

2. Бимолекулярное нуклеофильное замещение
(механизм SN2).
Второй механизм соответствует схеме 2 (стр. 151) и предполагает протекание одностадийной реакции, в ходе которой одновременно разрывается связь реакционного центра с нуклеофугом и образуется связь этого центра с нуклеофилом:
В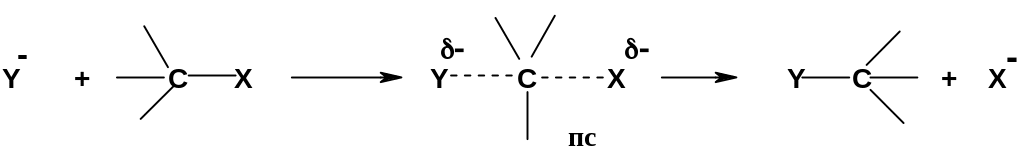 переходном состоянии (пс) связь С-Х
примерно наполовину разорвана, а связь
С-Н примерно наполовину образована;
отрицательный заряд распределен между
уходящей и вступающей группами (здесь
взят пример с анионным нуклеофилом –
для данного механизма это наиболее
частый случай). Далее связь С-Х окончательно
разрывается, а связь С-У окончательно
образуется. В отличие от механизма
SN1,
в переходном состоянии вокруг атома
углерода находится пять
групп (лигандов); реакционный центр
пентакоординирован. [Пять заместителей
вокруг атома углерода могут находиться
только в
переходном состоянии,
но не в
интермедиате
(см.выше)].
переходном состоянии (пс) связь С-Х
примерно наполовину разорвана, а связь
С-Н примерно наполовину образована;
отрицательный заряд распределен между
уходящей и вступающей группами (здесь
взят пример с анионным нуклеофилом –
для данного механизма это наиболее
частый случай). Далее связь С-Х окончательно
разрывается, а связь С-У окончательно
образуется. В отличие от механизма
SN1,
в переходном состоянии вокруг атома
углерода находится пять
групп (лигандов); реакционный центр
пентакоординирован. [Пять заместителей
вокруг атома углерода могут находиться
только в
переходном состоянии,
но не в
интермедиате
(см.выше)].
Очень важно отметить, что нуклеофил атакует реакционный центр только со стороны, обратной уходящей группе («атака с тыла»); более того, необходимым условием протекания этого механизма с достаточной скоростью является нахождение нуклеофила, реакционного центра и уходящей группы на одной прямой. Это – стереоэлектронное требование к данному механизму.
Поскольку в единственной (и, следовательно, скоростьопределяющей) стадии реакции принимают участие две молекулы (субстрат и реагент), данный механизм называют бимолекулярным нуклеофильным замещением и обозначают индексом SN2 (Substitution Nucleophilic bimolecular).
Энергетический профиль характерен для одностадийной реакции:
 G
G
Y---R---X
R-X R-Y
Координата реакции

Кинетические и стереохимические характеристики реакций SN2:
А. Кинетика. Скорость реакции зависит как от концентрации субстрата, так и от концентрации реагента; реакция имеет второй порядок – первый по субстрату и первый по реагенту: v=k[R-X][Y¯].
 Б.
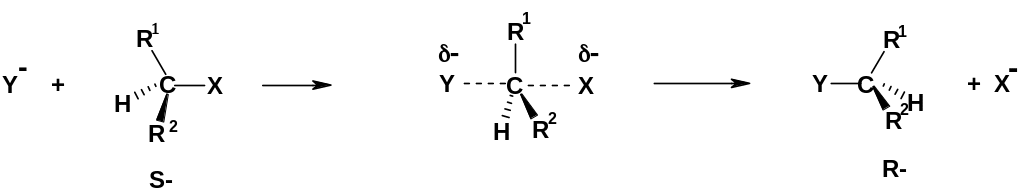
Стереохимия.
Если в реакцию
SN2
вводится оптически активный субстрат,
то конфигурация продукта реакции обратна
конфигурации субстрата;
т.е. результатом реакции является
обращение
конфигурации.
Это происходит вследствие того, что
нуклеофил подходит к реакционному
центру со стороны, обратной уходящей
группе: происходит «выворачивание»
конфигурации; этот процесс иногда
сравнивают с выворачиванием зонтика
при сильном ветре:
Б.
Стереохимия.
Если в реакцию
SN2
вводится оптически активный субстрат,
то конфигурация продукта реакции обратна
конфигурации субстрата;
т.е. результатом реакции является
обращение
конфигурации.
Это происходит вследствие того, что
нуклеофил подходит к реакционному
центру со стороны, обратной уходящей
группе: происходит «выворачивание»
конфигурации; этот процесс иногда
сравнивают с выворачиванием зонтика
при сильном ветре:
Таким образом, если исходный субстрат имеет S-конфигурацию, то конечный продукт будет иметь R-конфигурацию ( разумеется, если при замещении не меняется порядок старшинства заместителей).
