

Регионаправленность реакций электрофильного замещения в конденсированных ароматических системах.
Ранее рассмотренные примеры региоселективности и регионаправленности реакций электрофильного замещения относились к производным бензола. Для конденсированных аренов проблема региоселективности и регионаправленности появляется уже при введении первого заместителя в ядро, поскольку уже в незамещенных конденсированных аренах атомы водорода не эквивалентны.
Рассмотрим реакции электрофильного замещения в нафталине; здесь возможны два варианта введения одного заместителя – в положение 1 (-положение) и в положение 2 (-положение):
П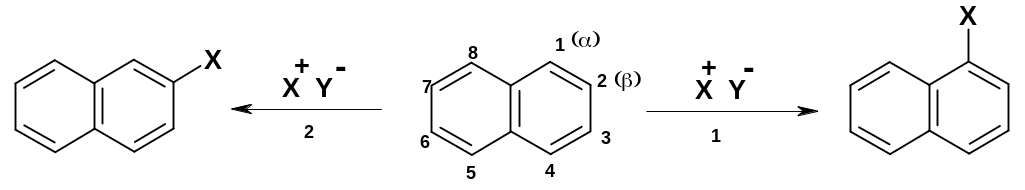 ри
кинетически контролируемых реакциях
электрофильного замещения заместитель
ориентируется в положение 1.
Это становится понятным при рассмотрении
устойчивости -аддуктов,
образующихся при протекании реакций
по обоим возможным направлениям:
ри
кинетически контролируемых реакциях
электрофильного замещения заместитель
ориентируется в положение 1.
Это становится понятным при рассмотрении
устойчивости -аддуктов,
образующихся при протекании реакций
по обоим возможным направлениям:
В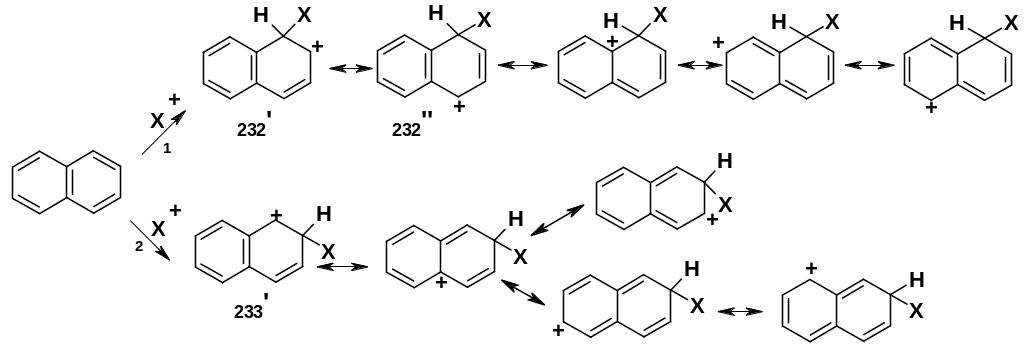 обоих случаях σ-аддукты включают пять
резонансных структур. При ориентации
в положение 1 (направление 1) две
из них -
(232’) и (232’’) - обладают повышенной
устойчивостью, так как в них сохраняется
ароматическое ядро;
в остальных трех ароматическая структура
полностью нарушена, и они заметно менее
устойчивы. При ориентации в положение
2 (направление 2) только
в одной
резонансной структуре (233’) сохраняется
ароматическое ядро, остальные четыре
структуры неустойчивы. Следовательно,
σ-аддукт,
образующийся при протекании реакции
по направлению 1, более устойчив, что и
обеспечивает доминирование этого
направления.
обоих случаях σ-аддукты включают пять
резонансных структур. При ориентации
в положение 1 (направление 1) две
из них -
(232’) и (232’’) - обладают повышенной
устойчивостью, так как в них сохраняется
ароматическое ядро;
в остальных трех ароматическая структура
полностью нарушена, и они заметно менее
устойчивы. При ориентации в положение
2 (направление 2) только
в одной
резонансной структуре (233’) сохраняется
ароматическое ядро, остальные четыре
структуры неустойчивы. Следовательно,
σ-аддукт,
образующийся при протекании реакции
по направлению 1, более устойчив, что и
обеспечивает доминирование этого
направления.
