

2.4. Реальная и химическая энергии сольватации
Предположим,
что металл
М погружен
в раствор соли этого металла и между
электродом и раствором установилось
электрохимическое равновесие.
Между точкой
1 вблизи металла и точкой
2 вблизи раствора (рис. 2.3) существует
измеряемая разность
потенциалов
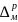 ψ
– так называемый вольта-потенциал
металл/раствор.
ψ
– так называемый вольта-потенциал
металл/раствор.









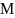 1 Рис.
2.3. Схема для расчета
1 Рис.
2.3. Схема для расчета



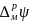 реальной энергии
сольватации
реальной энергии
сольватации




 2
иона
2
иона




 χ
χ

 3
3
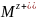
Испарим
1 моль
атомов металла,
что потребует
затраты энергии
 Ионизируем
эти атомы
в точке 1 вблизи поверхности
металла, затратив
на это
работу
Ионизируем
эти атомы
в точке 1 вблизи поверхности
металла, затратив
на это
работу
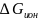 .
Образовавшиеся в результате ионизации
электроны
введем
обратно в металл.
Полученный за счет этого выигрыш
энергии
составит
z
.
Образовавшиеся в результате ионизации
электроны
введем
обратно в металл.
Полученный за счет этого выигрыш
энергии
составит
z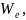 где
где
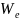 –
работа
выхода электрона
из металла. Ионы
металла
–
работа
выхода электрона
из металла. Ионы
металла
 из точки 1 переведем
в точку 2. Поскольку между этими точками
существует
разность
потенциалов
,
то при этом будет затрачена
работа
из точки 1 переведем
в точку 2. Поскольку между этими точками
существует
разность
потенциалов
,
то при этом будет затрачена
работа
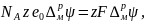 где F
=
где F
= – число Фарадея.
– число Фарадея.
Далее ионы
металла внесем
в раствор.
Соот-ветствующий этому процессу
энергетический
эффект
равен
так называемой реальной
энергии сольватации
ионов
–
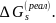 Для заверше-ния цикла перенесем
ионы
из
раствора в металл, что в условиях
равновесия
не требует совершения
работы.
По закону Гесса,
Для заверше-ния цикла перенесем
ионы
из
раствора в металл, что в условиях
равновесия
не требует совершения
работы.
По закону Гесса,

и, следовательно,
 (2.4.1)
(2.4.1)
Все величины
в правой части уравнения (2.4.1) доступны
экспериментальному определе-нию,
и это открывает
путь реальной энергии
сольватации
отдельного
вида ионов.
Идея этого метода нахождения
 была выдвинута
А.Н. Фрумкиным. В табл. 2.4 приведены
значе-ния
для
некоторых ионов, рассчитанные по наиболее
надежным данным Дж. Рэндлса.
была выдвинута
А.Н. Фрумкиным. В табл. 2.4 приведены
значе-ния
для
некоторых ионов, рассчитанные по наиболее
надежным данным Дж. Рэндлса.
В литературе используется также приведен-ная шкала, в которой за ноль принимаются термодинамические величины для иона водоро-да. Расчеты любых энергий в такой шкале основаны на последовательном рассмотрении сольватации кислот и солей. Например, из свободной энергии (или энтальпии) для HCl определяют парциальные величины для хлорид-иона. Затем используют их в комбинации с данными для NaCl и определяют таким образом парциальные величины для катиона натрия и т.д.
Таблица 2.4.
Свободные
энергии Гиббса гидратации для некоторых
ионов, рассчитанные по уравнению (2.4.1)
и (в скобках) полученные в приведенной
шкале с использование масс-спектрометрических
данных для иона
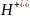
Катион |
|
|
|
|
|
|
– |
1089 (0) |
511 (–575) |
411 (–680) |
337 (–752) |
316 (–775) |
284 (–798) |