Выше были рассмотрены «чистые» механизмы SN1 и SN2; они наблюдаются не всегда: в ряде случаев реакция имеет параметры, промежуточные между этими механизмами, например, дробный порядок реакции – выше первого и ниже второго. В подобных случаях говорят о «промежуточных» или «пограничных» механизмах. Природа этих механизмов является предметом дискуссий; наиболее простое предположение состоит в том, что часть молекул субстрата реагирует по механизму SN1, а другая часть – по механизму SN2, однако есть и другие предположения. В данном курсе этот вопрос подробно обсуждаться не будет; обсуждение сосредоточится на «чистых» механизмах.
Зависимость протекания реакций нуклеофильного замещения при sp3-атоме углерода от различных факторов.
После рассмотрения общих схем двух типовых механизмов нуклеофильного замещения можно попытаться «привязать» эти механизмы к конкретным субстратам и реакциям и использовать их для объяснения экспериментальных результатов, планирования проведения реакций и управления их протеканием. Для этого нужно представлять влияние различных факторов, во-первых, на выбор механизма, а, во-вторых, - на активность реагирующей системы, в случае оптически активных субстратов – на стереоселективность, а в некоторых особых случаях– и на региоселективность. [Если посмотреть на приведенную выше общую формальную схему реакций SN, может показаться, что проблема региоселективности здесь вообще не стоит: нуклеофил становится на то место, откуда ушел нуклеофуг. Однако в некоторых случаях замещение сопровождается перегруппировками, а в некоторых – реагент содержит более одного нуклеофильного центра; при этом становится возможным образование двух или более продуктов реакции замещения].
Прежде всего необходимо оценить активность реагирующей системы; здесь решающую роль играют скорости реакций. Для оценки скорости реакций SN2 необходимо оценить энергию переходного состояния единственной стадии реакции, а для оценки скорости реакций SN1 – энергию переходного состояния первой, лимитирующей стадии; вместо переходного состояния в реакциях SN1 можно рассматривать интермедиат (карбокатион) – согласно постулату Хэммонда его структура близка к структуре переходного состояния. Чем ниже энергия переходного состояния или интермедиата,тем выше скорость реакции; в случае же высокой энергии активации реакция может полностью блокироваться. Энергия активации реагирующей системы зависит как от внутренних, так и от внешних факторов.
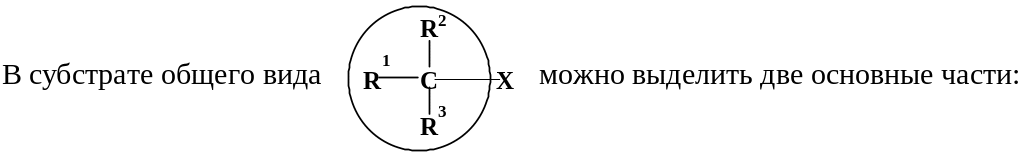 I.
Внутренние
факторы
– строение
субстрата и реагента.
I.
Внутренние
факторы
– строение
субстрата и реагента.
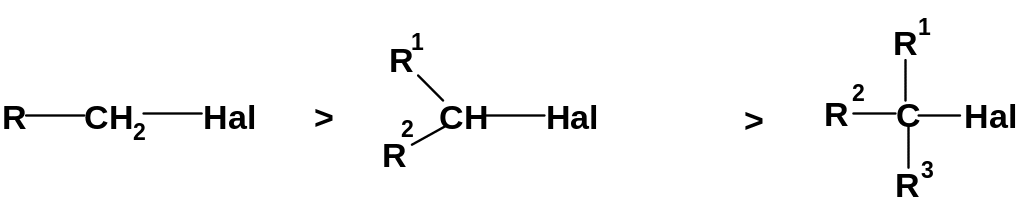
А. Строение субстрата.
1. Реакционный центр и связанные с ним группы R1, R2, R3; этот фрагмент можно назвать «основной частью» субстрата. 2. Уходящую группу Х (нуклеофуг).
1. Строение основной части субстрата. С реакционным центром могут быть связаны разные группы (водород, алкильные, алкенильные, арильные группы, гетероатомы). От их природы зависят электронные и пространственные факторы в субстрате.