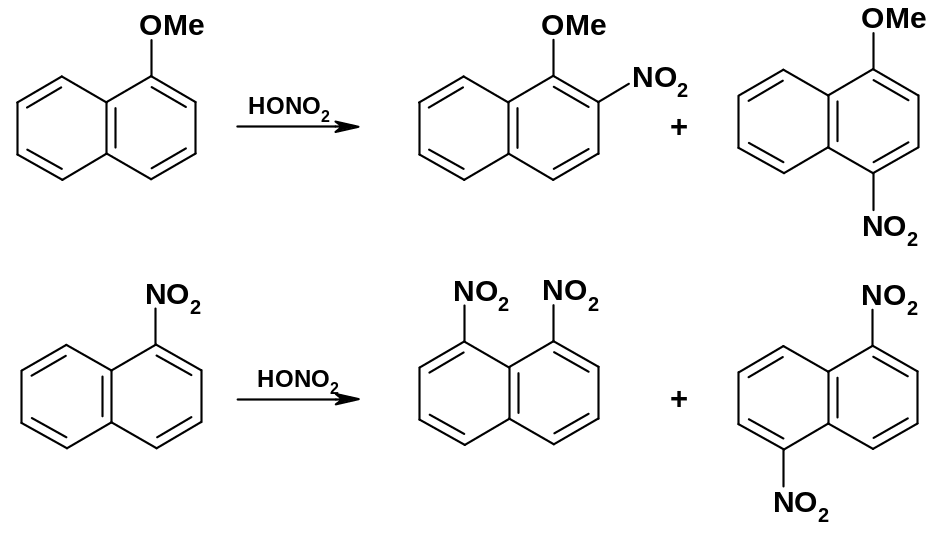
Если необходимо ввести в молекулу нафталина второй заместитель, то действует следующее правило: если первый заместитель донорный, то второй заместитель направляется в то же кольцо, в котором находится первый заместитель; если первый заместитель акцепторный , то второй заместитель направляется в другое кольцо:
 Для
антрацена (204) и фенантрена (234) вступающая
группа ориентируется в центральное
ядро, в положение 9 (10):
Для
антрацена (204) и фенантрена (234) вступающая
группа ориентируется в центральное
ядро, в положение 9 (10):
Т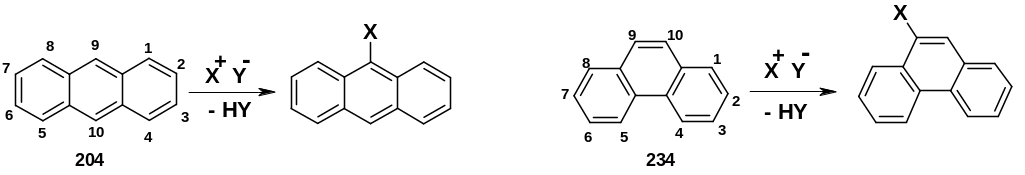 акое
направление реакций объясняется тем,
что -аддукты
в этих случаях включают устойчивые
резонансные структуры, содержащие два
изолированных бензольных
ядра, в которых электронная плотность
максимально делокализована; это дает
наибольший выигрыш в энергии. При
ориентации в другие положения резонансные
структуры аддуктов могут включать
только два конденсированных
бензольных ядра, что дает меньший выигрыш
в энергии.
акое
направление реакций объясняется тем,
что -аддукты
в этих случаях включают устойчивые
резонансные структуры, содержащие два
изолированных бензольных
ядра, в которых электронная плотность
максимально делокализована; это дает
наибольший выигрыш в энергии. При
ориентации в другие положения резонансные
структуры аддуктов могут включать
только два конденсированных
бензольных ядра, что дает меньший выигрыш
в энергии.
В конденсированных системах – нафталине и, тем более, антрацене и фенантрене – электронная плотность делокализована в несколько меньшей степени, чем в бензоле, поэтому их стабильность несколько меньше. С другой стороны, σ-аддукты не менее устойчивы, чем в реакциях бензола. Поэтому энергия активации для конденсированных систем ниже, чем для бензола; нафталин реагирует с электрофильными реагентами быстрее, чем бензол; антрацен и фенантрен еще легче вступают в электрофильные реакции.
Еще раз напомним: все рассмотренные выше правила ориентации (регионаправленности реакций) относятся к кинетически контролируемым реакциям. При термодинамическом контроле ориентация может быть иной. В качестве примера можно рассмотреть сульфирование нафталина:
К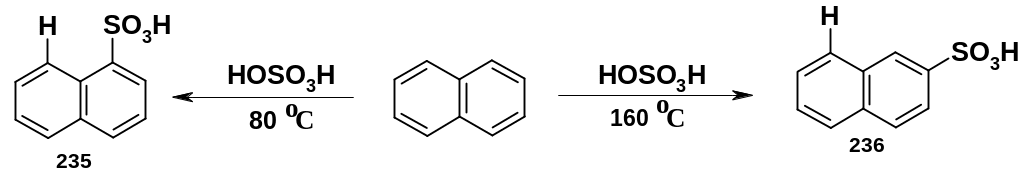 ак
уже упоминалось, реакция сульфирования
в достаточной степени обратима. При 80
оС реакция
контролируется кинетически
(равновесие
не достигается); сульфогруппа в
соответствии с рассмотренными выше
правилами ориентируется в положение 1
[продукт (235)]. При более высокой температуре
(160 оС)
равновесие достигается, реакция
контролируется термодинамически;
сульфогруппа ориентируется в положение
2 [продукт (236)]. Такая ориентация объясняется
тем, что 1-нафталинсульфокислота
(235) менее
устойчива,
чем 2-изомер
(236), потому что в продукте (235) происходит
отталкивание между сближенными в
пространстве сульфогруппой и атомом
водорода в положении 8 (напомним, что
при термодинамическом контроле образуется
более устойчивый конечный
продукт, а
не интермедиат).
