

Успадкування аутосомно-рецесивних ознак у людини
На відміну від аутосомно-домінантного типу успадкування аутосомно-рецесивне виявляється лише в результаті шлюбу двох гетерозигот. Тому такі ознаки частіше з’являються у близькородинних шлюбах. Чим менша концентрація аутосомно-рецесивного гена в популяції, тим більша вірогідність його реалізації при кровній спорідненості батьків. За аутосомно-рецесивним типом успадковуються більше 780 хвороб.
Деякі аутосомно-рецесивні ознаки наведено в табл. 1.
За аутосомно-рецесивним типом успадковується ген, що зумовлює здатність сприймати смак фенілтіокарбаміда або схожих з ним сполук.
Рецесивним є ген, що приводить до порушення будови внутрішнього вуха і, отже, до глухоти. Відсутність слуху з самого народження веде до німоти. Ген цей зустрічається досить часто, і шлюби між двома глухонімими — поширене явище. Цікаво відзначити, що від такого шлюбу іноді народжуються, всупереч сподіванням, здорові (з нормальним слухом) діти.
За аутосомно-рецесивним типом успадковуються багато захворювань з порушеннями обміну речовин. Ця велика група захворювань налічує близько 600 ензимопатій: відсутність або недостатність відповідного ферменту приводить до блокування біохімічних реакцій. Ензимопатії виникають в результаті мутацій генів, відповідальних за синтез певного ферменту.
У

 більшості випадків ензимопатії протікають
паралельно з розумовою
відсталістю. Виключенням із загального
правила є одне з порушень
амінокислотного обміну
— альбінізм — відсутність
ферменту тиразинази, яка перетворює
амінокислоту тирозин в пігмент меланін.
У окремих індивідуумів меланін зовсім
не утворюється або утворюється в таких
малих кількостях, що шкіра, волосся
виявляються дуже світлими, а очі —
червоними (відсутність пігменту
не маскує кровоносні судини сітківки).
Світлобоязнь і
незвичайний колір очей примушують таких
людей носити темні окуляри. В Екваторіальній
Америці збереглося ціле плем’я
альбіносів.
більшості випадків ензимопатії протікають
паралельно з розумовою
відсталістю. Виключенням із загального
правила є одне з порушень
амінокислотного обміну
— альбінізм — відсутність
ферменту тиразинази, яка перетворює
амінокислоту тирозин в пігмент меланін.
У окремих індивідуумів меланін зовсім
не утворюється або утворюється в таких
малих кількостях, що шкіра, волосся
виявляються дуже світлими, а очі —
червоними (відсутність пігменту
не маскує кровоносні судини сітківки).
Світлобоязнь і
незвичайний колір очей примушують таких
людей носити темні окуляри. В Екваторіальній
Америці збереглося ціле плем’я
альбіносів.
До порушень амінокислотного обміну відносяться також алкаптонурія, фенілкетонурія й інші «природжені дефекти метаболізму».
При алкаптонурії у хворих виділяється темна, майже чорна сеча. Пов’язано це з присутністю в сечі гомогентензинової кислоти — проміжного продукту метаболізму двох амінокислот — фенілаланіна та тирозина. У хворих забарвлені також хрящі і в літньому віці розвиваються артрити (від грец. артрон — суглоб).
Фенілкетонурія (хвороба Феллінга) — різке підвищення змісту в крові амінокислоти фенілаланіна і перетворення її в ряд продуктів, наприклад у фенілпіровиноградну і фенілмолочную кислоти. На відміну від гомогентензинової кислоти, яка не має явного несприятливого впливу на тканини мозку, продукти, що утворюються при фенілкетонурії, є дуже токсичними. Тому у дітей при цій патології спостерігається різко виражена розумова відсталість. Захворювання виражається також у зниженні кількості пігменту меланіну, тому хворі завжди виглядають, як блакитноокі блондини зі світлою шкірою. В даний час діагноз можна поставити при народженні дитини експрес-методом: на змочену сечею пелюшку наносять 5 крапель 10% розчину FеС1з, при цьому спостерігається почервоніння, яке швидко проходить).
З наймолодшого віку з їжі таких дітей слід виключити фенілаланін, рекомендується спеціальна дієта: каші на кобилячому молоці (бідному на амінокислоти), мед, масло, овочі, саго (фенілаланіну зовсім не містять) у поєднанні з лікуванням берлофеном (гідролізат білка). Цим попереджають розвиток порушень функцій мозку і діти розвиваються нормально. Без лікування у новонароджених при звичному годуванні розвивається ряд розладів, що приводять надалі до передчасної смерті. В деяких популяціях частота цього гена складає 1:7000. В 15% випадків це захворювання зареєстровано у нащадків, що походять від шлюбу кровно споріднених батьків. У плазмі крові таких гетерозигот спостерігається вдвічі більше норми фенілаланіна після стандартної дієти із звичним вмістом у ній цієї амінокислоти.