А. Для реакций SN1 очень важную роль играют электронные факторы. Реакции SN1 ускоряются электронодонорными группами; эти группы стабилизируют интермедиат – карбокатион. Наиболее типичные иллюстрации:
а. Если с реакционным центром связаны алкильные группы, то лучше всего реагируют по механизму SN1 третичные субстраты, хуже – вторичные и плохо – первичные:
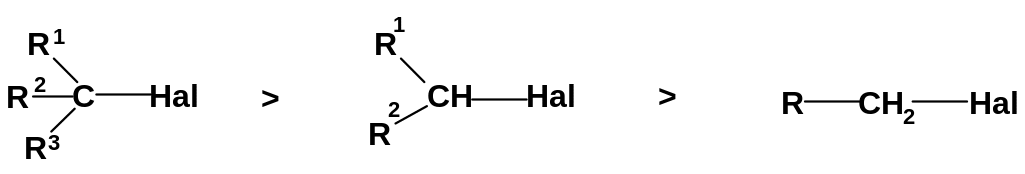 Алкильные
группы являются электронодонорными
(+I-эффект);
поэтому третичные карбокатионы являются
наиболее устойчивыми (с катионным
центром связаны три донорных группы);
вторичные (два донора) и тем более
первичные (один донор) карбокатионы
менее устойчивы. Разница в скоростях
может быть весьма значительной: например,
скорость взаимодействия трет-бутилбромида
(R1=R2=R3=CH3,
Hal=Br)
с водой на 8 порядков выше скорости
взаимодействия этилбромида с водой
(R=CH3,
Hal=Br).
[Тем не менее, не стоит думать, что
третичные алкилгалогениды – идеальные
субстраты для реакций SN1:
именно для этих субстратов реакции
замещения в наибольшей мере подавляются
конкурентными реакциями элиминирования;
это будет рассмотрено несколько позже].
Алкильные
группы являются электронодонорными
(+I-эффект);
поэтому третичные карбокатионы являются
наиболее устойчивыми (с катионным
центром связаны три донорных группы);
вторичные (два донора) и тем более
первичные (один донор) карбокатионы
менее устойчивы. Разница в скоростях
может быть весьма значительной: например,
скорость взаимодействия трет-бутилбромида
(R1=R2=R3=CH3,
Hal=Br)
с водой на 8 порядков выше скорости
взаимодействия этилбромида с водой
(R=CH3,
Hal=Br).
[Тем не менее, не стоит думать, что
третичные алкилгалогениды – идеальные
субстраты для реакций SN1:
именно для этих субстратов реакции
замещения в наибольшей мере подавляются
конкурентными реакциями элиминирования;
это будет рассмотрено несколько позже].
б. Хорошо реагируют по механизму SN1 аллильные и бензильные галогениды, потому что в этих случаях в интермедиатах –аллильных и бензильных карбокатионах - происходит делокализация заряда за счет сопряжения (в данных случаях связи С=С и арильные группы проявляют +М-эффект):
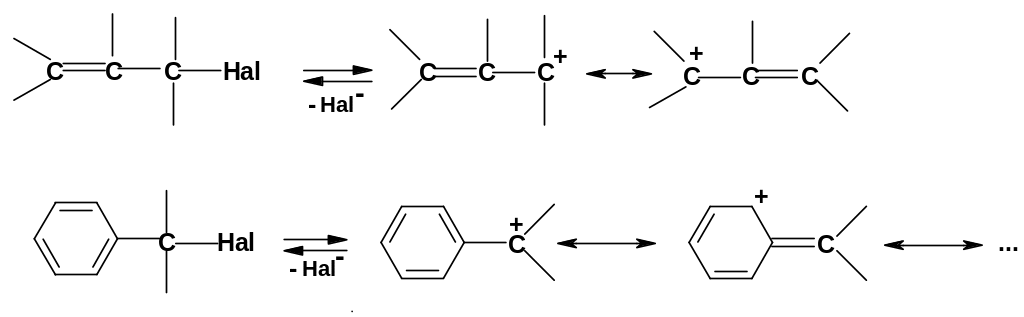 Даже
первичные аллил- и бензилгалогениды
реагируют по механизму SN1
быстрее вторичных алкилгалогенидов.
Исключительно
легко
реагируют по механизму SN1
диарилметилгалогениды Ar2CH-Hal
и особенно триарилметилгалогениды
Ar3C-Hal,
поскольку в карбокатионе заряд
делокализован с участием двух или трех
ароматических ядер; такие субстраты
реагируют на 3-8 порядков быстрее
бензилгалогенидов.
Даже
первичные аллил- и бензилгалогениды
реагируют по механизму SN1
быстрее вторичных алкилгалогенидов.
Исключительно
легко
реагируют по механизму SN1
диарилметилгалогениды Ar2CH-Hal
и особенно триарилметилгалогениды
Ar3C-Hal,
поскольку в карбокатионе заряд
делокализован с участием двух или трех
ароматических ядер; такие субстраты
реагируют на 3-8 порядков быстрее
бензилгалогенидов.