ак
уже упоминалось, реакция сульфирования
в достаточной степени обратима. При 80
оС реакция
контролируется кинетически
(равновесие
не достигается); сульфогруппа в
соответствии с рассмотренными выше
правилами ориентируется в положение 1
[продукт (235)]. При более высокой температуре
(160 оС)
равновесие достигается, реакция
контролируется термодинамически;
сульфогруппа ориентируется в положение
2 [продукт (236)]. Такая ориентация объясняется
тем, что 1-нафталинсульфокислота
(235) менее
устойчива,
чем 2-изомер
(236), потому что в продукте (235) происходит
отталкивание между сближенными в
пространстве сульфогруппой и атомом
водорода в положении 8 (напомним, что
при термодинамическом контроле образуется
более устойчивый конечный
продукт, а
не интермедиат).
Реакции электрофильного замещения с термодинамическим контролем – более редкий вариант, чем кинетически контролируемые реакции; вероятность термодинамического контроля увеличивается при повышении температуры реакции.
Коротко остановимся еще на двух моментах, касающихся региоселективности реакций электрофильного замещения в ароматическом ядре.
А. Степень региоселективности. Было бы ошибкой считать, что заместитель в ядре всегда ориентирует вступающую группу исключительно в то или иное положение (те или иные положения). В общем случае речь идет о преимущественной ориентации; во многих реакциях образуются некоторые количества продуктов «неправильной» ориентации, например, m-изомеров для ориентантов первого рода. [В этом отношении правила ориентации менее строги, чем рассмотренные ранее правила протекания перициклических реакций, имеющие абсолютный характер].
Степень региоселективности зависит: а). От силы ориентанта (степени его активирующей или дезактивирующей способности): чем сильнее ориентант, тем выше региоселективность; например, группа ОН ориентирует вступающий заместитель более «точно», чем группа СН3. б). От активности электрофильной частицы: чем она более активна, тем меньше степень региоселективности; это один из типичных примеров действия принципа «более активно – менее селективно». Из рассмотренных реакций наименее региоселективна реакция алкилирования по Фриделю-Крафтсу; например, при алкилировании толуола этилбромидом образуется до 20% «неправильного» m-этилтолуола. Дело в том, что электрофильная частица (R+) весьма активна и, следовательно, мало «разборчива». Ацилирование по Фриделю-Крафтсу и бромирование более региоселективны, т.к. электрофильные частицы – соответственно, RСО+ и Br+ - менее активны. В частности, при ацилировании того же толуола хлорангидридом уксусной кислоты образуется не более 2% т-ацетилтолуола.
Б. Соотношение о- и р-изомеров. Заместители первого рода ориентируют вступающую группу как в орто-, так и в пара-положение; естественен вопрос, в каком соотношении образуются орто- и пара-изомеры. Если замещение в этих положениях равновероятно, то соотношение орто-/пара- должно составлять 2:1, т.к. есть два орто-положения и только одно пара-положение. Реально это соотношение, как правило, не соблюдается: в большинстве случаев пара-изомера образуется больше, чем «по статистике», а орто-изомера – меньше. Можно назвать две причины этого.
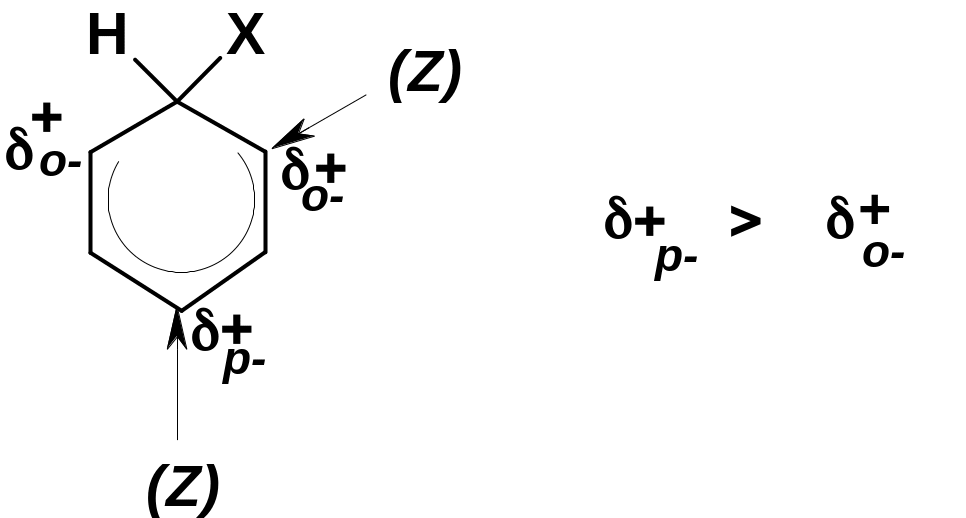 Первая,
несколько менее очевидная, заключается
в том, что в -аддукте
величина положительного заряда в
пара-положении
к вступающей
группе (Х)
несколько больше, чем в орто-положении.
