь при повышении Т, поскольку сильное межмолекулярное взаимодействие увеличивает Tg. При комнатной температуре эти полимеры находятся в стеклообразном состоянии.
Для некоторых полимеров подобного типа можно обнаружить «память», связанную с обратимостью деформаций. Так, из полимера можно вырезать какой либо предмет при Т < Tg, а затем, нагрев до Т > Tg, растянуть его в тонкую нить. Если эту нить охладить вновь в растянутом состоянии до Т < Tg, она может храниться в течение долгого времени, но при кратковременном нагреве до Т > Tg без нагрузки, она сразу же принимает первоначальную форму предмета в самопроизвольном процессе увеличения энтропии.
Увеличение полярности приводит к возрастанию τ вследствие уменьшения кинетической гибкости цепей; рост высоты активационного барьера замедляет возвращение системы в исходное состояние после снятия нагрузки.
Образование редкой пространственной сетки, например, в процессе вулканизации, не мешает проявлению высокоэластических свойств, но увеличивает модуль. Рост числа мостичных связей приводит к образованию жестких материалов, не способных к высокоэластической деформации (эбонит).
Пластические свойства появляются, как мы видели, при нагревании до Т > Tf и обусловлены последовательным перемещением цепей. Движение цепи как целого связано с большими затратами энергии на одновременное преодоление большого числа межмолекулярных связей. Поэтому течение полимеров следует представлять себе как последовательное перемещение отдельных звеньев цепи. Для этого необходимо локальное выпрямление цепей и, таким образом, пластичность связана с гибкостью цепей и с эластическими их свойствами. Факторы, вызывающие увеличение жесткости цепей (мостичные связи, полярные группы), уменьшают или полностью исключают пластичность. Так, пространственные, особенно сшитые, полимеры даже при редкой сетке теряют способность к необратимым деформациям, а следовательно, не могут переходить в вязкотекучее состояние.
Введение большого числа полярных групп резко увеличивает вязкость (η ≈ 1012 П), сильно снижая пластичность и текучесть. В практике для увеличения пластичности в линейные полимеры вводят специальные вещества — пластификаторы. Эти вещества внедряются между макромолекулами или блоками (пачками) и раздвигают их, ослабляя межмолекулярные силы и снижая Tg и Tf. Пластификаторы, взаимодействуя с макромолекулами, как бы « сольватируют» их. Поэтому для неполярных полимеров применяют неполярные пластификаторы типа СС14, для полярных— полярные, например дибутилфталат. В настоящее время используют десятки тысяч различных пластификаторов.
Таким образом, основные параметры, определяющие структурно-механические свойства полимерных материалов (модуль эластичности, пластическая вязкость, время релаксации и другие параметры), являются функциями строения полимеров. Изучив природу этой связи, химик, подобно архитектору, может в настоящее время, скрепляя или раздвигая цепи, вводя полярные группы, заместители больших размеров и т. д., конструировать новые материалы с требуемыми свойствами, заранее заданными, сообразно с целью их практического применения.
Изучив в основных чертах строение и свойства полимеров, рассмотрим взаимодействие их с растворителем, приводящее к образованию растворов ВМС.