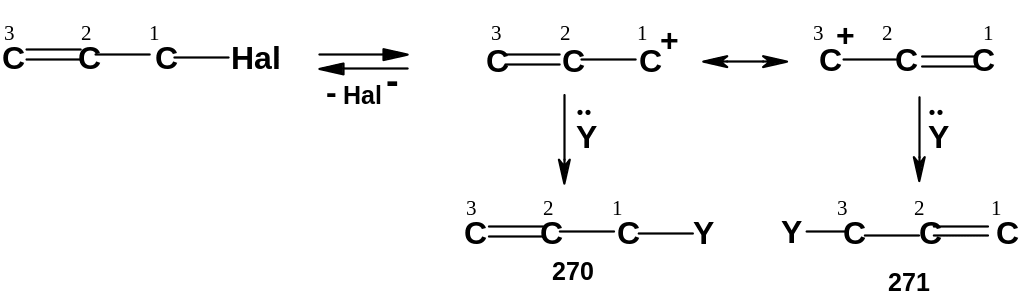
Нуклеофильное замещение в аллильных субстратах по механизму SN1 обычно сопровождается аллильной перегруппировкой: наряду с продуктом «нормального» замещения (270) образуется продукт замещения с перегруппировкой (271), в котором нуклеофил присоединяется к атому С3, а двойная связь перемещается в положение 1,2:
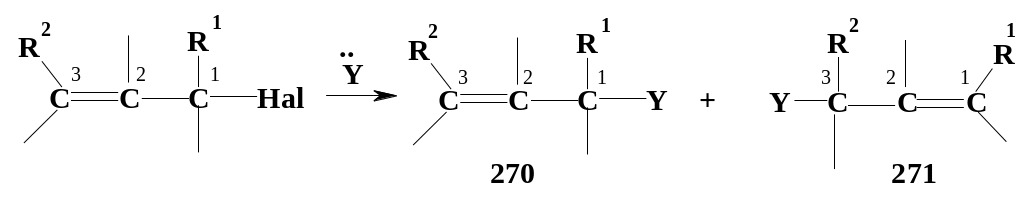 Причина
- делокализация заряда в промежуточном
карбокатионе между двумя атомами
углерода (С1и С3). Заряд
распределяется между ними довольно
равномерно; нуклеофил может присоединяться
и к одному и к другому атому, образуя
как продукт «нормального»замещения
(270), так и продукт замещения с
перегруппировкой (271):
Причина
- делокализация заряда в промежуточном
карбокатионе между двумя атомами
углерода (С1и С3). Заряд
распределяется между ними довольно
равномерно; нуклеофил может присоединяться
и к одному и к другому атому, образуя
как продукт «нормального»замещения
(270), так и продукт замещения с
перегруппировкой (271):
 Б.
Для реакций
SN2
влияние
электронных факторов выражено менее
заметно, чем
для реакций SN1;
к тому же оно не столь однозначно: в
случае «чистого» механизма SN2
реакция в определенной степени ускоряется
акцепторными
группами,
потому что они увеличивают частичный
положительный заряд на реакционном
центре. Если же механизм смешанный, то
реакция может ускоряться донорами, а в
отдельных случаях электронное влияние
даже отсутствует.
Б.
Для реакций
SN2
влияние
электронных факторов выражено менее
заметно, чем
для реакций SN1;
к тому же оно не столь однозначно: в
случае «чистого» механизма SN2
реакция в определенной степени ускоряется
акцепторными
группами,
потому что они увеличивают частичный
положительный заряд на реакционном
центре. Если же механизм смешанный, то
реакция может ускоряться донорами, а в
отдельных случаях электронное влияние
даже отсутствует.
Зато первостепенную роль приобретают пространственные факторы.
а. Напряжение в переходном состоянии. Этот фактор действует наиболее часто. Как уже указывалось, в переходном состоянии реакции SN2 вокруг реакционного центра находятся пять заместителей. Поэтому, если хотя бы часть из них имеет более или менее значительный объём, вокруг реакционного центра становится «тесно»; возникает отталкивание, т.е. пространственное напряжение, повышающее энергию переходного состояния; энергия активации возрастает и скорость реакции уменьшается. Вследствие этого первичные субстраты реагируют по механизму SN2 легче вторичных; третичные субстраты редко реагируют по этому механизму (тем более что для них легко идут конкурирующие реакции элиминирования):

 б.
Стереоэлектронный
фактор. Как
уже упоминалось, протекание реакции
SN2
возможно только тогда, когда нуклеофил
подходит к реакционному центру «в тыл»
уходящей группе так, чтобы образовалось
линейное
трехцентровое переходное состояние.
Если тыловая сторона пространственно
экранирована, то подход нуклеофила с
тыла затруднен; реакция сильно замедляется
или вообще блокируется.
б.
Стереоэлектронный
фактор. Как
уже упоминалось, протекание реакции
SN2
возможно только тогда, когда нуклеофил
подходит к реакционному центру «в тыл»
уходящей группе так, чтобы образовалось
линейное
трехцентровое переходное состояние.
Если тыловая сторона пространственно
экранирована, то подход нуклеофила с
тыла затруднен; реакция сильно замедляется
или вообще блокируется.