Поэтому, если электронодонорный ориентант
(Z)
стоит в пара-положении
к группе Х, то он стабилизирует -аддукт
в большей степени, чем если он стоит в
орто-положении
(чем больше заряд, тем сильнее
стабилизирующее действие донора).
Первая,
несколько менее очевидная, заключается
в том, что в -аддукте
величина положительного заряда в
пара-положении
к вступающей
группе (Х)
несколько больше, чем в орто-положении.
Поэтому, если электронодонорный ориентант
(Z)
стоит в пара-положении
к группе Х, то он стабилизирует -аддукт
в большей степени, чем если он стоит в
орто-положении
(чем больше заряд, тем сильнее
стабилизирующее действие донора).
Вторая,
более очевидная, состоит в том, что в
орто-изомере
две группы (Х и Z)
сближены, а в пара-изомере
они удалены. Поэтому, если ориентирующая
или вступающая группа (или они обе) имеют
достаточно большой объём, то при
образовании орто-изомера
возникает пространственное напряжение
(отталкивание), уменьшающее стабильность
-аддукта.
В качестве примера можно привести
нитрование толуола (237) и трет-б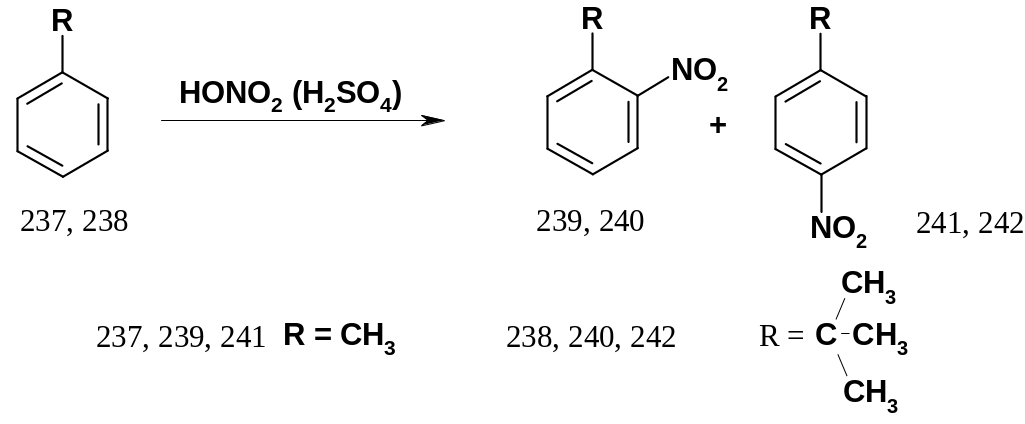 утилбензола
(238):
утилбензола
(238):
При нитровании толуола (237) образуется 58% о-изомера (239) и 37% р-изомера (241); при нитровании трет-бутилбензола (238) образуется лишь 16% о-изомера (240) и 73% р-изомера (242).
Таковы, в общем виде, закономерности влияния заместителей в ядре на активность и селективность реакций электрофильного замещения. Эти закономерности позволяют целенаправленно проводить модификацию ароматических структур – как наращивание углеродного скелета, так и функционализацию. Приведем два простых примера:
Пример 1. Как из толуола получить: А. пара-нитробензойную кислоту?
Б. мета-нитробензойную кислоту?
А. Вначале проводят нитрование толуола с образованием пара-нитротолуола; метильная группа – ориентант первого рода, поэтому нитрогруппа направляется в о- и р-положения. Затем окисляют метильную группу р-нитротолуола до карбоксильной (реакция будет рассмотрена ниже).
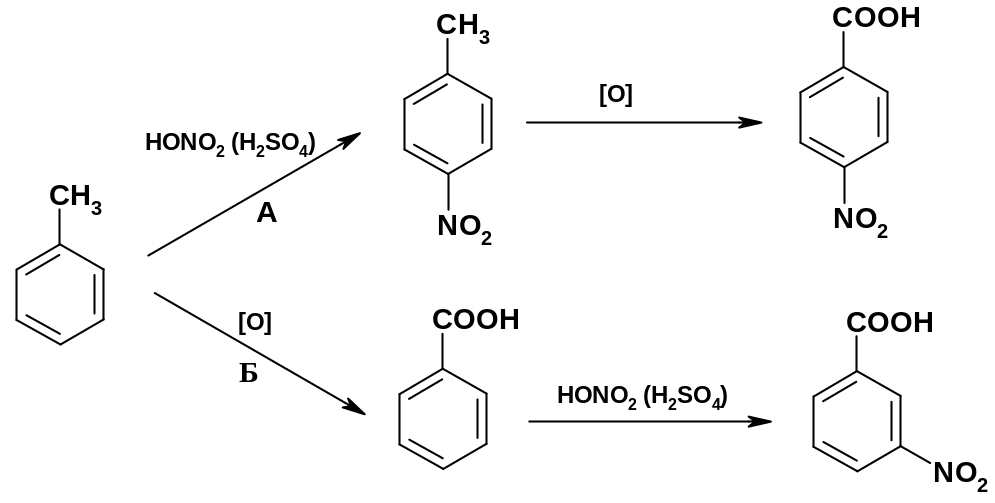 Б.