П орушення
ліпідного обміну — амавротична
ідіотія (хвороба Тея-Сакса),
пов’язана з відсутністю
ферменту гексосаміндази А,
що спричиняє важкий розлад нервової
системи. Цю хворобу можна виявити лише
в другій половині першого року життя
дитини, коли спостерігається прогресуюче
відставання фізичного розвитку, порушення
зору й інтелекту, надалі хворий сліпне,
розвивається недоумство
і повна безпорадність. Важкі симптоми
наростають, приводять до смерті дитини
у віці до 4–5 років.
орушення
ліпідного обміну — амавротична
ідіотія (хвороба Тея-Сакса),
пов’язана з відсутністю
ферменту гексосаміндази А,
що спричиняє важкий розлад нервової
системи. Цю хворобу можна виявити лише
в другій половині першого року життя
дитини, коли спостерігається прогресуюче
відставання фізичного розвитку, порушення
зору й інтелекту, надалі хворий сліпне,
розвивається недоумство
і повна безпорадність. Важкі симптоми
наростають, приводять до смерті дитини
у віці до 4–5 років.
Це захворювання рідкісне, зустрічається з частотою 1:250000 часто виявляється у подружжя, предками якого були вихідці з Бухари або інших областей Середньої Азії.
Галактоземія — порушення вуглеводневого обміну. Вона обумовлена порушенням діяльності печінки, накопиченням у тканинах (у тому числі і в крові) галактози, яка є у молоці. Без лікування розвивається цироз печінки; до патологічного процесу залучаються й інші життєво важливі органи. Зрештою хвороба приводить до недоумства і ранньої смерті. На початку життя, як тільки новонароджений починає одержувати молоко, спостерігаються жовтяниця, блювота, диспепсичні розлади, зниження маси тіла. При ранній діагностиці таких дітей до трирічного віку переводять на безмолочне вигодовування, тобто виключають продукти, що містять галактозу. Такі діти розвиваються нормально і відхилень у психіці у них не спостерігається. Носіями такого гена, що викликає захворювання (тобто гетерозиготи) є в середньому 1 : 70000.
Аномалії, пов’язані з порушеннями розпаду деяких вуглеводовмісних сполук, викликають розвиток мукополісахаридозів (гаргоїлізми). При цих захворюваннях уражена сполучна тканина, а отже, страждають опорно-трофічні функції і моторика. Для хворих мукополісахаридозом характерна потворна статура (діти нагадують потвор-гаргоїдів, які прикрашають паризький храм Нотр-Дам), наявність множинних вад внутрішніх органів (печінки, нирок, серця, аорти, нервової системи) і очей.
Н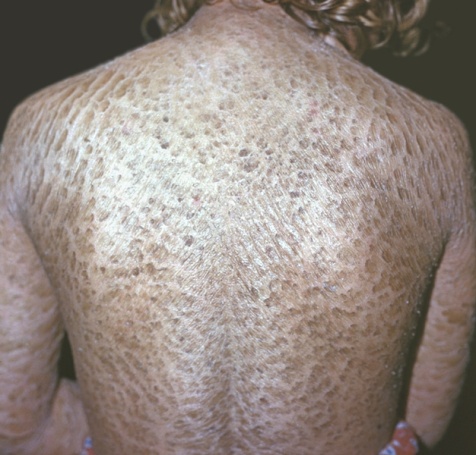 адзвичайно
рідкісне спадкове
злоякісне захворювання — природжений
іхтіоз
(від грец. іхтіс — риба). Вся шкіра такого
хворого покрита значними за розміром
зроговіли пластинами, що нагадують
луску риби; при цьому порушенні є
неможливим шкірне дихання. Дитина або
народжується мертвою, або вмирає
незабаром після народження.
адзвичайно
рідкісне спадкове
злоякісне захворювання — природжений
іхтіоз
(від грец. іхтіс — риба). Вся шкіра такого
хворого покрита значними за розміром
зроговіли пластинами, що нагадують
луску риби; при цьому порушенні є
неможливим шкірне дихання. Дитина або
народжується мертвою, або вмирає
незабаром після народження.
Як приклад злоякісних новоутворень, що передаються по аутосомно-рецесивному типу, можна назвати також гліому сітківки ока — пухлину глії (проміжної тканини центральної нервової системи).