Один из наиболее типичных примеров – неопентилгалогениды (272); весьма объёмная трет-бутильная группа не позволяет подойти к нуклеофилу точно в тыл атому галогена, и соединения (272) практически не реагируют по механизму SN2. Другой пример – мостиковые галогениды (273) (производные норборнана); здесь атака с тыла невозможна, так как нуклеофил не может проникнуть внутрь мостиковой системы; поэтому соединения (273) не реагируют по механизму SN2 [по механизму SN1 реакция здесь также не идет, т.к. не может образоваться плоский карбокатион; т.е. атом галогена здесь вообще не замещается].
2. Природа уходящей группы (нуклеофуга).
Реакции нуклеофильного замещения протекают тем быстрее, чем легче происходит отрыв уходящей группы. Легкость отрыва нуклеофуга изменяется в очень широком диапазоне; различают «хорошие» и «плохие» уходящие группы. Хорошие уходящие группы – те, которые уходят в виде устойчивых анионов или нейтральных молекул. Плохие группы уходят в виде малоустойчивых анионов. Для галогенпроизводных нуклеофуг отрывается в виде галогенид-аниона. Устойчивость анионов растет в ряду F¯< Cl¯ < Br¯ < I¯; фтор является плохой уходящей группой, йод – хорошей; хлор и бром занимают промежуточное положение. Действительно, при прочих равных условиях фторпроизводные обычно реагируют наиболее медленно, йодпроизводные – наиболее быстро. В качестве примера можно привести реакцию получения фторпроизводных из других галогенпроизводных путем обмена галогенов:
R-Br + K+F- R-F + K+Br-
Реакция идет слева направо; обратная реакция не идет, т.к. атом фтора – плохая уходящая группа.
Указанные закономерности относятся и к реакциям SN1 и к реакциям SN2; но все же большее значение имеют для реакций SN1; для их успешного протекания весьма желательна хорошая уходящая группа.
3. Природа нуклеофильного реагента.
Природа нуклеофила может влиять на скорость, а в некоторых специальных случаях – и на региоселективность нуклеофильного замещения.
а. Влияние на скорость.
В реакциях SN1 скорость не зависит от природы нуклеофила, т.к. нуклеофил присоединяется не на лимитирующей, а на быстрой стадии. Поэтому в реакциях SN1 широко используют слабые нуклеофилы (вода, спирты, карбоксилат-анионы). Такие нуклеофилы, как вода и спирты, обычно бывают растворителями в данных реакциях; реакции, в которых реагент одновременно является растворителем, называются реакциями сольволиза. [Надо, однако, иметь в виду, что галогенпроизводные нерастворимы в воде, поэтому для их гидролиза нужно использовать смесь воды с органическим растворителем, который смешивается с водой и не обладает нуклеофильными свойствами; обычно используют смеси воды с ацетоном].
Реакции SN2 идут тем быстрее, чем более сильным нуклеофилом является реагент. Если нуклеофил слабый, реакция SN2 идет крайне медленно. Такая зависимость вполне естественна, т.к. нуклеофил здесь участвует в скоростьопределяющей стадии.
Оценить силу нуклеофила не всегда просто; она, в частности, зависит от применяемого растворителя и от ряда других факторов. Поэтому можно сформулировать лишь некоторые закономерности, справедливые в первом приближении:
1. Анионы более нуклеофильны, чем их сопряженные кислоты (т.е. продукты их протонирования), если эти сопряженные кислоты содержат атомы с неподеленными парами электронов (т.е. обладают нуклеофильностью). Например, ОН¯ значительно сильнее, чем Н2О, а NH2¯ значительно сильнее NH3.
2. Чем устойчивее анион, тем слабее его нуклеофильность. Например, алкоголят-анионы RO‾ значительно сильнее карбоксилат-анионов RCOO‾, в которых заряд делокализован по двум атомам кислорода (стр. 33). Чрезвычайно стабильные сульфат- и перхлорат-анионы практически не обладают нуклеофильными свойствами.
3. Если нуклеофильные центры находятся на атомах элементов одного периода, то слева направо по периоду нуклеофильность уменьшается. Например, NH3 > H2O; NH2¯ > OH¯ > F¯.
4. Если нуклеофильные центры находятся на атомах элементов одной группы, то сверху вниз по группе нуклеофильность растет. Например, F¯ < Cl¯ < Br¯ < I¯ .
 б.
Влияние на
селективность.
Проблема региоселективности и
регионаправленности возникает в том
случае, когда в качестве нуклеофилов
выступают амбидентные
анионы. Это
– анионы, содержащие два или более
сильно взаимодействующих нуклеофильных
центра; при этом с субстратом может
реагировать только
один центр;
вступление его в реакцию «выключает»
остальные центры. [Не следует путать
амбидентные анионы с бинуклеофилами,
например, двухатомными спиртами или
диаминами – у них каждый нуклеофильный
центр может реагировать независимо от
другого].
б.
Влияние на
селективность.