Вначале окисляют толуол до бензойной
кислоты; далее проводят нитрование
кислоты; карбоксильная группа – ориентант
второго
рода – направляет нитрогруппу в
m-положение;
нитрование бензойной кислоты идет
заметно труднее, чем нитрование толуола,
но, применяя достаточно жесткие условия,
удается получить хороший выход продукта:
Б.
Вначале окисляют толуол до бензойной
кислоты; далее проводят нитрование
кислоты; карбоксильная группа – ориентант
второго
рода – направляет нитрогруппу в
m-положение;
нитрование бензойной кислоты идет
заметно труднее, чем нитрование толуола,
но, применяя достаточно жесткие условия,
удается получить хороший выход продукта:
Таким образом, применяя однотипные реакции, но в разной последовательности, удается получить различные целевые продукты.
Пример 2. Как из бензола получить н-бутилбензол?
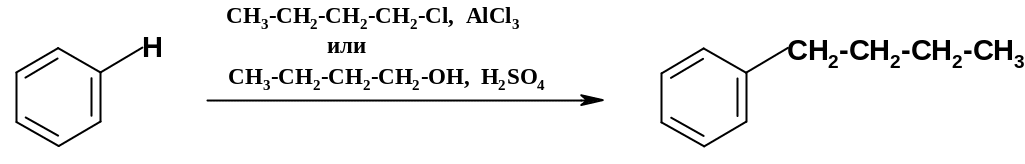 «На бумаге» ответ
кажется очевидным – алкилирование
бензола по Фриделю-Крафтсу с использованием
1-галогенбутана или 1-бутанола:
«На бумаге» ответ
кажется очевидным – алкилирование
бензола по Фриделю-Крафтсу с использованием
1-галогенбутана или 1-бутанола:
Действительно, таким путем можно получить желаемый продукт. Однако этот вариант связан по крайней мере с двумя сложностями: А. н-Бутильная группа (как и все алкильные группы) является электронодонорной (пусть и в небольшой степени); она ускоряет реакции электрофильного замещения, поэтому вторая н-бутильная группа должна входить в ядро легче, чем первая; реакция имеет тенденцию не останавливаться на стадии образования монозамещенного бензола, а идти дальше. Чтобы главным продуктом реакции все же было монозамещенное производное, необходимо брать заметный избыток бензола. Б. Другим осложнением является изомеризация электрофильной частицы при алкилировании по Фриделю-Крафтсу – карбокатиона: первичный карбокатион склонен перегруппировываться во вторичный; это приводит к получению не целевого продукта, а его изомера – втор-бутилбензола:
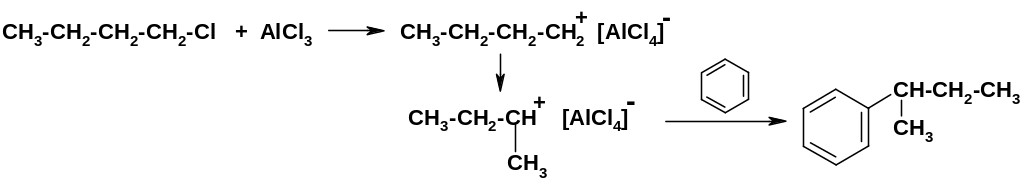 [Об
изомеризации карбокатионов упоминалось
в разделе «Алканы»; более подробно она
будет рассмотрена позднее].
[Об
изомеризации карбокатионов упоминалось
в разделе «Алканы»; более подробно она
будет рассмотрена позднее].
Обе эти трудности можно обойти, если избрать другой путь: ацилирование бензола по Фриделю-Крафтсу хлорангидридом масляной кислоты и последующее восстановление кетона (243) до углеводорода (например, гидразином и щелочью по Вольфу-Кижнеру):
А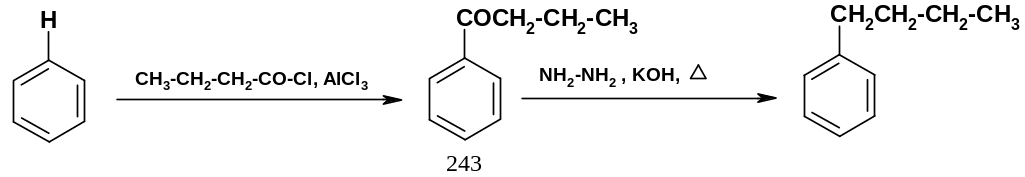 цилирование
идет однозначно, потому что: А. Ацильная
группа (точнее ее ядро – карбонильная
группа) – акцептор,
затрудняющий введение второй ацильной
группы, поэтому реакция останавливается
на образовании монозамещенного
производного; Б. Ацил-катион не
изомеризуется. Восстановление кетона
также протекает с хорошим выходом.
цилирование
идет однозначно, потому что: А. Ацильная
группа (точнее ее ядро – карбонильная
группа) – акцептор,
затрудняющий введение второй ацильной
группы, поэтому реакция останавливается
на образовании монозамещенного
производного; Б. Ацил-катион не
изомеризуется. Восстановление кетона
также протекает с хорошим выходом.