 кДж/моль
кДж/моль
Анион |
|
|
|
|
– кДж/моль |
414 (–675) |
296 (–800) |
271 (–826) |
239 (–861) |
Выбор в качестве
точки
отсчета именно
иона водорода определяется
тем обстоятельством, что для него
возможно относительно точное
экспе-риментальное определение
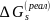 методом
масс-спектрометрии. Оно основано
на измерении
сво-бодных
энергий
и теплот
образования кластеров
методом
масс-спектрометрии. Оно основано
на измерении
сво-бодных
энергий
и теплот
образования кластеров
 (n
от 1 до 6) и последующей экстрапо-ляции
к n
(n
от 1 до 6) и последующей экстрапо-ляции
к n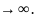 C
использованием этой величины приведенная
шкала
может быть трансформиро-вана
в абсолютную.
Соответствующие числен-ные значения
(в скобках) сопоставлены в табл. 2.4 с
результатами расчетов по уравнению
(2.4.1) Величина
,
полученная экстраполяцией
масс-спектрометрических данных для
(1104 кДж/моль), находится
в хорошем согласии с независимым
определением
работы
выхода про-тона
из разбавленных растворов (1096 кДж/моль)
C
использованием этой величины приведенная
шкала
может быть трансформиро-вана
в абсолютную.
Соответствующие числен-ные значения
(в скобках) сопоставлены в табл. 2.4 с
результатами расчетов по уравнению
(2.4.1) Величина
,
полученная экстраполяцией
масс-спектрометрических данных для
(1104 кДж/моль), находится
в хорошем согласии с независимым
определением
работы
выхода про-тона
из разбавленных растворов (1096 кДж/моль)
На границе
воздух/раствор из-за определен-ной
ориентации
диполей
растворителя возникает
некоторый скачок
потенциала χ
– так называ-емый поверхностный
потенциал.
Поэтому энер-гетический эффект,
сопровождающий перенос заряженной
частицы
через
границу воздух/ра-створ (из точки 2 в
точку 3 на рис. 2.3), отражает
не только ион-дипольное взаимодействие,
но и электрическую работу,
которая для моля i-х
ионов
равна
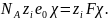 Изменение
свободной энергии,
обусловленное
только взаи-модействием
ионов с
диполями растворителя и
отнесенное
к молю ионов, называется
химиче-ской
энергией сольватации
Изменение
свободной энергии,
обусловленное
только взаи-модействием
ионов с
диполями растворителя и
отнесенное
к молю ионов, называется
химиче-ской
энергией сольватации
 .
Таким образом, реальная и химическая
энергии
сольватации связаны
соотношением
.
Таким образом, реальная и химическая
энергии
сольватации связаны
соотношением
=
+
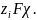 (2.4.2)
(2.4.2)
Экспериментально
можно
определить только
;
величины
и
 опытному
опреде-лению недоступны.
С другой стороны, все моде-льные методы
расчета дают
,
и, следова-тельно,
строгая
экспериментальная проверка
этих моделей невозможна.
Поэтому необходимо
добиться
согласованности результатов
в рамках различных модельных расчетов.
Один из спосо-бов
такого согласования
обычно основывается
на чисто модельном расчете
химической энергии
гидратации
иона. Другой
способ
состоит в разложении
экспериментальной величины
на
два
слагаемых,
причем правило,
по которому производится
такое разложение, осно-вывается
на другом модельном предположении.
Так, например,
в методе Бернала и Фаулера принимается,
что для ионов
опытному
опреде-лению недоступны.
С другой стороны, все моде-льные методы
расчета дают
,
и, следова-тельно,
строгая
экспериментальная проверка
этих моделей невозможна.
Поэтому необходимо
добиться
согласованности результатов
в рамках различных модельных расчетов.
Один из спосо-бов
такого согласования
обычно основывается
на чисто модельном расчете
химической энергии
гидратации
иона. Другой
способ
состоит в разложении
экспериментальной величины
на
два
слагаемых,
причем правило,
по которому производится
такое разложение, осно-вывается
на другом модельном предположении.
Так, например,
в методе Бернала и Фаулера принимается,
что для ионов
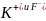
 =
=

поскольку эти ионы имеют приблизительно оди-наковые радиусы (см. табл. 2.2). Однако величи-ны для одного и того же иона, рассчи-танные разными методами, иногда различаются более чем на 100 кДж/моль. При постоянной и известной в соответствии с уравнением (2.4.2) это отвечает различиям в поверностных потенциалах, превышающим 1 В.
Эти различия
значительно превосходят
воз-можные ошибки
современных модельных оце-нок
χ.
Такие
оценки
для водных растворов показывают,
что χ
= +0,1 В. Это означают, что диполи
воды преимущественно
ориентированы
атомами
кислорода
в направлении воздушной фазы. Поэтому
в настоящее время наиболее точные
значения
можно рассчитать
по уравнению (2.4.2), используя
опытные
и значения χ,
полученные
на основе модельных оценок.
Для водных
растворов при
условии, что χ
 кДж/моль,
где
кДж/моль,
где
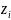 учитыва-ет
не только величину,
но и знак
заряда иона. Используя
данные
Рэндлса, табл. 2.4 и уравнение (2.4.2), можно
построить зависимость
от обратного кристаллографического
радиуса
кати-онов и анионов (рис. 2.4). В качественном
сог-ласии с моделью Борна, химическая
энергия
гид-ратации ионов
падает
с ростом радиуса
ионов.
Однако при одинаковых
размерах катионов
и анионов последние гидратируются
сильнее.
Это может быть объяснено
асимметрией
диполей
воды,
благодаря которой положительный
конец диполя
ближе
подходит
к центру
катиона (К.П.Мищенко). Эффект
асимметрии все же не
столь значителен,
чтобы можно было приравнять
химические энергии
гидратации
и
,
как это
предполагалось в одном из методов
разделения величин
учитыва-ет
не только величину,
но и знак
заряда иона. Используя
данные
Рэндлса, табл. 2.4 и уравнение (2.4.2), можно
построить зависимость
от обратного кристаллографического
радиуса
кати-онов и анионов (рис. 2.4). В качественном
сог-ласии с моделью Борна, химическая
энергия
гид-ратации ионов
падает
с ростом радиуса
ионов.
Однако при одинаковых
размерах катионов
и анионов последние гидратируются
сильнее.
Это может быть объяснено
асимметрией
диполей
воды,
благодаря которой положительный
конец диполя
ближе
подходит
к центру
катиона (К.П.Мищенко). Эффект
асимметрии все же не
столь значителен,
чтобы можно было приравнять
химические энергии
гидратации
и
,
как это
предполагалось в одном из методов
разделения величин
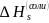 на
и
.
Для более стро-гого учета
эффекта асимметрии
в современных
на
и
.
Для более стро-гого учета
эффекта асимметрии
в современных
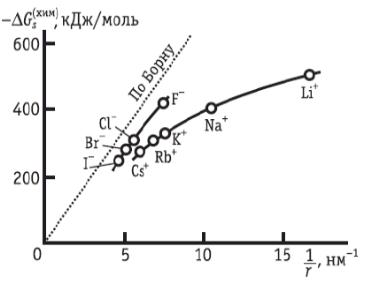
Рис. 2.4. Зависимость химической энергии
гидратации от обратного радиуса иона
моделях молекулы воды рассматривают не как диполи, а как квадруполи. Таким образом, проблема разделения общих термодинамических величин, характеризующих процесс растворения, требует использования нетермодинамических (микроскопических) предположений.