Проблема региоселективности и
регионаправленности возникает в том
случае, когда в качестве нуклеофилов
выступают амбидентные
анионы. Это
– анионы, содержащие два или более
сильно взаимодействующих нуклеофильных
центра; при этом с субстратом может
реагировать только
один центр;
вступление его в реакцию «выключает»
остальные центры. [Не следует путать
амбидентные анионы с бинуклеофилами,
например, двухатомными спиртами или
диаминами – у них каждый нуклеофильный
центр может реагировать независимо от
другого].
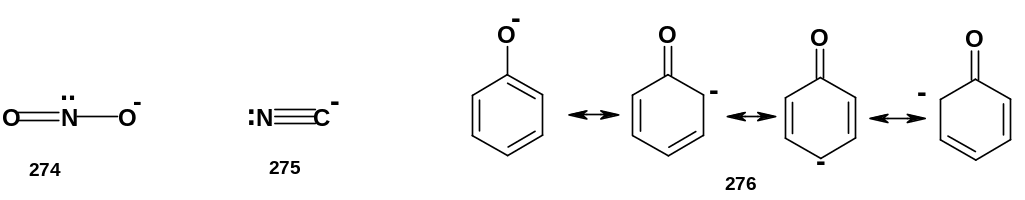
Ниже приведены некоторые примеры амбидентных анионов:
В нитрит-анионе (274) два нуклеофильных центра – отрицательно заряженный атом кислорода и атом азота, имеющий неподеленную пару электронов. Аналогичная ситуация (анионный центр и нейтральный атом с неподеленной парой) в цианид-анионе (275). В фенолят-анионе (276) четыре нуклеофильных центра – атом кислорода и атомы углерода в орто- и пара-положениях к кислороду; в резонансном гибриде, каковым является фенолят-анион, частичный отрицательный заряд на атоме кислорода, естественно, значительно больше, чем на атомах углерода. Известно еще несколько типов амбидентных анионов; одними из наиболее важных (если не самыми важными) являются анионы 1,3-дикарбонильных соединений и эфиров 3–оксокарбоновых кислот; они будут рассмотрены позднее.
При вступлении в реакцию разных центров образуются разные продукты:
Т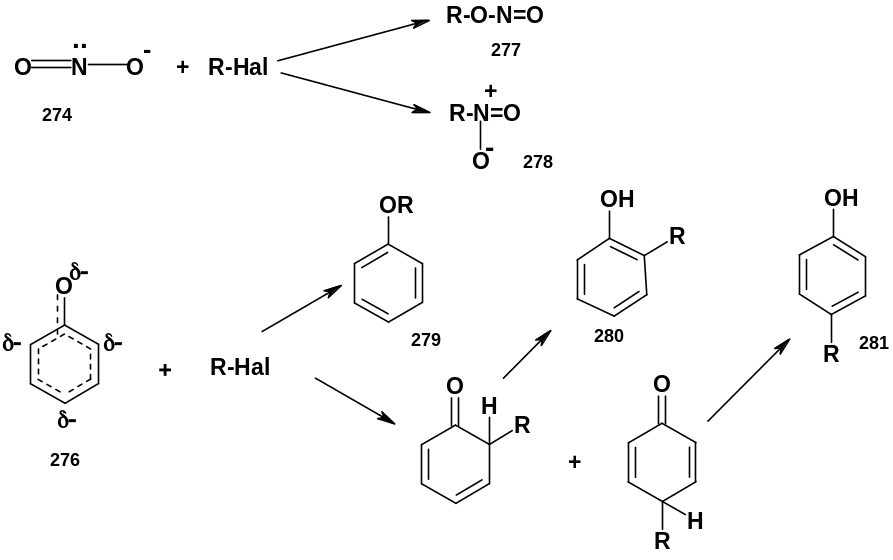 ак,
при взаимодействии галогенидов с
нитрит-анионом возможно образование
эфиров азотистой кислоты – алкилнитритов
(277) (в реакцию вступает О-центр амбидентного
аниона) или нитросоединения (278) (в реакцию
вступает N-центр).
Аналогично, из фенолят-аниона возможно
получение простого эфира фенола (279)
(О-алкилирование) или замещенных
циклогексадиенонов, которые
перегруппировываются в фенолы (280, 281)
(С-алкилирование).
ак,
при взаимодействии галогенидов с
нитрит-анионом возможно образование
эфиров азотистой кислоты – алкилнитритов
(277) (в реакцию вступает О-центр амбидентного
аниона) или нитросоединения (278) (в реакцию
вступает N-центр).
Аналогично, из фенолят-аниона возможно
получение простого эфира фенола (279)
(О-алкилирование) или замещенных
циклогексадиенонов, которые
перегруппировываются в фенолы (280, 281)
(С-алкилирование).
Вопрос заключается в том, какой из центров амбидентного аниона вступает в реакцию с определенным субстратом в определенных условиях.