Таким образом, в данном случае проигрыш в числе стадий вполне компенсируется чистотой их протекания, и «длинный» путь оказывается в какой-то степени предпочтительнее «короткого».
Реакции с нуклеофилами для самих аренов не характерны. Однако они легко протекают с некоторыми их функциональными производными; подобные реакции будут рассмотрены в дальнейшем.
I.6.2.2.Реакции присоединения к ароматическому ядру.
Как уже упоминалось, арены обладают пониженной склонностью к реакциям присоединения (это энергетически невыгодно); тем не менее, такие реакции известны.
1. Присоединение хлора. При взаимодействии бензола с хлором при интенсивном освещении (на солнечном свету или лучше при УФ-облучении) происходит присоединение трех молекул хлора с образованием гексахлорциклогексана (244). Реакция, как и многие другие, протекающие при освещении, имеет радикальный характер:
П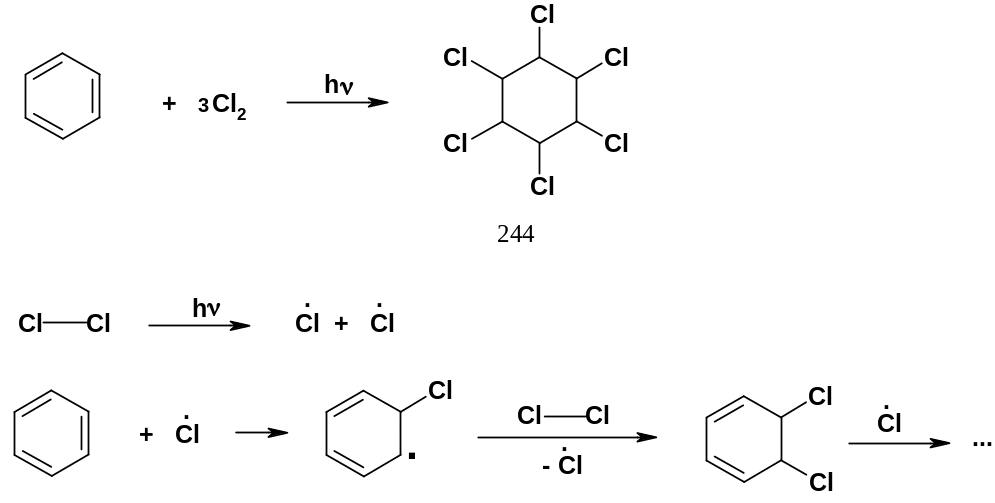 родукт
(244) получается в виде смеси нескольких
стереоизомеров. Ранее он использовался
в сельском хозяйстве в качестве
инсектицида под названием «гексахлоран»;
наряду с известным ДДТ этот инсектицид
оказался одним из наиболее известных
загрязнителей окружающей среды ввиду
своей токсичности и в еще большей мере
– своей стойкости.
родукт
(244) получается в виде смеси нескольких
стереоизомеров. Ранее он использовался
в сельском хозяйстве в качестве
инсектицида под названием «гексахлоран»;
наряду с известным ДДТ этот инсектицид
оказался одним из наиболее известных
загрязнителей окружающей среды ввиду
своей токсичности и в еще большей мере
– своей стойкости.
2. Диеновый синтез. Формально ароматические ядра содержат 1,3-диеновые системы; для последних характерна реакция диенового синтеза. Бензол и его гомологи вступают в эту реакцию чрезвычайно трудно, поскольку они наиболее устойчивы, а реакция диенового синтеза нарушает ароматическую структуру. Нафталин и его гомологи, где делокализация не столь совершенная, как в бензоле, вступают в эту реакцию легче производных бензола, но также достаточно трудно. Антрацен, напротив, хорошо вступает в реакцию Дильса-Альдера с разнообразными диенофилами; в роли 1,3-диеновой системы выступают двойные связи среднего ядра (на схеме выделены); при этом в образующемся аддукте (245) сохраняются два изолированных бензольных ядра, что выгодно энергетически:
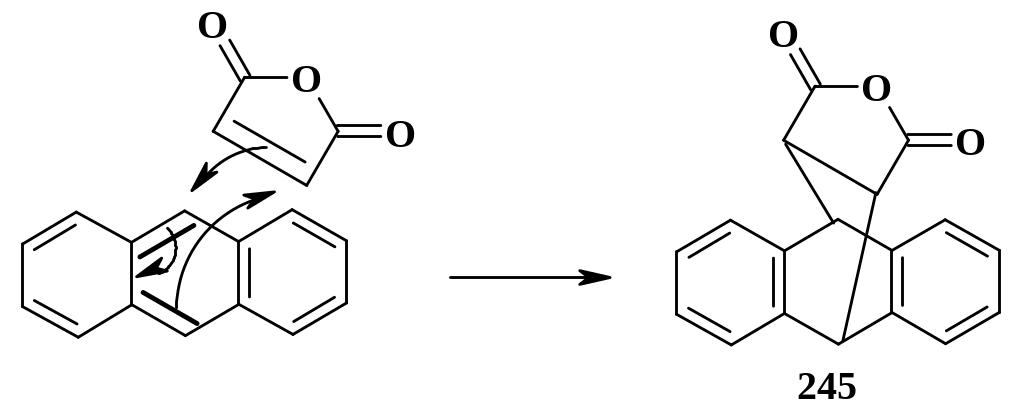 К
реакциям присоединения относятся также
гидрирование и озонирование ароматических
ядер; эти реакции будут рассмотрены в
разделе «окислительно-восстановительные
превращения».
К
реакциям присоединения относятся также
гидрирование и озонирование ароматических
ядер; эти реакции будут рассмотрены в
разделе «окислительно-восстановительные
превращения».
I.6.2.3. Реакции боковых цепей, связанных с ароматическими ядрами.
В боковых цепях, связанных с ароматическими ядрами, особую химическую активность проявляет положение, непосредственно связанное с бензольным ядром; оно называется бензильным положением. Причина – активация бензильного положения ароматическим ядром; эта активация происходит потому, что ароматическое ядро стабилизирует переходные состояния и интермедиаты реакций по бензильному положению за счет сопряжения. Такая активация наблюдается для многих типов реакций в зависимости от того, какие атомы связаны с бензильным атомом углерода. Для самих аренов, т.е. углеводородов, бензильное положение активировано чаще всего в радикальных реакциях – аналогично аллильному положению в алкенах (стр. 93). Как и для аллильных реакций, здесь образуется резонансно стабилизированный радикал с делокализованным неспаренным электроном:
О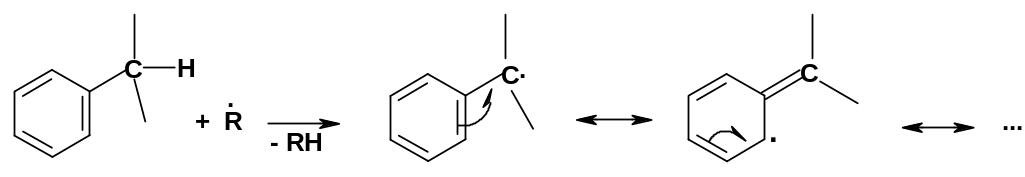 бразование
достаточно устойчивых бензильных
радикалов облегчает протекание
радикальных реакций.
бразование
достаточно устойчивых бензильных
радикалов облегчает протекание
радикальных реакций.
1. Галогенирование бензильного положения.
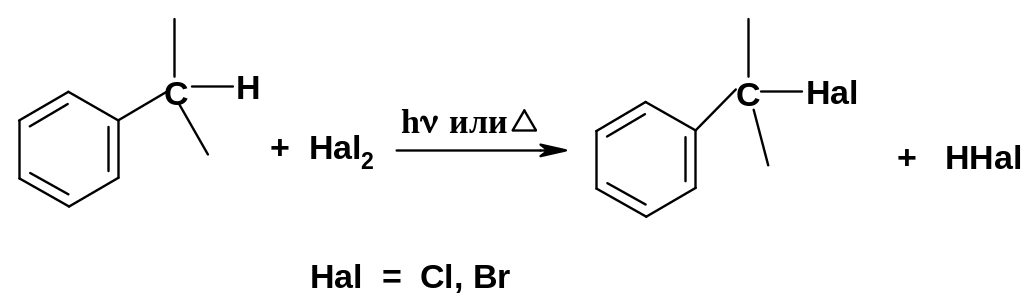 Эту
реакцию часто называют «галогенированием
в боковую цепь», в отличие от ранее
рассмотренного электрофильного
«галогенирования в ядро». Взаимодействие
с галогенами протекает при нагревании
или освещении (но
без катализатора
– Fe
или FeHal3:
катализаторы
направляют галоген в ядро!):
Эту
реакцию часто называют «галогенированием
в боковую цепь», в отличие от ранее
рассмотренного электрофильного
«галогенирования в ядро». Взаимодействие
с галогенами протекает при нагревании
или освещении (но
без катализатора
– Fe
или FeHal3:
катализаторы
направляют галоген в ядро!):
Механизм этой реакции ничем не отличается от механизма галогенирования алканов (стр. 72), но реакции идут значительно легче, чем для алканов. Поэтому, если боковые цепи содержат более одного атома углерода, галогенирование идет региоселективно – преимущественно по бензильному положению, например, при хлорировании р-диэтилбензола хлорируются два бензильных положения; образуется дихлорпроизводное (246):
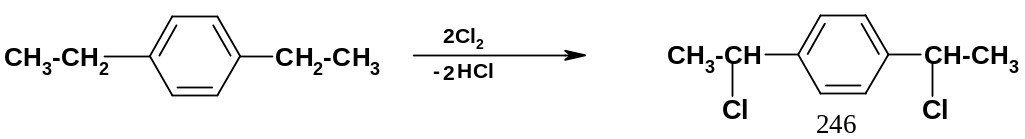 При
действии избытка галогена (в первую
очередь, хлора) можно заместить все
бензильные атомы водорода; например,
из толуола можно получить бензилхлорид
(247), бензилиденхлорид (248) и бензотрихлорид
(249):
При
действии избытка галогена (в первую
очередь, хлора) можно заместить все
бензильные атомы водорода; например,
из толуола можно получить бензилхлорид
(247), бензилиденхлорид (248) и бензотрихлорид
(249):
![]() Для
бензильного галогенирования можно
также использовать «аллильный»
галогенирующий реагент -–NBS
в присутствии пероксидов (стр. 93).