Однозначный ответ для всех случаев реакций подобных анионов вряд ли возможен, т.к. результат зависит от сочетания многих факторов. Однако можно сформулировать основные закономерности, позволяющие с определенной степенью вероятности предсказывать селективность реакций. Эти закономерности выведены на основе принципа жестких и мягких кислот и оснований (принципа ЖМКО).
Как уже упоминалось, взаимодействие нуклеофила и электрофила есть взаимодействие основания Льюиса и кислоты Льюиса. Основания и кислоты Льюиса можно разделить на жесткие и мягкие (Р.Пирсон, 1963):
Жесткие основания – основания Льюиса, у которых атом-донор электронной пары имеет высокую электроотрицательность и низкую поляризуемость. К этому типу оснований относятся, в частности, Н2О, ОН¯, ROH, RO¯, Cl¯, NH3, RNH2
Мягкие основания – основания, у которых донорный атом имеет низкую электроотрицательность и высокую поляризуемость. Наиболее известные основания этого типа – Н2S, RSH, RS¯, J¯, CN¯, алкены, арены.
Жесткие кислоты – кислоты Льюиса, у которых атомы – акцепторы электронной пары имеют малые размеры, высокий положительный заряд и низкую поляризуемость. К ним относятся Na+, Li+, BF3, AlCl3; наиболее известная жесткая кислота – Н+.
Мягкие кислоты – кислоты, у которых атомы-акцепторы имеют большие размеры, малый положительный заряд и высокую поляризуемость.К этой группе относятся Cu+, Ag+, Hg2+, Br2, I2, карбонильные соединения.
Основное правило, характеризующее кислотно-основные взаимодействия: жесткие кислоты преимущественно реагируют с жесткими основаниями, а мягкие кислоты – с мягкими основаниями.
Понятия «жесткий» и «мягкий» – сугубо качественные; основное правило не имеет абсолютного характера; тем не менее, с его помощью удается удовлетворительно объяснить значительное число экспериментальных данных и предсказать с достаточной надежностью целый ряд новых результатов.
Если вернуться к амбидентным анионам, легко заметить, что их нуклеофильные (основные) центры различаются по степени жесткости (мягкости) – один из них всегда более жесткий, другой – более мягкий. Например, у нитрит-аниона (274) анионный атом кислорода – жесткий центр, нейтральный атом азота – мягкий центр. У цианид-аниона (275) атом азота более жесткий, чем атом углерода. У фенолят-аниона атом кислорода – заметно более жесткий центр, чем атомы углерода в о- и р-положениях.
Субстраты, с которыми реагируют нуклеофилы, – кислоты Льюиса. При реакции SN1 нуклеофил взаимодействует с карбокатионом, который является жесткой кислотой (высокий положительный заряд). Поэтому в реакциях SN1 преимущественно реагирует более жесткий центр амбидентного аниона.
При реакции SN2 нуклеофил атакует атом углерода, имеющий частичный, небольшой по величине положительный заряд, т.е. мягкую кислоту. Поэтому в реакциях SN2 преимущественно реагирует более мягкий центр амбидентного аниона.
В качестве примера можно рассмотреть реакцию галогенпроизводных с нитрит-анионом (см. выше). Eсли реакция идет по механизму SN1 (например, хорошая уходящая группа, бензильный субстрат, протонный растворитель ), то преимущественно образуются эфиры азотистой кислоты (277) – как результат атаки субстрата жестким кислородным центром амбидентного аниона. Если реакция идет по механизму SN2 (первичный субстрат, диполярный апротонный растворитель), то преимущественно образуются нитросоединения 278, поскольку здесь предпочтительно реагирует мягкий азотный центр.
II. Внешние факторы. Здесь наиболее важно рассмотреть влияние растворителя. Растворители играют заметную роль в любых гетеролитических реакциях; в реакциях нуклеофильного замещения их влияние весьма значительно и достаточно хорошо изучено.
Растворители сольватируют участвующие в реакции частицы – исходные и конечные продукты, переходные состояния, интермедиаты; сольватация понижает энергию сольватируемых частиц. Если переходное состояние или интермедиат сольватируются сильнее исходных соединений, то энергия активации снижается, и реакция ускоряется; если сильнее сольватируются исходные частицы, реакция замедляется.