Для
бензильного галогенирования можно
также использовать «аллильный»
галогенирующий реагент -–NBS
в присутствии пероксидов (стр. 93).
2. Образование бензильных гидропероксидов.
Э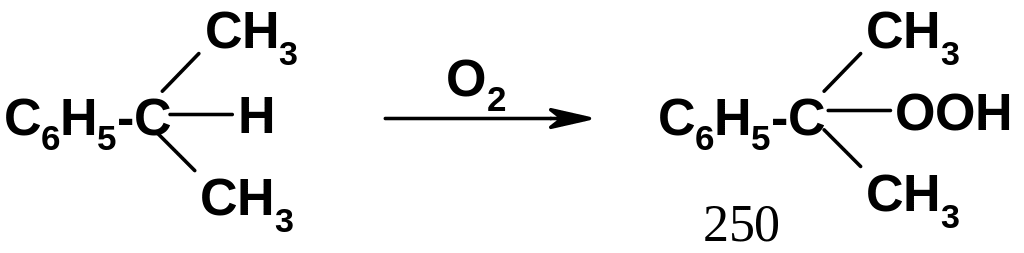 то
–один из вариантов окисления боковых
цепей аренов. Наиболее известный пример
– окисление изопропилбензола (кумола)
кислородом воздуха:
то
–один из вариантов окисления боковых
цепей аренов. Наиболее известный пример
– окисление изопропилбензола (кумола)
кислородом воздуха:
Механизм реакции радикальный; он вполне аналогичен механизму окисления алканов (стр.75), но реакция идет значительно легче. Образующийся при этом гидропероксид кумола (250) является полупродуктом в промышленном синтезе фенола и ацетона (будет рассмотрено в дальнейшем).
Другие реакции окисления боковых цепей будут рассмотрены при описании окислительно-восстановительных превращений аренов.
Стабильные триарилметильные радикалы.
 Как
отмечалось выше, бензильные радикалы
имеют повышенную устойчивость. Если же
радикальный центр связан с тремя
ароматическими ядрами, устойчивость
радикалов возрастает настолько, что
они способны к длительному существованию,
т.е. становятся долгоживущими.
Простейшим устойчивым триарилметильным
радикалом является трифе- нилметильный
радикал (интермедиат при галогенировании
и окислении трифенилметана):
Как
отмечалось выше, бензильные радикалы
имеют повышенную устойчивость. Если же
радикальный центр связан с тремя
ароматическими ядрами, устойчивость
радикалов возрастает настолько, что
они способны к длительному существованию,
т.е. становятся долгоживущими.
Простейшим устойчивым триарилметильным
радикалом является трифе- нилметильный
радикал (интермедиат при галогенировании
и окислении трифенилметана):
Этот радикал впервые был получен американским химиком М.Гомбергом в 1900 г. при действии серебра на трифенилхлорметан; он явился первым стабильным радикалом. В растворах этот радикал способен к длительному существованию и находится в равновесии со своим димером (продуктом рекомбинации). Некоторые триарилметильные радикалы в растворах вообще не димеризуются. Столь высокая стабильность обусловлена во-первых сопряжением радикального центра с тремя бензольными ядрами, т.е. делокализацией неспаренного электрона по девяти положениям этих ядер; во-вторых, пространственным экранированием радикального центра, что затрудняет рекомбинацию. Роль пространственного экранирования проявляется в том , что при рекомбинации двух трифенилметильных радикалов образуется не гексафенилэтан, а соединение (251):
В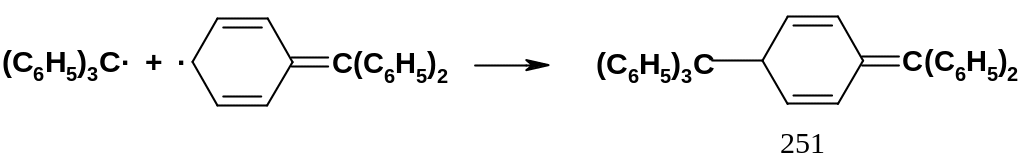 рекомбинации участвует резонансная
структура с неспаренным электроном не
на центральном атоме углерода, а в
пара-положении
одного из ядер – в этом случае структура
менее напряжена, чем гексафенилэтан.
рекомбинации участвует резонансная
структура с неспаренным электроном не
на центральном атоме углерода, а в
пара-положении
одного из ядер – в этом случае структура
менее напряжена, чем гексафенилэтан.
Кроме триарилметильных, известны другие типы стабильных свободных радикалов (триарилгидразильные, нитроксильные и др.)