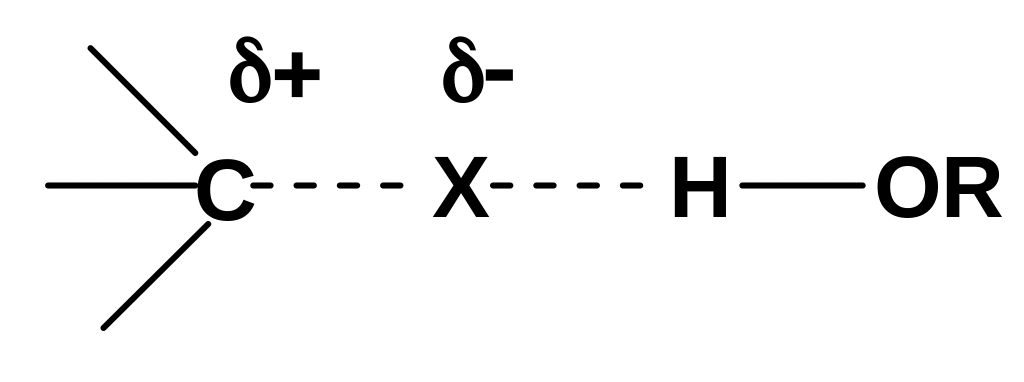 Для
проведения реакций SN1
наилучшими
являются
полярные
протонные растворители
– вода, спирты, карбоновые кислоты. В
этих реакциях переходные состояния и
интермедиаты (ионы) более
полярны,
чем исходные субстраты, и поэтому они
сильнее, чем субстраты, сольватируются
полярными
растворителями. Особо сильно сольватируется
уходящая группа путем образования
водородных связей с протонным
растворителем; таким образом, протонный
растворитель помогает уходу нуклеофуга:
Для
проведения реакций SN1
наилучшими
являются
полярные
протонные растворители
– вода, спирты, карбоновые кислоты. В
этих реакциях переходные состояния и
интермедиаты (ионы) более
полярны,
чем исходные субстраты, и поэтому они
сильнее, чем субстраты, сольватируются
полярными
растворителями. Особо сильно сольватируется
уходящая группа путем образования
водородных связей с протонным
растворителем; таким образом, протонный
растворитель помогает уходу нуклеофуга:
В некоторых случаях полярные протонные растворители одновременно являются и нуклеофильными реагентами – это реакции сольволиза (см. выше).
При проведении реакций SN2 с участием анионного нуклеофила (этот случай наиболее типичный) полярные протонные растворители, напротив, затрудняют протекание реакции, т.к. они сильно сольватируют исходный нуклеофил: Y‾-----H-OR и, тем самым, снижают его активность. Здесь можно использовать неполярные апротонные растворители: бензол, гексан, диэтиловый эфир, диоксан. Но наилучшие результаты дает использование диполярных апротонных растворителей, таких как диметилформамид HCON(CH3)2, диметилсульфоксид (СН3)2SО и некоторых других. Молекулы этих растворителей представляют собой диполи, положительный полюс которых пространственно экранирован (закрыт), а отрицательный открыт:
П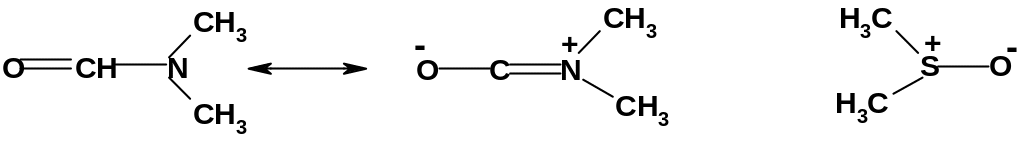 оэтому
такие растворители хорошо сольватируют
катионы, но практически не сольватируют
анионы. Заряженный нуклеофил (анион)
всегда имеет противоион, с которым
взаимодействует (например, Na+
ОН-);
это взаимодействие понижает активность
аниона в реакции с субстратом. Диполярный
апротонный растворитель сольватирует
противоион и высвобождает анион, делая
его более активным; скорость реакции
SN2
заметно возрастает.
оэтому
такие растворители хорошо сольватируют
катионы, но практически не сольватируют
анионы. Заряженный нуклеофил (анион)
всегда имеет противоион, с которым
взаимодействует (например, Na+
ОН-);
это взаимодействие понижает активность
аниона в реакции с субстратом. Диполярный
апротонный растворитель сольватирует
противоион и высвобождает анион, делая
его более активным; скорость реакции
SN2
заметно возрастает.
Иногда роль растворителя заключается не в сольватации, а просто в растворимости исходных и конечных продуктов. В качестве примера можно привести реакцию замещения хлора или брома на иод (реакция Финкельштейна):
R-Cl + NaJ R-J + NaCl
Реакцию проводят в ацетоне, в котором NaJ растворим, а NaCl и NaBr нерастворимы; они выпадают в осадок, и равновесие смещается вправо.
Совокупность приведенных закономерностей позволяет сориентироваться в реакциях нуклеофильного замещения при sp3-атоме углерода. Обычная логическая схема при этом бирают подходящий механизм (моно- или бимолекулярный); далее подбирают оптимальные условия для проведения реакции по выбранному механизму.
Ниже суммированы признаки, характерные для протекания реакции замещения по тому или иному механизму:
SN1 |
SN2 |
Полярные протонные растворители Третичные субстраты Хорошая уходящая группа Слабый нуклеофил Жесткий центр амбидентного аниона. |
Апротонные (лучше диполярные) растворители Первичные субстраты Сильный нуклеофил Мягкий центр амбидентного аниона |